

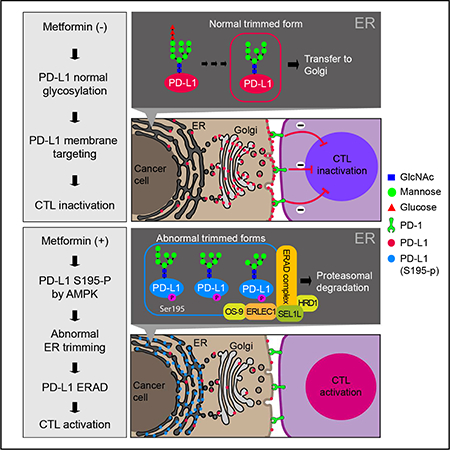
Summary
Metformin has been reported to possess antitumor activity and maintain high cytotoxic T lymphocyte (CTL) immune surveillance. However, the functions and detailed mechanisms of metformin’s role in cancer immunity are not fully understood. Here we show metformin increases CTL activity by reducing the stability and membrane localization of programmed death ligand-1 (PD-L1). Furthermore, we discover that AMP-activated protein kinase (AMPK) activated by metformin directly phosphorylates S195 of PD-L1. S195 phosphorylation induces abnormal PD-L1 glycosylation, resulting in its ER accumulation and ER-associated degradation (ERAD). Consistently, tumor tissues from metformin-treated breast cancer patients exhibit reduced PD-L1 levels with AMPK activation. Blocking the inhibitory signal of PD-L1 by metformin enhances CTL activity against cancer cells. Our findings identify a new regulatory mechanism of PD-L1 expression through the ERAD pathway and suggest that the metformin-CTLA4 blockade combination has the potential to increase the efficacy of immunotherapy.
eTOC blurb
Cha et al. elucidated a mechanism that metformin-activated AMPK phosphorylates PD-L1 at S195 to induce abnormal glycosylation and degrades PD-L1 through ERAD pathway. This study suggests the potential to use metformin as an adjuvant with various non-PD-L1/PD-1 targeting immune therapies.
Graphical Abstract
Introduction
Metformin is a widely used oral medication to treat type 2 diabetes (T2D) and recognized as a safe and well-tolerated drug from several decades of clinical experience. In addition, metformin has been reported to exert antitumor effects as several case-control studies of T2D patients showed metformin treatment reduced the incidence of various cancer types (Evans et al., 2005; Viollet et al., 2012). Moreover, metformin also maintains high cytotoxic T lymphocyte (CTL) activity in tumor tissues (Eikawa et al., 2015). Those findings imply that the antitumor effects of metformin may play a role in immune response against tumor progression.
During the process of immune editing, cancer cells cleverly escape immune surveillance by manipulating immune checkpoint molecules, which are critical to maintaining a balanced immune activity to prevent autoimmunity and limit collateral tissue damage (Watanabe and Nakajima, 2012). Programmed death ligand-1 (PD-L1) is a critical immune checkpoint molecule exploited by cancer cells to escape immune surveillance (Pardoll, 2012). When PD-L1 on cancer cells and macrophages binds to programmed cell death protein-1 (PD-1) on activated CTLs infiltrating into tumors, PD-L1-induced inhibitory signal shuts down their antitumor activity (Chen, 2004). Furthermore, PD-L1 is expressed at relatively much higher levels in tumor tissues compared with normal tissues (Topalian et al., 2016). Thus, blocking the PD-L1/PD-1 axis is recognized as the attractive target for cancer immunotherapy (Chen and Han, 2015).
Glycosylation is an important post-translational modification, which facilitates the folding (Shental-Bechor and Levy, 2008), intracellular transport (Vagin et al., 2009), and function of immunogenic glycoproteins (Wolfert and Boons, 2013). Specifically, N-glycosylation of PD-L1 was recently demonstrated to be essential for its function (Li et al., 2016). This type of modification is a stepwise reaction carried out from the endoplasmic reticulum (ER) to the Golgi apparatus. Normally, the precursor glycan (Glc3Man9GlcNAc2) covalently attached to NXT motif (-Asn-X-Ser/Thr-) of glycoprotein is trimmed to Man8GlcNAc2 in the ER followed by glycan remodeling in the Golgi (Breitling and Aebi, 2013). However, when the protein is misfolded or when abnormal glycan structure is present, the ER α−1,2 mannosidase I (ERMan I), ER degradation-enhancing α-mannosidase-like protein 1 (EDEM1), EDEM2, and EDEM3 inordinately remove mannose from Man9GlcNAc2, resulting in Man7–5GlcNAc2 that is recognized by the ERAD complex (Ferris et al., 2014; Xu and Ng, 2015). The ERAD complex consists of various components for substrate recognition, ubiquitination, and retro-translocation (Xu and Ng, 2015). N-linked glycoproteins with abnormal Man7–5GlcNAc2 are recognized by the ERAD substrate recognition factors, OS-9 and ERLEC1, and ERAD E3 ligase, such as HRD1, is recruited to the substrate via the adaptor protein SEL1L (Vembar and Brodsky, 2008; Xu and Ng, 2015). Following poly-ubiquitination by SEL1L/HRD1, the substrate is retro-translocated from the ER into the cytoplasm and subsequently degraded by the cytoplasmic proteasome (Sun et al., 2015; van den Boomen and Lehner, 2015).
Previous diabetes studies indicated that the AMP-activated protein kinase (AMPK) is a main effector of metformin (Zhou et al., 2001), and that metformin may indirectly activate AMPK by inducing stress conditions, such as mitochondrial malfunction that depletes ATP (Hawley et al., 2010). AMPK is a heterotrimeric Ser/Thr kinase consisting of one catalytic (α) and two regulatory (β and γ) subunits and functions as a key regulator of cellular metabolism and energy homeostasis (Hardie et al., 2016). Under various stress conditions consuming ATP, several upstream kinases activate AMPK via phosphorylation of Thr172 in the activation loop of its αsubunit. In addition, AMPK is known to suppress tumor progression (Kuhajda, 2008), implying that the antitumor effect of metformin may be mediated by AMPK signaling.
The detailed molecular mechanism of metformin-mediated antitumor immunity is not well understood. Here we show that AMPK activated by metformin decreases the expression of PD-L1 in the cancer cells by altering PD-L1 glycan structure, which promotes PD-L1 degradation, and subsequently blocking immune inhibitory signaling. We further evaluate the addition of metformin to boost the efficacy of immunotherapy as a potentially effective combination therapy for cancer treatment.
Results
Metformin increases CTL activity through the AMPK/PD-L1 axis
To investigate the role of metformin in antitumor immunity, we compared the antitumor effects of metformin between immunocompetent and severe combined immune deficiency (SCID) mice in a 4T1 breast tumor model (Figure 1A). Interestingly, significant tumor regression was only observed in immunocompetent mice but not those with immune deficiency after one week of metformin treatment (Figure 1B, C), suggesting that the antitumor effect of metformin is closely related to immune response. Because antitumor immunity is accompanied by apoptosis in tumor tissues (Chen et al., 2004), we compared the levels of cleaved caspase 3 (CCA3) and found that although a small number of cells underwent apoptosis in tumor tissues of the control mice, strong clustered apoptotic signals were extensively observed in that of the metformin-treated group (Figure 1D). CD8+CTL, the major effector of antitumor immunity, eliminate cancer cells by secreting granzyme B (GB), an apoptosis trigger (Chowdhury and Lieberman, 2008). Thus, we examined the CD8+CTL population as well as CTL activity by measuring GB release and showed that metformin treatment indeed increased the CD8+ CTL population and GB release by 2.2 (± 0.58) and 3.45 (± 0.81) fold, respectively, compared to control (Figure 1E). Together, these data suggested that metformin increases CTL activity, which may contribute to its antitumor effects.
Figure 1. Metformin increases CTL activity through the AMPK/PD-L1 axis.

(A) 4T1_luc2 cells were injected into mice (n = 6 mice per group) on day 0, and metformin administered as indicated. (B) Tumor volume was measured on the indicated time points. Data represent mean ± SD. (C) Quantification (top) of bioluminescence imaging. Bottom, endpoint images shown (D) Immunostaining of cleaved caspase 3 in the 4T1 tumor mass. Hoechst: nuclear counter staining (pseudo-color: green). Scale bar, 200 μm (inset, 50 μm). (E) Immunostaining of CD8 (CTL marker) and granzyme B (activity of T cell) in the 4T1 tumor mass. Scale bar, 200 μm (inset, 50 μm). For (D) and (E), data represent mean ± SD. n = 12; 3 tissue slides per tumor, 4 mice per group. Unit = 466,038 μm2 in (D) and 262,144 μm2 in (E). (F) T cell-mediated cancer cell killing assay. MDA-MB-231 WT and AMPKα KO cells co-cultured with activated T cell for 48 hr with or without metformin (5 mM) were subjected to crystal violet staining. MDA-MB-231 to T-cell ratio, 1:5. Data represent mean ± SD. n = 3. (G) Correlation analysis between PD-L1 and AMPKα T172-p expression in 14 breast cancer cell lines. (H) BT-549 and MDA-MB-231 were treated with increasing concentrations of metformin (1.25 to 5 mM) for 24 hr. (I) MDA-MB-231 parental, AMPKα WT and KO cells were treated with metformin (5 mM) for 24 hr. Compound C (Com C) AMPK inhibitor was pretreated 6 hr before metformin treatment. (J) Membrane PD-L1 expression by flow cytometric analysis after metformin (5 mM) treatment for 24 hr. (K) Right, quantitation of binding of green fluorescent-labeled PD-1/Fc on MDA-MB-231 treated with or without metformin for 24 hr. Data represent mean ± SD. n = 3. Left, representative images shown. Scale bar, 100 μm. (L) MDA-MB-231 WT and PD-L1 KO cells co-cultured with activated T cells for 48 hr with or without metformin (5 mM) were subjected to crystal violet staining. MDA-MB-231 to T- cell ratio, 1:3.
*P, 0.01~0.05, **P, 0.001~0.01, #P, < 0.001, and NS, not significant, Student’s t test.
Because AMPK is a major effector of metformin in diabetes (Zhou et al., 2001), we speculated that AMPK may also be involved in the increased T-cell activity induced by metformin. Thus, we performed T-cell killing assay using wild-type (WT) and AMPKα knockout (KO) MDA-MB-231 or AMPKα knockdown (KD) BT-549 breast cancer cells (Figure 1F, S1A and S1B). Knocking out AMPKα significantly neutralized metformin-induced T cell-mediated cancer cell death compared with WT cells (Figure 1F and S1B; red highlight). Because cancer cells exploit immune checkpoints to inhibit CTL activity (Pardoll, 2012), we next investigated the association between AMPKα and immune checkpoint proteins. To this end, we found that metformin treatment decreased the expression of PD-L1 among several major checkpoint proteins (Figure S1C). Therefore, we examined the expression and activity level of AMPKα and PD-L1 in a panel of breast cancer cell lines and found they correlated negatively (Figure 1G and S1D). To further validate the association between metformin, AMPKα, and PD-L1, we treated BT-549 and MDA-MB-231 cells with increasing concentrations of metformin. The expression of PD-L1 was reduced with a clear band shift (red asterisk) in a dose- and time-dependent manner whereas the activated form of AMPKα was increased (Figure 1H and S1E). Such reduction in PD-L1 by metformin was also demonstrated in non-small cell lung cancer (H1975 and H358) and colon cancer (RKO) cells (Figure S1F). The addition of a more specific AMPK activator, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), also decreased the levels of PD-L1 with a band shift (Figure S1G, red asterisk). Furthermore, when AMPK was pharmacologically and genetically inhibited, such reduction in PD-L1 expression was not observed (Figure 1I). Functionally, the reduction in PD-L1 expression on the membrane (Figure 1J) led to substantial decrease in PD-1 binding (Figure 1K). Knocking out PD-L1 in the MDA-MB-231 and BT-549 (PD-L1 KO) abrogated metformin-induced T cell-mediated cell killing compared with control (WT) cells (Figure 1L and S1H). Collectively, these results suggested that AMPK activated by metformin decreases PD-L1 levels in cancer cells, which in turn increases CTL activity against cancer cells.
AMPK activated by metformin directly phosphorylates serine 195 of PD-L1
To determine whether AMPK interacts directly with PD-L1, we performed a Duolink (Figure 2A and S2A) and co-immunoprecipitation (IP) assay (Figure 2B) with or without metformin. AMPK-PD-L1 binding peaked at 3 hr after metformin treatment and maintained for more than 24 hr. Consistently, GST pull-down assay showed that AMPK bound to PD-L1 directly (Figure S2B). We also demonstrated that PD-L1 is a substrate of AMPK as AMPK directly phosphorylated PD-L1 by in vitro kinase assay (Figure 2C). The kinetic assay showed that the substrate affinity of PD-L1 to AMPK was higher than that of acetyl-CoA carboxylase (ACC), a known AMPK substrate (Fullerton et al., 2013) (Figure 2D). Based on the reported phospho-motif of AMPK (Hardie et al., 2016), S195 of PD-L1 was predicted as the phospho-site for AMPK (Figure S2C), which was also detected by mass spectrometric analysis (Table S1). Alanine mutation of PD-L1 S195 (S195A; non-phosphorylated form) abrogated its phosphorylation by AMPK (Figure 2E). To further support the existence of PD-L1 S195 phosphorylation (S195-p) in vivo, we generated a specific antibody against PD-L1 S195-p (Figure S2D). Interestingly, to detect S195-p, it was necessary to remove the glycan structure by PNGase F after IP purification (Figure S2E), indicating that the glycan structures near S195 interfered with the binding of the PD-L1 S195-p antibody. Phosphorylation of S195 of wild-type (WT) PD-L1 increased at 2 hr after metformin treatment and peaked around 6 hr. In contrast, no phosphorylation was observed in the S195A mutant (Figure 2F), and knocking out AMPKα abolished S195-p of PD-L1 (Figure 2G).
Figure 2. AMPK activated by metformin directly phosphorylates serine 195 of PD-L1.

(A) BT-549 cells were treated with metformin (5 mM) for the indicated time. Detection of endogenous AMPKα and PD-L1 binding (red dots) by Duolink II assay. Three different positions were randomly selected at each point, and the number of red dots were divided by the number of nuclei. Data represent mean ± SD. n = 3. *P, 0.01~0.05, **P, 0.001~0.01, and #P, < 0.001, Student’s t test. Scale bar, 20 μm. Right, MDA-MB-231 WT, PD-L1 KO and AMPKα KO cells were used as negative controls. Scale bar, 25 μm. (B) MDA-MB-231 cells were cultured for 6 hr with or without metformin (5 mM) and MG132 (10 μM). Endogenous PD-L1 and AMPKα were immunoprecipitated and their binding was analyzed with immunoblotting. (C) In vitro kinase activity of AMPK toward PD-L1 with 32P-labeled ATP. (D) Kinetics of PD-L1 phosphorylation by AMPK. Acetyl-CoA carboxylase (ACC) was used as positive control. Km and Vmax were calculated using the Michaelis-Menten equation. (E) In vitro phosphorylation assay and phospho-tag gel shifting assay. W, PD-L1/WT. A, PD-L1/S195A. (F) PD-L1/S195 phosphorylation was examined using anti-PDL1/S195-p antibody at different time points after metformin (5 mM) treatment. (G) Western blot analysis of MDA-MB-231 WT and AMPKα KO cells after metformin treatment (5 mM) for 8 hr. Endogenous PD-L1 purified by IP was subjected to immunoblotting with PDL1 S195-p antibody after PNGase F reaction. (H) PD-L1 and AMPK subcellular localization of MDA-MB-231 WT and AMPKα KO cells. (I) Trypsin digestion of ER fractions with or without permeabilization. (J, K) BT-549 cells were treated with metformin (5 mM) for 3 hr. (J) BT-549 cells were subjected to Duolink II assay combined with immunofluoresence staining using markers for ER (HSP90B1), Golgi (TNG46), and nuclei (Hoechst). Scale bar, 20 μm (inset, 10 μm). (K) Duolink assay with antibodies specific for ECD (Ab205921 and 86744S) and ICD (13684S and GTX104763) of PD-L1. Scale bar, 50 μm (inset, 25 μm). Red dots: AMPK-PD-L1 binding in (J) and (K).
Since Ser 195 is present on the extracellular domain (ECD), its phosphorylation by AMPK should occur in the ER or/and Golgi lumen. PD-L1 is located in the ER, Golgi, cytoplasmic membrane, and nucleus (Satelli et al., 2016), and AMPK is known to be present in the ER, Golgi, cytoplasm and nucleus (Qi et al., 2008). Moreover, a recent study reported high AMPK activity in the ER and Golgi (Miyamoto et al., 2015). Consistently, cellular fractionation (Figure 2H) also indicated that AMPK is located in the ER region. To further determine whether AMPK resides in the ER lumen, we carried out trypsin digestion of the ER fractions with or without pre-permeabilization and then examined the levels of AMPK, ER transmembrane protein IRE1α (cytosolic part), and ER lumen protein HSP90B1 using the indicated antibodies (Figure S2F). In the permeable fraction, no signals were detected with antibodies against both cytosolic and luminal proteins after trypsinization (Figure 2I, Triton X100 (+) and S2F, panel b). In the non-permeable fraction, signal for the cytosolic domain of IRE1α were rapidly reduced after trypsinization, whereas signals for AMPK and HSP90B1 were maintained (Figure 2I, Triton X100 (−) and S2F, panel c). These results strongly supported localization of AMPK inside the ER lumen.
Data from Duolink® assay indicated that the binding between PD-L1 and AMPK (Duo: red) occurred in the ER (HSP90B1; green), Golgi (TNG46; magenta), and nucleus (Hoechst; blue) (Figure 2J). To further validate the binding between PD-L1 and AMPK on the luminal side, we performed Duolink assay (AMPK/PD-L1) with antibodies specific for ECD (86744S and ab205921) and the intracellular domain (ICD) (13684S and GTX104763) of PD-L1. Prior to detection of PD-L1/AMPK interaction by Duolink assay, these four antibodies were first verified for IF staining (Figure S2G) as control in parental BT-549 and PD-L1 KO BT-549 cells. Our results indicated that the antibodies against ECD, but not those against ICD, exhibited strong Duolink signals (Figure 2K) which are co-localized with ER lumen marker HSP90B1 (Figure S2H; white arrow), suggesting that PD-L1 binds to AMPK primarily on the luminal side of ER (Figure S2I). Together, these results indicated that metformin-activated AMPK locates in the lumen of ER and directly phosphorylates PD-L1 S195.
Phosphorylation of PD-L1 S195 induces its abnormal glycosylation and blocks its ER-to-Golgi translocation
To validate the function of S195-p, we generated BT-549 and MDA-MB-231 stable cell lines expressing WT, S195A (non-phosphomimetic), S195D (phosphomimetic), or S195E (phosphomimetic) PD-L1. To exclude endogenous signals from PD-L1, we knocked down endogenous PD-L1 in these stable cell lines (Figure S3A, Left). Interestingly, while bands of similar size representing WT and S195A PD-L1 were detected, the S195E and S195D mutant showed a significant band shift (Figure 3A and S3A, Right_red asterisk) similar to that in the presence of metformin (Figure 1H and S1E_red asterisk). PD-L1 contains four N-glycosylation sites, N35, N192, N200, and N219 (Li et al., 2016) (Figure 3B), and is heavily glycosylated (Figure 3A, WT PD-L1 vs 4NQ PD-L1_non-glycosylated form). Based on the reduction in size, this band shifting may be attributed to the changes in the glycan structure. Indeed, when cells were treated with glycosylation inhibitor, tunicamycin, protein bands corresponding to WT, S195A, S195D, and S195E PD-L1 shifted down to a size similar to the non-glycosylated form (4NQ; red arrow), and none of the bands was below the size of 4NQ (Figure. 3D). These results indicated that the difference in molecular weight between S195D and S195E PD-L1, and WT and S195A PD-L1 was due to alterations in the glycan structure. To validate this, we performed mass spectrometric analyses and showed that the glycan structure of the S195E mutant PD-L1 was different from that of WT PD-L1. Diverse glycan structures consisting of various saccharides, e.g., fucose, galactose, N-acetylneuraminic acid, and N-acetylglucosamine, can be found on the WT PD-L1, but only the mannose-rich structures (M5, M6, M7, M8 and M9) were detected at the four glycosylation sites on S195E (Figure 3C, S3B and Table S2). Such mannose-rich structures are attributed to excessive ER mannose trimming (Ferris et al., 2014; Xu and Ng, 2015), and S195-p is expected to occur before ER trimming. Metformin treatment caused a band shift of WT PD-L1, but not S195A, to a size similar to that of the S195E mutant (red asterisk) (Figure 3D). These results suggested that metformin-activated AMPK directly phosphorylates PD-L1 at S195, which induces abnormal ER mannose trimming during glycosylation.
Figure 3. Phosphorylation of PD-L1 S195 induces its abnormal glycosylation and blocks its ER-to-Golgi translocation.

(A) WT, S195A S195D, S195E, and 4NQ PD-L1 stable cells were treated with or without tunicamycin (5 μg/ml) for 24 hr. (B) Schematic diagram of PD-L1 showing the position of S195 and the 4 N-glycosylation sites. (C) Comparison of the glycan structure between WT and S195E PD-L1 by IP/Mass analysis. (D) BT-549 and MDA-MB-231 stable cells expressing WT, S195E, or S195A PD-L1 were treated with metformin (5 mM) for 24 hr. (E) Expression pattern of PD-L1 in MDA-MB-231 WT, S195A and S195E PD-L1 stable cells by IF staining. (F) MDA-MB-231 stable cells co-stained with antibodies against PD-L1 and Golgi markers (GM130: cis, Giantin: medial, TNG46: trans). (G) IF staining with antibodies against PD-L1 and ER marker (HSP90B1) (H) Flow cytometric analysis of membrane PD-L1 in MDA-MB-231 WT, S195A and S195E PD-L1 stable cells. Data represent mean ± SD. n = 3. (I) Binding of green fluorescent-labeled PD-1/Fc to MDA-MB-231 WT, S195A and S195E PD-L1 stable cells was quantified. Data represent mean ± SD. n = 3. (J) PD-L1 localization in MDA-MB-231 expressing WT, S195E or NXT motif mutant (glycosylation site mutant) PD-L1 by IF staining. For experiments shown in (E), (F), (G) and (J), MG132 (10 μM) was added 6 hr prior to fixation to prevent degradation of PD-L1. Hoechst: nuclear counter staining. Scale bar, 20 μm (inset, 10 μm). *P, 0.01~0.05, **P, 0.001~0.01, and #P, < 0.001, Student’s t test. NS, not significant.
During intracellular transport of membrane glycoproteins, specific glycosylation in one compartment is closely related to the cargo transfer to the next (Vagin et al., 2009). Since S195-p induces abnormal glycosylation of PD-L1, we asked whether S195-p by metformin affects localization of PD-L1 in the cells. To this end, we performed IF staining of WT, S195A and S195E PD-L1 stable cell lines. Both WT and S195A PD-L1 were located primarily on the cell membrane (white arrowheads) and within the perinuclear cisternae (red arrowheads) (Figure 3E; see Figure 3F, G for more details). In contrast, S195E PD-L1 was detected throughout the cytosol with reduced signals on the membrane (Figure 3E; yellow arrow). Co-staining of PD-L1 (white; 1 and 5) and three Golgi markers (2 and 6), GM130 (cis; blue), giantin (medial; green), and TNG46 (trans; magenta), showed that WT (3 and 4) but not S195E (7 and 8) PD-L1 colocalized with the Golgi markers (Figure. 3F). In contrast, co-staining of PD-L1 (red; 1 and 5) and the ER marker HSP90B1 (green; 2 and 6) indicated that S195E (7 and 8) but not WT (3 and 4) PD-L1 co-localized with the ER (Figure 3G). These data suggested that S195-p induces ER accumulation of PD-L1 by blocking ER-to-Golgi translocation. Flow cytometric analysis also showed significantly lower cell membrane PD-L1 in S195E than in WT or S195A (Figure 3H) PD-L1, which led to decreased binding with PD-1 (Figure 3I).
We next mutated the NXT motif to determine the glycosylation sites of PD-L1 affected by S195-p (Figure S3C). Individual PD-L1 mutations (N35Q, N192Q, N200Q and N219Q) did not affect subcellular localization of PD-L1 compared with the WT protein (Figure 3J, WT and NXT single mutants). In contrast, mutation at all four sites exhibited the same ER accumulation as did the S195E mutant (Figure 3J, 4NQ and S195E), implying that only severe abnormality in the collective glycan structure from S195-p leads to ER accumulation of PD-L1.
To validate the role of PD-L1 S195-p on the antitumor effect of metformin, stable cells expressing WT, S195A, or S195E PD-L1 were treated with metformin and assayed to examine PD-L1 localization (Figure S3D) and its activity in T-cell inhibition (Figure S3E, S3F and S3G). In the presence of metformin, WT PD-L1 accumulated in the ER similar to that observed for the S195E mutant (Figure S3D, WT and WT + Metf). ER localization of the S195A mutant, however, was not affected by metformin as shown by IF staining (Figure S3D, S195A and S195A + Metf). Functionally, the S195E PD-L1 stable cells induced higher T cell-mediated cytolytic activity compared with those expressing WT or S195A PD-L1 (Figure S3E, S3F and S3G). Although metformin increased T cell-mediated killing of both WT and S195A stable cells, the reduction in cell survival of S195A stable cells was relatively less than that of WT PD-L1 (Figure S3F and S3G, red bars). Together, these data suggested that S195-P by metformin prevents the translocation of PD-L1 to the cell membrane and subsequent PD-1 binding, and therefore, increasing the activity of CTL.
Phosphorylation of S195 induces ER-associated degradation of PD-L1
The above findings indicated that PD-L1 S195-p induces a mannose-rich glycan structure and ER accumulation (Figure 3), which are common characteristics of ERAD substrates (Ferris et al., 2014; Vembar and Brodsky, 2008). We compared the binding partners of S195A and S195E PD-L1 by immunoprecipitation/mass spectrometry (IP-MS) analysis and found that eight of the top 10 candidates with increased binding to S195E were ERAD components (Figure 4A). Ingenuity pathway analysis based on this mass data showed the binding of PD-L1 with components of unfolding protein response (UPR) and ubiquitination pathway was increased by S195-p (Figure 4B). In addition, although the exogenous PD-L1 protein level driven by the CMV promoter was decreased by metformin (Figure S4A), the endogenous RNA level of PD-L1 was not significantly changed (Figure S4B). These results strongly suggested that the reduction in PD-L1 level via metformin-induced S195-p is associated with the ERAD pathway.
Figure 4. Phosphorylation of S195 induces ER-associated degradation of PD-L1.

(A) IP-MS analysis showing candidates with increased binding to PD-L1 S195E compared to PD-L1 S195A. ERAD components are shown in red. (B) IPA based on the mass data from panel (A) showing unfolding protein response and ubiquitination pathways closely related to ERAD were activated (C) MDA-MB-231 stable cells were treated with 50 μM cycloheximide (CHX) for the indicated time. The band intensity was quantified by Image J analysis. Data represent mean ± SD. n = 3. **P, 0.001~0.01, and NS, not significant, Student’s t test. (D) MDA-MB-231 stable cells were treated with proteasome inhibitor MG132 (10 μM) and ERAD inhibitor eeyarestatins (Eer I, 20 μM) for the indicated time. (E, F) MDA-MB-231 cells were treated with or without metformin (5 mM) for 24 hr and MG132 (10 μM) for 6 hr. Endogenous PD-L1 (E) or Flag-tagged WT, S195A, or S195E PD-L1 (F) was pulled down by the PD-L1 antibody (E) or Flag M2 magnetic bead (F), respectively, followed by Western blotting to detect ERAD components (SEL1L, HRD1, ERLEC1, and OS9). (G) MDA-MB-231 cells expressing exogenous HA-ubiquitin were cultured with or without metformin (5 mM) for 24 hr followed by MG132 (10 μM) for 6 hr. Ubiquitination of endogenous PD-L1 was examined by HA immunoblotting after IP with antibody against PD-L1. (H) Ubiquitination of WT, S195A, and S195E PD-L1 was examined by HA immunoblotting after IP with Flag M2 magnetic bead. (I) Control or HRD1 siRNA was transfected into MDA-MB-231 cells expressing exogenous HA-ubiquitin. Ubiquitination of endogenous PD-L1 in each transfectant was examined by HA immunoblotting after IP with PDL1 antibody. (J) Control or HRD1 siRNA was transfected into MDA-MB-231 WT and S195E PD-L1 stable cells expressing exogenous HA-ubiquitin. Ubiquitination of WT and S195E PD-L1 was examined by HA immunoblotting after IP with Flag M2 magnetic bead. (K) siRNA targeting SEL1L, HRD1, OS9 or ERLEC1 was transfected into MDA-MB-231 (L) Each siRNA for ERAD components was transfected into MDA-MB-231 S195E PD-L1 stable cells followed by immunoblotting with the indicated antibodies. M, mock. W, WT PD-L1. A, S195A PD-L1. E, S195E PD-L1.
As expected, S195E was less stable than WT or S195A PD-L1, and the stability of S195E was similar to that of the non-glycosylated 4NQ mutant (Figure 4C and S4C). Consistently, treatment of metformin or AICAR significantly reduced the stability of endogenous PD-L1 (Figure S4D). Indeed, inhibition of the ERAD pathway by MG132 or eeyarestatin I (Eer I) restored S195E protein level (Figure 4D). Metformin treatment induced the binding of endogenous PD-L1 to the ERAD components, SEL1L, HRD1, ERLEC1, and OS9 (Figure 4E, lines 2 vs 3). Yet, the phosphorylation-mimicking S195E PD-L1 bound strongly to these ERAD components independently of metformin treatment (Figure 4F, lines 7 and 8). Metformin also increased the binding of WT PD-L1 (Figure 4F, lines 3 vs. 4), but not the S195A mutant (Figure 4F, lines 5 vs. 6), to ERAD components. Since ERAD is accompanied by poly-ubiquitination (Lemus and Goder, 2014), we also examined the changes in ubiquitination of PD-L1 by metformin. Ubiquitination of endogenous PD-L1 was substantially increased after metformin treatment (Figure 4G, lines 2 vs 3). Without metformin, the S195E mutant was highly ubiquitinated compared with WT PD-L1 (Figure 4H, line 2 vs. 4). In the presence of metformin, WT PD-L1 exhibited similar ubiquitination to S195E (Figure 4H, line 6 vs. 8) whereas no changes were observed for the S195A mutant (Figure 4H, line 3 vs. 7). When the K48R mutant ubiquitin (non-polymerized form) was used for assay, no changes in ubiquitination were observed (Figure S4E) for all three forms of PD-L1, indicating that increased ubiquitination of PD-L1 is poly-ubiquitination associated with proteasomal degradation. Since the ERAD E3 ligase HRD1 (Ferris et al., 2014) bound to PD-L1, we examined the possibility that HRD1 also functions as an E3 ligase of PD-L1. Knocking down HRD1 clearly reduced the ubiquitination of endogenous PD-L1 (Figure 4I, lines 2 and 3 vs. 4 and 5) induced by metformin and abrogated the strong ubiquitination of the S195E mutant (Figure 4J, lines 2 and 3 vs. 5 and 6). Consistently, knocking down HRD1 increased the stability of PD-L1 S195E and 4NQ mutants, suggesting that HRD1 functions as an E3 ligase during ERAD of PD-L1 (Figure S4F). Finally, knocking down the ERAD components induced the accumulation of both endogenous PD-L1 (Figure 4K, blue arrow, normal glycosylated; red arrow, abnormal glycosylated form) and S195E PD-L1 (Figure 4L). These data suggested that metformin-induced S195 phosphorylation of PD-L1 promotes its degradation via the ERAD pathway.
The reduction of PD-L1 by metformin-activated AMPK is physiologically significant and clinically relevant
To validate these results that metformin reduces PD-L1 level via S195 phosphorylation in vivo, we generated mouse 4T1 stable cell lines expressing WT and human S195-equivalent mutants, S194A, or S194E PD-L1 as Ser194 of mouse (m) corresponds to Ser 195 of human (h) PD-L1 based on sequence homology analysis (Figure S5A). A band shift (Figure S5B) and ER accumulation (Figure S5C) similar to S195E hPD-L1 was also observed for S194E mPD-L1. Tumor growth of S194E mPD-L1 4T1 stable cells was significantly reduced compared to those expressing WT mPD-L1 in an immunocompetent mouse model (Figure 5A). Furthermore, the antitumor effect of metformin was lower on S194A mPD-L1 than on WT mPD-L1 4T1 stable cells (Figure 5B and C). Consistently, S194E mPD-L1 4T1 tumors exhibited a significant increase in the CD8+ CTL populations and their activity compared with WT mPD-L1 4T1 tumors (Figure 5D, S195E + PBS and WT + PBS). Although metformin highly improved the CTL immunity in WT mPD-L1 4T1 tumors (Figure 5D, WT + PBS and WT + Metf), such effects were relatively lower in S194A mPD-L1 4T1 tumors (Figure 5D, S195A + PBS and S195A + Metf). These data strongly support the notion that PD-L1 S195 phosphorylation is an important event for the antitumor effect of metformin.
Figure 5. The reduction of PD-L1 by metformin-activated AMPK is physiologically significant and clinically relevant.

(A) 4T1 WT or S194E mPD-L1 stable cells (5 × 104) were injected into BALB/c mice (n = 7 mice per group) (B) 4T1 WT or S194A mPD-L1 stable cells (5 × 104) were injected into BALB/c mice (n = 7 mice per group) on day 0, and metformin (200 mg/kg) administered by i.p. injection starting at day 4 for 18 days. Tumor volume was measured on the indicated time points. Data represent mean ± SD. (C) The relative tumor volume (%) at the end point (day 22) of panel (B). (D) Left, representative images of immunostaining of CD8 and GB in the 4T1 tumor mass. Hoechst: counter staining. Right, CD8 and GB were quantified using Image J. Data represent mean ± SD. n = 12. Three tissue slides per tumor, 4 mice per group. Unit = 262,144 μm2. (E) AMPKα T172-p and PD-L1 levels before and after metformintreatment in tumor tissues of breast cancer patients. −, score 0; +, score 1; ++, score 2; +++, score 3; ↑, up; ↓, down; X, no change. (F) Representative IHC images of the cases shown in panel (E). Scale bar, 50 μm. (G) Plot of IHC scores of AMPKα T172-p and PD-L1 expression levels in responders before and after metformin treatment. n = 7 (H) PBS and metformin were administered to a pair of SCID mice with same patient tumor for 7 days. Immunostaining of AMPKα T172-p and PD-L1 in the PDX tumor mass. Hoechst:counter staining. Scale bar, 200 μm. (I) Western blotting of lysates from PDX tumors in (H) with the indicated antibodies.
*P, 0.01~0.05, **P, 0.001~0.01, and #P, < 0.001, Student’s t test.
To validate the above findings that metformin reduces PD-L1 level in human tumor tissues, we analyzed the association of AMPKα T172-p and PD-L1 levels in tumor tissues from breast cancer patients treated with metformin (Hadad et al., 2015). Tumor tissues were collected by needle biopsy before and after metformin treatment (see Methods) and analyzed in pairs (before and after). Among 17 patients, seven who showed elevated levels of AMPKα T172-p after metformin treatment were considered to be responders. Six of the seven responders had decreased PD-L1 levels after metformin treatment (Figure. 5E, cases 1~7; Fig. 5F, G). In contrast, the other ten non-responders did not show a clear correlation between AMPKα T172-p and PD-L1 levels (Figure 5E_case 8~17). The strong negative correlation in responders is in agreement with our hypothesis that AMPK activated by metformin induces PD-L1 degradation via ERAD pathway. We also observed a negative correlation between AMPKα T172-p and PDL1 in breast cancer PDX mouse model (Figure 5H and I). A pair of SCID mice with the same patient tumor was treated with PBS or metformin. Seven days after metformin treatment, the levels of AMPKα T172-p and PD-L1 in the tumors were examined by IF staining and Western blotting. Metformin increased AMPKα T172-p and reduced PD-L1 expression (Fig. 5H and I). These results suggested that the reduction of PD-L1 by metformin is physiologically significant and clinically relevant.
The combination of metformin and CTLA4 blockade effectively suppresses tumor growth in vivo
PD-L1 and CTLA4 immune inhibitory checkpoints function by different mechanisms, and recent studies have attempted to block both pathways to improve efficacy (Callahan et al., 2014; Curran et al., 2010). Because we show that metformin blocks PD-L1/PD-1 signaling in the current study, we selected CTLA4 blockade for combination therapy with metformin in mouse 4T1 breast tumor model. Mice were treated with metformin and CTLA4 antibody as indicated (see Methods). Metformin treatment by both intraperitoneal injection and oral gavage reduced tumor size (Figure 6A, B and S6A) and increased survival rate (Figure 6C) similar to anti-CTLA4. Consistent with the above mechanistic findings, IF and flow cytometric analysis of tumor tissues showed that metformin reduced PD-L1 levels (Figure 6D, E) and increased the CD8+CTL population and their activity (Figure 6D, F and S6B). Regarding T-cell activation, the effects of metformin as indicated by the levels of GB or IFN-γ appeared to be similar to or better than that of CTLA4 blockade (Figure 6D, F). In addition, the metformin and anti-CTLA4 combination demonstrated significant improvement in tumor burden, survival rate, and CTL activity (Figure 6A–F and S6A) with no significant changes in body weight or detectable toxicity in the kidney and liver (Figure S6C). Similar results from the combination treatment were also observed in B16F10 melanoma (Figure 6G) and CT26 colon cancer (Figure 6H, I) immunocompetent mouse models. These findings suggested that metformin has the potential to enhance the efficacy of anti-CTLA4 therapy without additional toxicity.
Figure 6. The combination of metformin and CTLA4 blockade effectively suppresses tumor growth in vivo.

(A) Tumor growth of 4T1 cells in immunocompetent BALB/c mice treated with control, metformin, anti-CTLA4, or the combination. Data represent mean ± SD. n = 8 mice per group. (B) Quantification (top) of bioluminescence imaging (endpoint images shown). The maximum and minimum values were excluded for more accurate analysis. Data represent mean ± SD. n = 6 mice per group. (C) Survival of mice bearing 4T1-derived tumor following treatment with control, metformin, anti-CTLA4, or the combination. n = 8 mice per group, log-rank test. (D) Top, immunostaining of PD-L1, CD8, and GB in the 4T1 tumor mass. Hoechst: nuclear counter staining. Scale bar, 50 μm (inset, 200 μm). Bottom, quantification of PD-L1, CD8, and GB using Image J. Data represent mean ± SD. n = 12 (4 mice per group). Three tissue slides per tumor. Unit = 182,047 μm2. (E) Cells were extracted from 4T1-tumors by Percoll density gradient centrifugation. PD-L1 levels on extracted cells were evaluated by FACS. Cells were gated based on the FSC (forward scattered light) vs. SSC (side-scattered light). Membrane PD-L1 is shown as median fluorescence intensity (MFI). Data represent mean ± SD. n = 5 per group. (F) FACS analysis of CD45+CD3+CD8+ CTL activity as indicated by the activity intracellular IFN-γ in the leukocyte fractions from Percoll density gradient separation. Detailed gating information is shown in Figure S6B. Data represent mean ± SD. n = 7 per group. Two or three tumors were collected for one sample for the combination group. (G) Tumor growth of B16F10 melanoma cells in immunocompetent C57BL/6J mice treated with control, metformin, anti-CTLA4, or the combination. Data represent mean ± SD. n = 6 mice per group. (H) Tumor growth of CT26 colorectal cancer cells in immunocompetent BALB/C mice treated with control, metformin, anti-CTLA4, or the combination. Data represent mean ± SD. n = 6 mice per group. (I) Survival rate of mice bearing CT26-derived tumor following treatment with control, metformin, anti-CTLA4, or the combination. n = 10 mice per group, log-rank test.
*P, 0.01~0.05, **P, 0.001~0.01, and #P, < 0.001, Student’s t test.
Discussion
Metformin has been shown to directly induce antitumor effects by inhibiting the PI3K-Akt-mTOR and Ras-MAPK signaling pathways critical for cancer progression or indirectly by systemically reducing glucose and insulin metabolism to attenuate cancer cell growth (Hajjar et al., 2013; Pernicova and Korbonits, 2014). In addition, studies more recently showed that metformin regulates the differentiation and activity of T cells through an intrinsic pathway (Eikawa et al., 2015; Zhao et al., 2015), suggesting that the antitumor effects of metformin may also be linked to immune response. Here, our results further reinforced this possibility by demonstrating a new regulatory mechanism of PD-L1 expression.
We identified S195-p of PD-L1 as a critical event that determines whether it is transported to the membrane or degraded by ERAD. In addition, we identified HRD1 as an ERAD E3 ligase of PD-L1 (Figure 4I, 4J and S4F), implying that SEL1/HRD1, known to have a central role in ER quality control (van den Boomen and Lehner, 2015), may have important functions in anti-tumor immunity by regulating immune checkpoints. However, there are several possibilities how S195-p induces ERAD of PD-L1. First, the conformational changes induced by S195-p may mimic the unfolded form or simply facilitate the binding with ERAD complex. Second, ER accumulation due to malfunction during transfer to Golgi can turn on the ERAD pathway (Nishikawa et al., 1994), and S195-p may block access of PD-L1 to the ER-to-Golgi transport system, such as the COPII complex (Zanetti et al., 2011). Third, the structural changes induced by S195-p can increase the affinity of PD-L1 to the factors related to ER glycan trimming, e.g., ERMan I or EDEMs. Indeed, overexpression of ERMan I or EDEMs has been reported to increase the degradation of ERAD substrates with excessive glycan trimming in cells (Xu and Ng, 2015). Moreover, high levels of ERMan I produces more mannose 5, 6, 7 glycan structures through excessive trimming (Ferris et al., 2014), suggesting that increased encounters with these trimming factors can lead to abnormal glycan structures recognized by ERAD. Future studies are warranted to validate these hypotheses.
Consistent with previous studies reported the localization and kinase activity of AMPK to the ER and Golgi (Miyamoto et al., 2015; Qi et al., 2008), our data showed that AMPK, which phosphorylates S195 of PD-L1, was located in both the ER and Golgi, and the binding between AMPK and PD-L1 was detected in those compartments as well (Figure 2H and 2J). However, S195E PD-L1 (phosphorylation mimic form) was primarily detected in the ER but not Golgi (Figure 3F and 3G). Moreover, abnormal glycan trimming (Figure. 3C) and ERAD (Figure 4) of PD-L1 induced by S195-p occur in the ER prior to transport to Golgi. These results suggest that S195-p can take place before glycan trimming in the ER. Importantly, S195 is located in ECD of PD-L1, known to be exposed to lumen of ER and Golgi, not to the cytosol during transport. Moreover, the ER trypsinization assay clearly showed that AMPK practically locates in the lumen of ER (Figure 2I and S2F) and the advanced duo-link assay indicated that that PD-L1 binds to AMPK primarily on the luminal side of ER (Figure 2K, S2H and S2I). Together with these clear evidences, most reasonable interpretation is that AMPK can directly phosphorylate S195 of PD-L1 in the ER lumen. Further study is needed to explain how metformin-activated AMPK is located in the ER lumen.
Interestingly, the effects of metformin on tumor burden and CTL activity were much lower in S194A mPD-L1 than in WT mPD-L1 tumors, but the effects were not completely blocked (Figure 5B, 5C and 5D). Consistently, T cell-mediated cancer cell killing indicated that increased killing effect by metformin was significantly decreased in AMPKα KO cells (Figure 1F and S1B) and S195A PD-L1 stable cells (Figure S3F and S3G), but not completely neutralized, suggesting that metformin enhances T cell activity by not only blocking the inhibitory signal from cancer cells but also stimulating intrinsic T cell activation. A recent study indicated that metformin improves immune surveillance by reducing tumor hypoxia (Scharping et al., 2017). Consistent with those findings, we showed that metformin can enhance antitumor immunity by many approaches. The multiple functions of metformin may explain why it highly activates T cells similar to CTLA4 immunotherapy (Figure 6).
Several clinical studies indicated that metformin exhibits substantial antitumor effects in breast cancer patients, suggesting its therapeutic potential (Camacho et al., 2015). Recently, increased activation of AMPK through phosphorylation at T172 was reported in tumor tissues from non-diabetic breast cancer patients who received metformin treatment (Hadad et al., 2015). Result from IHC staining of tumor tissues from metformin-treated breast cancer patients are in agreement with those clinical data (Hadad et al., 2015) and provide a mechanism by which metformin induces antitumor effects in breast cancer (Figure 5E, F and G). Control case study of breast cancer patients with diabetes showed that the pathological complete response rate after chemotherapy was higher in the metformin-treated group (24%) than in non-treated group (8%), implying that the prognosis after chemotherapy is improved by metformin (Hajjar et al., 2013). Because metformin blocks the PD-L1/PD-1 pathway (Figure. 1), chemotherapy for patients taking metformin can be considered a form of combined immunotherapy. Interestingly, tumor tissues from patients with triple-negative breast cancer, a breast cancer subtype that does not respond to targeted therapies due to the lack of estrogen receptor, progesterone receptor, and HER2 expression (Plasilova et al., 2016), were found to have increased number of tumor-infiltrating lymphocytes accompanied by increased PD-L1 level (Wimberly et al., 2015). In this regard, metformin-mediated activation of CTL by blocking PD-L1/PD-1 signaling may be a potential treatment option for TNBC patients. Indeed, we showed that metformin can increase the killing effects of T cells in two TNBC cell lines, MDA-MB-231 and BT-549 (Figure. 1F, 1L, S1B, S3F, and S3G). Our data showed that metformin also reduced PD-L1 level in the non-small cell lung cancer (H1975 and H358) and colon cancer (RKO) cells (Figure S1F). In B16F10 melanoma and CT 26 colon cancer mouse models (Figure 6G, H, and I), metformin also led to improvement in tumor burden and survival rate. Future studies are warranted to extend current study to other cancer types.
Inhibitory immune checkpoint blockade by anti-CTLA-4, anti-PD-1, or anti-PD-L1 has provided substantial benefits to advanced cancer patients. Various approaches are under way to expand the benefits and improve the efficacy of these immune checkpoint inhibitors, including ongoing clinical trials to evaluate the combined blockade of CTLA4 and PD-L1 or PD-1 (Hassel, 2016; Wolchok et al., 2013). Another approach is combining immunotherapy with existing anticancer therapies like chemotherapy, radiation therapy, or targeted therapy (Bernstein et al., 2016; Parra et al., 2017). Despite these combinations having the potential to improve the outlook for cancer immunotherapy, they can also aggravate immune-related adverse events (irAEs) accompanied by checkpoint inhibition (Callahan et al., 2014; Champiat et al., 2016) or induce severe side effects with inherent toxicity of the anticancer therapies (Florento et al., 2012). In this regard, our findings indicated that metformin significantly improved the antitumor effects by CTLA4 blockade without detectable toxicity (Figure 6 and S6) and suggested that metformin has strong potential to be used in combination with immunotherapy.
STAR Methods
Animal and toxicity studies
All animal experiments were performed in accordance with guidelines approved by MD Anderson’s Institutional Animal Care and Use Committee. NOD SCID, BALB/c and C57BL/6 mice (6- to 8-week-old females) were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Mouse 4T1-Luc2 (Figure 1B, C, D, and E; 5 × 104 cells, Figure. 5A, B, C, and D; 1 × 105 cells, Figure. 6A, B, C, D and Fig. S6; 1 × 105 cells), B16-F10 (5 × 104 cells) or CT26 cells (1 × 105 cells) in 50 μL of medium mixed with 50 μL of matrigel basement membrane matrix (BD Biosciences, San Jose, CA, USA) were injected into the mammary fat pad (4T1) or subcutaneous tissue (B16F10 and CT26). Three (animal experiments in Figure 1) or four days (other animal experiments) after inoculation, metformin (200 mg/kg in 100 μL PBS) was administered to mice daily by i.p. injection for 2 weeks (animal experiments in Figure 1) or until end point (other animal experiments). CTLA-4 antibody (100 μg/100 μL, Bio X Cell) or control hamster IgG (100 μg/100 μL, Bio X Cell) was administered to mice by i.p. injection on day 7, 10, and 13 after inoculation. Tumor size was measured as indicated in the figures, and tumor volume was calculated by using the formula: π/6 × length × width2. Tumor growth was quantified by bioluminescence imaging at end point. Mice received D-luciferin (2.5mg/100 μL; Sigma-Aldrich, St. Louis, MO, USA) via i.p. injection after anesthesia and were imaged using IVIS100 (Perkin Elmer, Waltham, MA, USA). Mice with tumors greater than 1,500 mm3 were sacrificed.
Blood (300 μl) was collected from the orbital sinus of mice using a microhematocrit blood tube at the end point of experiment. The blood was stored in the heparinized blood tube and subjected to biochemical analysis for liver marker enzymes alanine transaminase (ALT) and aspartate transaminase (AST) and kidney marker by-products creatinine and blood urea nitrogen (BUN) to evaluate treatment toxicity by COSBA INTERGRA 400 plus (Roche Diagnostics, Rotkreuz, Switzerland) at The Department of Veterinary Medicine and Surgery, The University of Texas MD Anderson Cancer Center.
Cell culture and treatment
All cell lines used were obtained from American Type Culture Collection (Manassas, VA, USA) and independently confirmed by STR DNA fingerprinting at Texas MD Anderson Cancer Center and tests for mycoplasma were negative. Human breast cancer cell lines and mouse cancer cell lines, were cultured in DMEM/F12, DMEM, MEM respectively, supplemented with 10% FBS and 1% antibiotic mixture. Human lung cancer cell lines and RKO were cultured in RPMI-1640, and MEM respectively, supplemented with 10% FBS and 1% antibiotic mixture. Metformin was added to complete medium at the indicated concentrations and times.
Plasmids
The pGIPZ-sh PD-L1/Flag-hPD-L1 dual vector expressing Flag-PD-L1 with endogenous PD-L1 knockdown was constructed as described previously (Li et al., 2016). Briefly, pGIPZ-sh (h)PD-L1 clone #5 (3´-UTR region of (h)PD-L1: TTGACTCCATCTTTCTTCA, Thermo Scientific, Pittsburgh, PA, USA) and Flag-(h)PD-L1 WT (shRNA/ORF Core Facility UT MD Anderson Cancer Center, Houston, TX, USA) was constructed by the removal of GFP sequence in pGIPZ shPD-L1 clones #5 by Cla1 and Nhe1 restriction enzyme digestion and insertion of Flag-(h)PD-L1 WT. The pGIPZ-sh(h)PD-L1/Flag-PD-L1 S195A, S195D and 4NQ were acquired through site–direct mutagenesis of the pGIPZ-sh(h)PD-L1/Flag-PD-L1WT. pGIPZ-sh(m)PD-L1 clone #3 (Thermo Scientific, Pittsburgh, PA, USA) and Flag-(m)PD-L1 WT (shRNA/ORF Core Facility UT MD Anderson Cancer Center, Houston, TX, USA) were constructed for pGIPZ-sh(m)PD-L1/Flag-(m)PD-L1, which used as the template for pGIPZ-sh(m)PD-L1/Flag-(m)PD-L1 S194A and S194E. The pPET21a His-PD-L1 WT was constructed by inserting the extracellular domain (aa 19~238) of PD-L1 into pPET21a through Nde1 and Xho1 restriction enzyme digestion. The pPET21a His-PD-L1 S195A were acquired through site–direct mutagenesis of the pPET21a His-PD-L1 WT. All plasmids were confirmed using enzyme digestion and DNA sequencing.
Generation of PD-L1 stable and knockout (KO) stable cells
Using a pGIPZ-shPD-L1/Flag-PD-L1 dual expression construct, we established MDA-MB-231 and BT-549 stable cell lines expressing Flag-PD-L1 WT, S195A, S195E or 4NQ mutants with endogenous PD-L1 knockdown. Human PD-L1 KO MDA-MB-231 cells were generated using Pdcd-1L1 CRISPR/Cas9 KO vector (sc-401140, Santa Cruz, Dallas, TX, USA). To generate AMPKα KO MDA-MB-231, three different regions of AMPKα 1(NM_006251.5) and AMPKα 2 (NM_006252.3) were targeted using pLentiCRISPRv2 vectors, respectively. The targeting sequences are as follows;
AMPKα1–1 [CCAGGAACTGAGTCTGCGCA (−5 to −27);
AMPKα1–2 [TGGCGCTATATACGCTGTTG (4008 to 4030);
AMPKα1–3 [CATATTATTTGCGTGTACGA (1325 to 1347);
AMPKα2–1 [AGAAGCAGAAGCACGACGGG (79 to 101);
AMPKα2–2 [TACGTGCTGGGCGACACGCT (117 to 139);
AMPKα2–3 [TCTTCAGTTTCACCTCGCCT (1647 to 1669).
To package lentivirus, the dual expression or LentiCRISPR KO constructs were transfected into HEK 293T cells with two packaging plasmids (VSV-G and delta 8.2). The medium was changed at 24 hr after transfection and supernatant collected twice with 24-hr intervals. The centrifuged supernatant was filtered using a 0.45-μm filter. MDA-MB-231 and BT-549 cells (30% confluency) were incubated in lentivirus-containing medium with polybrene (10 μg/ml; EMD Millipore, Billerica, MA, USA). Forty-eight hr following infection, cells were subjected to puromycin selection (1 μg/ml; InvivoGen, San Diego, CA, USA).
Transfection of siRNA and expressing vectors
Commercial siRNAs were used to knock down SEL1L (#1 SASI_Hs01_00233054 and #2 SASI_Hs01_00233056; Sigma-Aldrich), ERLEC1 (#1 SASI_Hs02_00329444 and #2 SASI_Hs02_00329447; Sigma-Aldrich), OS9 (#1 SASI_Hs01_00097558 and #2 SASI_Hs01_00097559, Sigma-Aldrich) and HRD1 (SYVN1) (SignalSilence® SYVN1 siRNA I #13204; Cell Signaling). siRNAs were transfected into MDA-MB-231 naïve cells and MDA-MB-231 PD-L1/S195E stable cells using Electroporator (Nucleofector II; Amaxa Biosystems) according to the manufacturer’s instructions. pCMV HA-Ubiquitin (WT) and pCMV HA-Ubiquitin (K48R) were transfected into MDA-MB-231 PD-L1 WT, S195A, S195E, or mock stable cells with Lipofectamine® LTX with Plus™ Reagent (15338100; Life Technologies, Grand Island, NY, USA)
qRT–PCR assays
Cells were lysed with TRIzol (Life Technologies) immediately after washing with PBS. Total RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA was synthesized from purified RNA using SuperScript III First-Strand cDNA synthesis system (Life Technologies) according to the manufacturer’s instructions. qPCR was performed using real-time PCR machine (iQ5, BioRad). The comparative Ct method was used for the data analysis. HuPD-L1 mRNA were normalized to huGAPDH mRNA. The sequences of primers used for qRT PCR are as follows:
huPD-L1-F: 5´-ACAGCTGAATTGGTCATCCC-3´
huPD-L1-R: 5 ´-TGTCAGTGCTACACCAAGGC-3´
huGAPDH-F: 5´-AAGGTGAAGGTCGGAGTCAA-3´
huGAPDH-R: 5´-AATGAAGGGGTCATTGATGG-3´.
Identification of N-glycopeptides
The gel band containing either PD-L1WT or S195E was excised for in-gel trypsin digestion. The extracted peptide mixtures were then analyzed by nanospray LC-MS/MS on an Orbitrap Fusion Tribrid (Thermo Scientific, Waltham, MA, USA) coupled to an UltiMate 3000 RSLCnano System (Dionex, Thermo Scientific). The detailed sample preparation procedures and parameters used for MS and MS/MS analysis have been described previously (Yagi et al., 2016). For N-glycopeptide identification, the raw data file was processed and analyzed directly by Byonic (v 2.7.4) and the resulting assignments were further examined manually.
Immunofluorescence
Under anesthesia, the tumor mass was isolated from mice after perfusion with 0.1 M PBS (pH 7.4) and embedded into OCT block and frozen for cryostat section. Cryostat sections (8-μm thick) were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature (RT). After PBS washing, cryostat sections were incubated in the blocking solution (PBS including 3% donkey serum, 1% BSA, 0.3% Triton X-100, pH 7.4) for 30 min at RT. For cell staining, cells on round cover glass were fixed in 4% PFA at RT for 15 min after PBS washing. Cells were permeabilized in 0.5% Triton X-100 for 10 min and then in the blocking solution for 30 min at RT. In antibody reaction buffer (PBS plus 1% BSA, 0.3% Triton X-100, pH 7.4), samples were stained with primary antibodies against active caspase 3 (9661L, 1:300; Cell Signaling Technology, Danvers, MA, USA ), CD8 (MCA609G, 1:100; BioRad, Hercules, CA, USA), GranzymeB (AF1865, 1:500; R&D Systems, Minneapolis, MN, USA), PD-L1 (13684, 1:200; Cell Signaling Technology), HSP90B1 (NBP2–42379, 1:300; Novus Biologicals, Littleton, CO, USA), GM130 (610823, 1:200; BD Biosciences, San Jose, CA, USA), Alexa fluor 488 Giantin (908701, 1:200; BioLegend, San Diego, CA, USA), TNG46 (NB110–62093, 1:400; Novus Biologicals) overnight at 4 °C, followed by Alexa 350, 488, 546, and 647 (1:3,000, Life Technologies) secondary antibodies at RT for 1 hr. Hoechst 33342 (Life Technologies) was used for nuclear staining. To show the binding between AMPKα and PD-L1 in vivo, primary antibodies for PD-L1 (338364, 1:200; LSBio, Seattle, WA, USA), PD-L1 (ab205921, 1:200; Abcam), PD-L1 (86744S, 1:200; Cell Signaling Technology), PD-L1 (13684S, 1:200; Cell Signaling Technology), PD-L1 (GTX104763, 1:200; GeneTex), and AMPKα (AF2850, 1:300; R&D Systems) were applied to duo-link assay (DUO92101; Sigma). The confocal microscope (LSM700; Carl Zeiss, Thornwood, NY, USA) were used for image analysis.
Western blotting and co-immunoprecipitation
For Western blot analysis, cells were lysed in lysis buffer (1.25M urea and 2.5% SDS) after PBS washing. The viscosity of the lysate was removed by sonication, and protein concentration measured by bicinchoninic acid (BCA) reaction. Immunoblotting was performed with primary antibodies against PD-L1 (13684, 1:1,500; Cell Signaling Technology), PD-L1 (338364, 1:1,000; LSBio), AMPKα (5831S, 1:2,000; Cell Signaling Technology), AMPKα1 (AF3197, 1:2000; R&D Systems), AMPKα2 (AF2850, 1:2000; R&D Systems), AMPKα Thr172-p (2535S, 1:1,000; Cell Signaling Technology), Flag-tag (14793S, 1:2,000; Cell Signaling Technology), SEL1L (S3699, 1:5,000; Sigma-Aldrich), HDR1 (14773S, 1:2000; Cell Signaling Technology), ERELC1 (ab181166, 1:3,000; Abcam, San Francisco, CA, USA), OS9 (12497S, 1:1,000; Cell Signaling Technology), HIS-tag (A00186, 1:5000; GenScript), GST-tag (A190–122A, 1:10000; Bethyl Lab), HA-tag (3724S, 1:3,000; Cell Signaling Technology), GAPDH (2118, 1:3,000; Cell Signaling Technology), α-Tubulin (B-5–1-2, 1:5,000; Sigma-Aldrich), β-Actin (A2228, 1:10,000; Sigma-Aldrich). For Co-IP, cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin and protease inhibitor cocktail). Lysates (1 mg) were mixed with anti-PD-L1 (13684, 1:200; Cell Signaling Technology) or AMPKα (5831S, 1:200; Cell Signaling Technology) overnight at 4 °C and then pulled-down with protein G magnetic bead (9006, 1:30; Cell Signaling Technology) at 4 °C for 6 hr. For Flag-IP, lysates (500 μg) were incubated with M2 magnetic bead (M8823, 1:25; Sigma-Aldrich) at 4 °C for 6 hr. The magnetic beads bound with target proteins were washed with same lysis buffer and eluted with 3X LSB sample buffer or through flag-peptide competition.
GST pull-down assay
Commercial recombinant human GST (ab70456; Abcam), PD-L1/GST (ag12432; Proteintech), and AMPK holo complex (PV6238; Invitrogen) were subjected to GST pull-down assay. The conjugation of GST and PD-L1/GST with glutathione beads and the pull-down assay were performed using Pierce GST Protein Interaction Pull-Down Kit (21516; Thermo Scientific) according to manufacturer’s instructions.
Antibody generation and detection
The anti-phospho-huPD-L1 Ser195 antibody was raised against the region near Ser195 phosphorylation site of PD-L1. The phosphorylated synthetic peptide [C-KKLFNVTS(p)TLRINK] was used for immunization in the mice. The antibody was generated as previously described (Li et al., 2016). For detection, PD-L1 protein was purified through immunoprecipitation as described above. All glycan structures on purified PD-L1 protein were removed by PNGase F (New England BioLabs, Ipswich, MA, USA) at 37 °C for 1 hr and then the immunoblotting was performed with anti-phospho-huPD-L1 Ser195 (1:500).
PD-L1 and PD-1 binding assay
To measure interaction between PD-1 and PD-L1 proteins, cells were fixed in 4% PFA at RT for 15 min and then incubated with recombinant human PD-1 Fc protein (R&D Systems) for 1 hr, followed by anti-human Alexa Fluor 488 secondary antibodies (Life Technologies) at RT for 1 hr. Nuclei were stained with Hoechst 33342 (Life Technologies). The fluorescence intensity of Alexa Fluor 488 was measured with a microplate reader Synergy Neo (BioTeK, Winooski, VT, USA) and the cells were visualized using a confocal microscope (LSM700, Carl Zeiss).
T cell-mediated tumor cell killing assay
To acquire activated T cells, human peripheral blood mononuclear cells (PBMC; STEMCELL Technologies, Vancouver, BC, Canada) were cultured in ImmunoCult™-XF T cell expansion medium (10981; STEMCELL Technologies) with ImmunoCult™ Human CD3/CD28/CD2 T cell activator (10970; STEMCELL Technologies) and IL-2 (10 ng/mL; PeproTech, Rocky Hill, NJ, USA) for one week according to the manufacturer’s protocol. The experiments were performed in DMEM/F12 medium with anti-CD3 antibody (100 ng/ml; 16–0037; eBioscience, Thermo Scientific), IL-2 (10 ng/ml). Cancer cells were allowed to adhere to the plates overnight and then incubated for 48 hr with activated T cells in the presence or absence of metformin (5 mM). The ratios between cancer cells and activated cells (1:3, 1:5, and 1:10) were modified according to the purpose of each experiment (see Figure Legends). T cells and cell debris were removed by PBS wash, and living cancer cells were then quantified by a spectrometer at OD (570 nm) followed by crystal violet staining.
CTL profile analysis by FACS
Mouse Tumor Dissociation Kit (130–096-730, Miltenyl Biotec) and gentle MACS Octo Dissociator (130–096-427, Miltenyl Biotec) were used to digest the excised tumors from mice. Percoll gradient assay (17–5445-01, GE Healthcare) was then performed to separate cancer cells and enriched leukocytes. After blocking with CD16/CD32 (40477, 1:50; BioLegend, San Diego, CA, USA) antibody, cancer cell fractions were stained using PD-L1-APC (564715, 1:100; BD) antibody. Enriched leukocyte fractions were stained using CD3-PreCP (100325, 1:20; BioLegend, San Diego, CA, USA), CD8-APC/Cy7 (100713, 1:200; BioLegend), and CD45-APC (103111, 1:100; BioLegend) antibodies. After fixation (421401, BioLegend) and permeabilization (421402, BioLegend), intracellular INFγ was stained using INFγ-Pacific Blue (505817, 1:20; BioLegend) antibody. Stained T cells were analyzed by BD FACSCanto II (BD Biosciences) cytometer. Data was processed by the FlowJo software.
In vitro kinase assay
Recombinant His-PD-L1 WT and His-PD-L1 S195A (Extracellular domain; amino acids 19–238) were expressed in E-coli (BL21) by isopropyl β-D-1-thiogalactopyranoside (IPTG) induction and purified with Ni-NTA Superflow Agarose (Thermo Scientific) from bacterial lysates. Protein kinase assay was performed in a solution consisting of HEPES-Brij Buffer, active AMPK holoenzyme (10 ng; Thermo Scientific), 0.3 mM AMP, 0.2 mM ATP (with 0.5 mCi/mL γ−32P-ATP for radioactive assay), and/or 1 μg of protein substrate at 30 °C for 30 min. The kinase reaction was stopped by heating at 100 °C for 5 min in LSB sample buffer, and samples were subjected to SDS–PAGE, Coomassie staining and autoradiography.
ADP-Glo kinase assay
ADP-Glo™ Kinase assay kit (V9101, Promega, Corporation, Madison, WI, USA) was used to measure kinetics of PD-L1 (ab12688, Abcam) phosphorylation by AMPK (PV6238, Life Technologies) through a luminescent ADP detection assay. The well-known substrate of AMPK, ACC (ab157829, Abcam) was performed in this assay as a positive control. The experiments were carried on according to the manufacturer’s protocol. The values of Km and Vmax for AMPK were calculated using the Michaelis-Menten equation using the Graphpad Prism software.
Cell fractionation and endoplasmic reticulum enrichment
Cell fractionation kit (9038, Cell Signaling Technology, Danvers, MA, USA) was used to extract membrane, cytosolic and nuclear protein. Endoplasmic reticulum (ER) microsomal fractions were prepared by ER enrichment kit (NBP2–29482, Novus Biologicals, Littleton, CO). All experiments were performed according to per manufacturer’s instruction. Protein concentration was determined using the BCA protein assay kit (23225, Thermo Scientific, Waltham, MA, USA). Proteins (30 μg) from each fraction was analyzed by Western blotting.
Trypsinization of endoplasmic reticulum microsomal fractions
ER microsomal fraction (300 μl) was collected from 0.4 g cell pellet by ER enrichment kit (NBP2–29482, Novus Biologicals, Littleton, CO). After pretreatment with or without 1% triton X100 for 3 min, trypsin/EDTA (0.625 g/L trypsin and 0.05 g/L EDTA in PBS) solution was added to each ER microsomal fraction (20 μl). Samples were mixed and incubated for the indicated time. After trypsinization, mixed samples were subjected to Western blot analysis using primary antibodies against IRE1α (3294, 1:2000; Cell Signaling Technology), HSP90B1 (NBP2–42379, 1:3000; Novus), and AMPKα (5831S, 1:2000; Cell Signaling Technology).
Immunohistochemical staining of human breast tumor tissues
Human breast tumor tissue collection and study approval were described previously (Hadad et al., 2011). This trial (07/S1402/19) were approval by the Tayside Local Research Ethics Committee. Non-diabetic women with operable invasive breast cancer were recruited in this trial. The patient tumor tissues were collected by 14-gauge core needle biopsy before and after metformin treatment. The regimen of oral metformin treatment as following: 500 mg daily for one week, increased to 1 g twice daily for another week. Twenty-five pairs of tissues (before and after metformin treatment) were obtained as previously described (Hadad et al., 2011), and among those, 17 pairs were available for IHC staining and analysis. Immunohistochemical staining was performed as previously described (Li et al., 2016) using PD-L1 (205921, 1:150; Abcam) and AMPKα Thr172-p (2535S, 1:30; Cell Signaling Technology) antibodies. Briefly, tissue specimens were incubated with antibodies and a biotin-conjugated secondary antibody and then mixed with an avidin-biotin-peroxidase complex. Amino-ethylcarbazole chromogen was used for visualization. According to the immunoreactive score method, the intensity of staining (protein expression) was ranked into one of four groups: high (score 3), medium (score 2), low (score 1), and negative (score 0).
PDX model
The PDX model was obtained from the Cazalot Breast Cancer Model Resource at MD Anderson Cancer Center. This resource was established, in part, through a generous gift from the Cazelot family. Fragments form same tumor mass were implanted into BALB/c SCID mice as a pair. Metformin (200 mg/kg) was administered via i.p. injection for 7 days which began when tumor size reached around 500 mm3. The dissected tumor tissues were prepared for lysis and cryosection.
Statistical analysis
Data of bar graphs represents as percentage or fold change relative to control or untreated groups with standard deviation of three independent experiments. SPSS (Ver. 20, SPSS, Chicago, IL) was used for statistical analyses. Student’s t test was performed to compare two groups of independent samples. The correlation between AMPKα and PD-L1 was analyzed using Pearson Chi-Square test and Mann-Whitney test. A P value < 0.05 was considered statistically significant.
Data and Software Availability
https://data.mendeley.com/datasets/g66zzcbvfx/draft?a=e6eae081–06f3–44f2–96fc-2fb7fce63a37
Supplementary Material
Highlights.
Metformin enhances antitumor CTL immunity by blocking PD-L1/PD-1 axis.
Metformin-activated AMPK directly binds to and phosphorylates PD-L1 at S195.
Abnormal PD-L1 glycosylation induced by pS195 leads to PD-L1 degradation by ERAD.
Combination therapy with metformin and anti-CTLA4 has a synergistic antitumor effect.
Acknowledgments
We would like to thank the Academia Sinica Common Mass Spectrometry Facilities located at the Institute of Biological Chemistry for mass spectrometry data acquisition. This work was funded in part by the following: National Institutes of Health (CCSG CA16672); Cancer Prevention & Research Institutes of Texas (RP160710); Breast Cancer Research Foundation; National Breast Cancer Foundation, Inc.; Patel Memorial Breast Cancer Endowment Fund; The University of Texas MD Anderson-China Medical University and Hospital Sister Institution Fund (to M.-C.H.); Ministry of Health and Welfare, China Medical University Hospital Cancer Research Center of Excellence (MOHW107-TDU-B-212-114024 and MOHW107-TDU-B-212-112015); Center for Biological Pathways; Ministry of Science and Technology Oversees Project for Post Graduate Research (MOST; 104-2917-I-564-003; to W.-H.Y); Ministry of Science and Technology, Academia Sinica in Taiwan (105-0210-01-13-01; to K.-H. K); the National Research Foundation of Korea grant for the Global Core Research Center funded by the Korean government (MSIP; 2011-0030001; to J.-H.C).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernstein MB, Krishnan S, Hodge JW, and Chang JY (2016). Immunotherapy and stereotactic ablative radiotherapy (ISABR): a curative approach? Nature reviews. Clinical oncology 13, 516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling J, and Aebi M (2013). N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harbor perspectives in biology 5, a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, and Wolchok JD (2014). CTLA-4 and PD-1 Pathway Blockade: Combinations in the Clinic. Frontiers in oncology 4, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho L, Dasgupta A, and Jiralerspong S (2015). Metformin in breast cancer - an evolving mystery. Breast cancer research : BCR 17, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, Cauquil C, Chanson P, Collins M, Durrbach A, et al. (2016). Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Annals of oncology : official journal of the European Society for Medical Oncology 27, 559–574. [DOI] [PubMed] [Google Scholar]
- Chen CM, Song W, Kao JY, Zheng QD, and Chen JJ (2004). Expression of Fas ligand is not a main mechanism used by tumors to counteract antitumor immunity. Frontiers in bioscience : a journal and virtual library 9, 448–456. [DOI] [PubMed] [Google Scholar]
- Chen L (2004). Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nature reviews. Immunology 4, 336–347. [DOI] [PubMed] [Google Scholar]
- Chen L, and Han X (2015). Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. The Journal of clinical investigation 125, 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, and Lieberman J (2008). Death by a thousand cuts: granzyme pathways of programmed cell death. Annual review of immunology 26, 389–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, and Allison JP (2010). PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences of the United States of America 107, 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, and Udono H (2015). Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proceedings of the National Academy of Sciences of the United States of America 112, 1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, and Morris AD (2005). Metformin and reduced risk of cancer in diabetic patients. Bmj 330, 1304–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris SP, Kodali VK, and Kaufman RJ (2014). Glycoprotein folding and quality-control mechanisms in protein-folding diseases. Disease models & mechanisms 7, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florento L, Matias R, Tuano E, Santiago K, Dela Cruz F, and Tuazon A (2012). Comparison of Cytotoxic Activity of Anticancer Drugs against Various Human Tumor Cell Lines Using In Vitro Cell-Based Approach. International journal of biomedical science : IJBS 8, 76–80. [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nature medicine 19, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, Jellema G, Deharo S, Hardie DG, Pusztai L, et al. (2011). Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast cancer research and treatment 128, 783–794. [DOI] [PubMed] [Google Scholar]
- Hadad SM, Coates P, Jordan LB, Dowling RJ, Chang MC, Done SJ, Purdie CA, Goodwin PJ, Stambolic V, Moulder-Thompson S, et al. (2015). Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast cancer research and treatment 150, 149–155. [DOI] [PubMed] [Google Scholar]
- Hajjar J, Habra MA, and Naing A (2013). Metformin: an old drug with new potential. Expert opinion on investigational drugs 22, 1511–1517. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, and Brunet A (2016). AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends in cell biology 26, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassel JC (2016). Ipilimumab plus nivolumab for advanced melanoma. The Lancet. Oncology 17, 1471–1472. [DOI] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM, et al. (2010). Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell metabolism 11, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhajda FP (2008). AMP-activated protein kinase and human cancer: cancer metabolism revisited. International journal of obesity 32 Suppl 4, S36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemus L, and Goder V (2014). Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 3, 824–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, et al. (2016). Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 7, 12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T, Rho E, Sample V, Akano H, Magari M, Ueno T, Gorshkov K, Chen M, Tokumitsu H, Zhang J, et al. (2015). Compartmentalized AMPK signaling illuminated by genetically encoded molecular sensors and actuators. Cell reports 11, 657–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Hirata A, and Nakano A (1994). Inhibition of endoplasmic reticulum (ER)-toGolgi transport induces relocalization of binding protein (BiP) within the ER to form the BiP bodies. Molecular biology of the cell 5, 1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nature reviews. Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra K, Valenzuela P, Lerma N, Gallegos A, Reza LC, Rodriguez G, Emmenegger U, Di Desidero T, Bocci G, Felder MS, et al. (2017). Impact of CTLA-4 blockade in conjunction with metronomic chemotherapy on preclinical breast cancer growth. British journal of cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernicova I, and Korbonits M (2014). Metformin--mode of action and clinical implications for diabetes and cancer. Nature reviews. Endocrinology 10, 143–156. [DOI] [PubMed] [Google Scholar]
- Plasilova ML, Hayse B, Killelea BK, Horowitz NR, Chagpar AB, and Lannin DR (2016). Features of triple-negative breast cancer: Analysis of 38,813 cases from the national cancer database. Medicine 95, e4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, and Li P (2008). Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. The EMBO journal 27, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, Batth IS, Brownlee Z, Rojas C, Meng QH, Kopetz S, and Li S (2016). Potential role of nuclear PD-L1 expression in cell-surface vimentin positive circulating tumor cells as a prognostic marker in cancer patients. Scientific reports 6, 28910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Whetstone RD, Zeng X, and Delgoffe GM (2017). Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer immunology research 5, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shental-Bechor D, and Levy Y (2008). Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proceedings of the National Academy of Sciences of the United States of America 105, 8256–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, Ma H, Inoue T, Gao B, Kim H, et al. (2015). IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nature cell biology 17, 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Taube JM, Anders RA, and Pardoll DM (2016). Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nature reviews. Cancer 16, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin O, Kraut JA, and Sachs G (2009). Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. American journal of physiology. Renal physiology 296, F459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boomen DJ, and Lehner PJ (2015). Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting of MHC class I. Molecular immunology 68, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, and Brodsky JL (2008). One step at a time: endoplasmic reticulum-associated degradation. Nature reviews. Molecular cell biology 9, 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, and Andreelli F (2012). Cellular and molecular mechanisms of metformin: an overview. Clinical science 122, 253–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, and Nakajima H (2012). Coinhibitory molecules in autoimmune diseases. Clinical & developmental immunology 2012, 269756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly H, Brown JR, Schalper K, Haack H, Silver MR, Nixon C, Bossuyt V, Pusztai L, Lannin DR, and Rimm DL (2015). PD-L1 Expression Correlates with Tumor-Infiltrating Lymphocytes and Response to Neoadjuvant Chemotherapy in Breast Cancer. Cancer immunology research 3, 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. The New England journal of medicine 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfert MA, and Boons GJ (2013). Adaptive immune activation: glycosylation does matter. Nature chemical biology 9, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, and Ng DT (2015). Glycosylation-directed quality control of protein folding. Nature reviews. Molecular cell biology 16, 742–752. [DOI] [PubMed] [Google Scholar]
- Yagi H, Kuo CW, Obayashi T, Ninagawa S, Khoo KH, and Kato K (2016). Direct Mapping of Additional Modifications on Phosphorylated O-glycans of alpha-Dystroglycan by Mass Spectrometry Analysis in Conjunction with Knocking Out of Causative Genes for Dystroglycanopathy. Molecular & cellular proteomics : MCP 15, 3424–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Pahuja KB, Studer S, Shim S, and Schekman R (2011). COPII and the regulation of protein sorting in mammals. Nature cell biology 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Zhao D, Long XD, Lu TF, Wang T, Zhang WW, Liu YX, Cui XL, Dai HJ, Xue F, and Xia Q (2015). Metformin decreases IL-22 secretion to suppress tumor growth in an orthotopic mouse model of hepatocellular carcinoma. International journal of cancer 136, 2556–2565. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. (2001). Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation 108, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
https://data.mendeley.com/datasets/g66zzcbvfx/draft?a=e6eae081–06f3–44f2–96fc-2fb7fce63a37