

Abstract
Proliferating cell nuclear antigen (PCNA) is essential for DNA replication and repair, and cell growth and survival. Previously, we identified a novel class of small molecules that bind directly to PCNA, stabilize PCNA trimer structure, reduce chromatin-associated PCNA, selectively inhibit tumor cell growth, and induce apoptosis. The purpose of this study was to investigate the combinatorial effects of lead compound PCNA-I1S with DNA damaging agents on cell growth, DNA damage, and DNA repair in four lines of human prostate and lung cancer cells. The DNA damage agents used in the study include ionizing radiation source cesium-137 (Cs-137), chemotherapy drug cisplatin (cisPt), ultraviolet-C (UV-C), and oxidative compound H2O2. DNA damage was assessed using immunofluorescent staining of γH2AX and the Comet assay. The homologous recombination repair (HRR) was determined using a plasmid-based HRR reporter assay and the nucleotide excision repair (NER) was indirectly examined by the removal of UV-induced cyclobutane pyrimidine dimers (CPD). We found that PCNA-I1S inhibited cell growth in a dose-dependent manner and significantly enhanced the cell growth inhibition induced by pretreatment with DNA damaging agents Cs-137 irradiation, UV-C, and cisPt. However, the additive growth inhibitory effects were not observed in cells pre-treated with PCNA-I1S, followed by treatment with cisPt. H2O2 enhanced the level of chromatin-bound PCNA in quiescent cells, which was attenuated by PCNA-I1S. DNA damage was induced in cells treated with either PCNA-I1S or cisPt alone and was significantly elevated in cells exposed to the combination of PCNA-I1S and cisPt. Finally, PCNA-I1S attenuated repair of DNA double strand breaks (DSBs) by HRR and the removal of CPD by NER. These data suggest that targeting PCNA with PCNA-I1S may provide a novel approach for enhancing the efficacy of chemotherapy and radiation therapy in treatment of human prostate and lung cancer.
Introduction
Proliferating cell nuclear antigen (PCNA) is an evolutionally very well conserved multifunctional protein [1, 2] and a non-oncogenic protein essential for tumor cell growth and survival. It is overexpressed in all tumors [2]. Overexpression of PCNA in prostate cancer [3, 4] and non-small cell lung carcinoma (NSCLC) [5] is associated with advanced disease and metastasis, and is a reliable biomarker predicting poor prognosis of cancers of various tissue types [3, 4, 6–8]. Given that tumor cells are more active in replication and contain much higher levels of damaged DNA [9, 10] than normal cells, they are more vulnerable to the stress of downregulation or inhibition of PCNA function. Therefore, targeting PCNA could be an effective approach for treatment of cancer.
Native PCNA, present predominantly in the nucleoplasm as “free-form PCNA”, is a ring-shaped homotrimeric protein joined together through head to tail interaction [11, 12]. To be functional, PCNA must be linearized or monomerized, and relocalized. Upon being loaded onto the primer-template junctions of DNA, PCNA encircles DNA, serves as a platform for and interacts with proteins involved in DNA replication and repair and other cellular processes [2, 13–16]. When monomerized and exported to cytoplasm, PCNA was shown to interact with procaspases to inhibit apoptosis [17] and with glycolytic enzymes to promote glycolysis [18]. PCNA also interacts with some cell signaling proteins, such as PI3K proteins, and regulates cell signaling processes [19]. On cell membrane, PCNA interrupts the recognition of tumor cells by natural killer cells [20]. PCNA interacts with its partner proteins containing PIP (PCNA interaction protein)-box, KA-box, APIM (AlkB homologue 2 PCNA-interacting motif), and other motifs [2, 16, 19].
Great efforts have been made to develop novel approaches targeting PCNA for cancer therapy. Peptides mimicking the APIM or a sequence of caPCNA (“cancer associated PCNA”) selectively inhibit tumor cell growth, induce apoptosis, and enhance cytotoxicity of chemotherapy drugs on tumor cells [19, 21–23]. The selective inhibitory effects were also observed in cancer cells treated with small molecule T2AA targeting the PIP-box [24, 25] and small molecule AOH1160 targeting caPCNA [26]. Targeting PCNA in replisomes with monoclonal antibodies triggers lethal DNA replication stress in tumor cells [27]. The PCNA-targeting peptides and small molecule (AOH1160) are well tolerated in animals and show the therapeutic effects against various types of tumors, especially when combined with DNA damage drugs [19, 21, 23, 26, 28].
Targeting PCNA with peptides and small molecules on a single motif described above only interrupts PCNA interactions with certain partner proteins and, hence, compromises some functions of PCNA. Given that PCNA must be relocalized to execute its functions, we hypothesized that targeting PCNA relocalization could be a more effective approach. We have identified a novel class of small molecules (termed PCNA inhibitors, PCNA-Is) that bind directly to PCNA trimers at the interfaces of two monomers, stabilize the trimer structure, and interfere with PCNA relocalization [29]. The two lead compounds PCNA-I1 and PCNA-I1S selectively inhibit tumor cell growth and induce apoptosis in tumor cells at nanomolar concentrations. Tumor cells of various tissue types, including cells with p53 deletion or mutation and cells with multidrug resistant phenotype, are all susceptible to the PCNA inhibitors [29–31]. Moreover, PCNA-I1 therapy significantly retards the growth of human prostate cancer xenografts in mice and does not cause apparent toxicity to the hosts [31]. Induction of DNA damage by PCNA-I1 and PCNA-I1S reveals the underlying molecular mechanism by which the compounds inhibit cell growth. The purpose of this study was to investigate the combinatorial effects of PCNA-I1S with DNA damaging agents on cell growth, DNA damage, and DNA repair. We found that treatment of PCNA-I1S in combination with DNA damaging agents Cs-137, UV-C, or cisplatin (cisPt) produced additive inhibitory effects on growth of prostate cancer and NSCLC cells. PCNA-I1S attenuated PCNA association to chromatin stimulated by H2O2 in quiescent cells, induced DNA damage, promoted DNA damage induced by cisPt, and inhibited DNA repair mediated by the homologous recombination repair (HRR) and the nucleotide excision repair (NER). Thus, targeting PCNA with PCNA-I1S may provide a novel approach for enhancing the efficacy of DNA damaging chemotherapy and radiation therapy in treatment of cancer.
Materials and methods
Reagents
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide], H2O2, and cisPt were purchased from Sigma Aldrich (St. Louis, MO). Antibodies against PCNA, α-tubulin, histone H1, and phospho-histone H2AX (Ser139) named as γH2AX were purchased from Cell Signaling Technologies (Danvers, MA). OxiSelect™ Cellular UV-Induced DNA Damage Staining kit and OxiSelect™ Comet assay kit were purchased from Cell Biolabs, Inc (San Diego, CA). HRR reporter pDR-GFP plasmid and transfection control pDsRed plasmid [32] were generously provided by Drs. EL Mustapha Bahassi and Peter Stambrook (University of Cincinnati, Cincinnati, OH). Lipofectamine 3000 and Alexa Flour secondary antibodies were obtained from ThermoFisher Scientific (Waltham, MA).
Cells and culture
Prostate cancer cells (LNCaP, PC-3, and 22Rv1) and A549 lung cancer cells were obtained from ATCC (Manassas, VA). The cells were expanded and kept in cryogenic storage for long-term safekeeping. The cells were authenticated genetically with PCR identifying the short tandem repeat (STR) and cell-specific profiling against ATCC database at the University of Arizona Genetics Core. The cells were cultured in RPMI-1640 medium supplemented with 10% FBS at 37°C in 5% CO2. Cells in exponential growth phase were harvested by treatment for 1–3 minute with a 0.25% trypsin-0.02% EDTA solution and resuspended in the medium. Only suspensions of single cell with viability exceeding 95% (ascertained by trypan blue exclusion) were used in the study.
Gamma- and ultraviolet-C (UV-C)-irradiation
For irradiation with a gamma-emitting cesium (Cs)-137 source, cells were detached, washed, resuspended in culture medium, and irradiated at desired doses in a Gamma Cell 40 Exactor research irradiator (Nordion, Ontario, Canada). For UV-C irradiation, cells were plated into 96-well plates in phenol red-free medium, allowed to attach overnight, and exposed to UV-C light from a germicidal lamp (G64T5L, Sankyo Denki, Japan) with an output of 25W at 253.7 nm and irradiance of 0.25 mW/cm2 at 1 m from the lamp in a tissue culture hood at the doses indicated in Results.
MTT assay
Cells were plated into 96-well plates and treated with PCNA-I1S and/or other DNA damaging agents as indicated and cultured for 96 hours. At the end of the incubation, the cells in control and treated cultures were stained with MTT as described in our previous study [33]. Growth inhibition (%) in the treated cells relative to the untreated control cells was calculated using the formula: (1-A570 of treated cells/A570 of control cells) x 100, which reflects the effects of the treatments on cell growth.
Immunofluorescent staining
Cells were seeded into chamber slides at 2 x 104 cells/well and allowed to adhere overnight. The cells were then treated with PCNA-I1S and/or other DNA damaging agents for the times indicated. For chromatin-bound PCNA staining, PC-3 cells were treated with ice-cold buffer A [10 mM Tris-HCl, pH 7.4, 2.5 mM MgCl2, 0.5% Nonidet P-40 (NP-40), 1 mM dithiothreitol, 1 mM PMSF, and protease inhibitor cocktail] for 5 min, fixed with cold methanol, and rinsed with PBS containing 0.1% Tween-20. For γH2AX staining, A549 cells were fixed with 2% paraformaldehyde for 20 minutes, followed by washing twice with PBS containing 0.1% Tween-20 and permeabilized with methanol for 10 minutes at -20°C. The cells were then blocked using 5% normal goat serum for 1 hour at room temperature. Primary antibodies were diluted per the manufacturer’s recommendation and incubated overnight at 4°C. After washing, the cells were incubated with a fluorochrome conjugated secondary antibody for 1 hour at room temperature and mounted for analysis under an Olympus fluorescent microscopy. DAPI (4′,6-Diamidino-2-Phenylindole, dihydrochloride) was used as a nuclear counterstain to reveal all cells. Images were captured with a cooled CCD camera using Spot Advanced software (Spot Imaging Solutions, Sterling Heights, MI).
Immunoblotting
Chromatin-associated PCNA was analyzed by immunoblotting as described in our previous studies [29, 30]. Cells were collected by trypsinization, pelleted (300 g, 5 min, 4°C), washed in PBS with 1 mM PMSF, and lysed in buffer A for 10 minutes. The samples were then pelleted by centrifugation (1,500 g, 5 min, 4°C) and the supernatant fraction collected as the NP-40-extractable (NP-E) fraction that contains free-form PCNA. The resulting pellet was washed in buffer B (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM PMSF, and protease inhibitor cocktail), resuspended and digested in buffer C (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 5 mM MgCl2, 0.2 mM PMSF, and protease inhibitor cocktail) with 200 units/107 cells of DNase I for 30 min with agitation at room temperature. After centrifugation (13,000 g, 5 min, 4°C), the supernatant was collected as NP-40-resistant (NP-R) fraction that contains chromatin-bound PCNA. The two fractions of proteins were resolved by SDS-PAGE and analyzed by immunoblotting using PCNA antibody as well as antibodies to α-tubulin and histone H1 as loading controls for NP-E and NP-R proteins, respectively.
The alkaline Comet assay
DNA damage in single cells was determined with a gel electrophoresis assay (the Comet assay). The alkaline Comet assay detecting DNA with both single-strand breaks (SSBs) and double-strand breaks (DSBs) [34, 35] was performed using the OxiSelect™ Comet assay kits following the manufacture’s instruction. After the staining with Vista Green DNA Dye (Cell Biolabs), the slides were examined under an Olympus fluorescent microscopy and images were captured. Images of the Comet assay from two experiments with 60–100 cells each treatment group were analyzed using OpenComet (v1.3.1), a plugin [36] for the imaging processing software Image J, to automatically calculate Tail DNA% (= 100 x Tail DNA Intensity/DNA Intensity) and Extent Tail Moment (= Length of Tail x Tail DNA%).
Determination of HRR in a GFP reporter assay
The pDR-GFP HRR reporter is a mammalian expression vector containing a green fluorescent protein (GFP) expression cassette. pDR-GFP plasmid has 2 tandem but inactive GFP repeats, one of which contains the unique I-SCE1 site. pDR-GFP was linearized by cleavage with I-SCE1 endonuclease and purified. Upon transfection into cells, GFP expression is detected when a functional GFP protein is reconstituted by HRR-mediated homologous recombination between the 2 non-functional repeats [32]. PC-3 and A549 cells were plated into 96-well plates at 2 x 104 cells/well in antibiotics-free media and allowed to adhere overnight. The cells were transfected with the linearized pDR-GFP HRR reporter or pDsRed (control vector) plasmids using Lipofectamine 3000 following the manufacturer’s instructions. Four hours later, PCNA-I1S was added to the cultures to treat the cells for 48 hours. Expression of GFP from pDR-GFP and red fluorescent protein (RFP) from control vector were examined and counted under an Olympus fluorescent microscopy, and images were captured.
Assessment of UV-C-induced DNA damage
Cells in phenol red-free medium were plated into 96-well plates at 2 x 104 cells/well and allowed to adhere overnight. The cells were exposed to UV-C, then cultured for 24 hours in the absence or presence of PCNA-I1S. The presence of cyclobutane pyrimidine dimers (CPD) induced by UV-C irradiation was immunofluorescently stained using the OxiSelect™ Cellular UV-Induced DNA Damage Staining Kits following the manufacture’s instruction and examined under an Olympus fluorescent microscopy and images were captured. DAPI (4′,6-Diamidino-2-Phenylindole, dihydrochloride) was used as a nuclear counterstain to reveal all cells. A reduction of CPD-positive cell population and/or CDP-staining intensity indicates the removal of CPD through NER in the cells.
Statistical analysis
Data from each assay were expressed as means ± SD. Statistical differences between 2 groups were determined by the Student’s t test. Data from Comet assay was analyzed by one-way Analysis of Variance (ANOVA). All statistical analyses were carried out using Prism software (GraphPad, San Diego, CA). P < 0.05 was considered significantly different.
Results
The combinatorial effects of PCNA-I1S and Cs-137 irradiation on cell growth inhibition
The measurement of in vitro tumor cell inhibition by PCNA-Is in MTT assay correlates closely with the measurement of cytotoxic activity of the compounds in colony formation assay and apoptosis assay [29, 31]. We assessed the combinatorial effects of PCNA-I1S with DNA damage agents on tumor cell growth using the MTT assay. First, we determined the combinatorial effects of PCNA-I1S with ionizing irradiation that mainly induces DNA SSBs and DSBs. PC-3, LNCaP, 22Rv1, and A549 cells were irradiated with Cs-137 at 2 or 4 Gy and plated into 96-well plates. Three hours later, the cells were treated with 0.125, 0.25, or 0.5 uM PCNA-I1S for 24 hours. The cells were then cultured in fresh medium for additional 72 hours in the absence of PCNA-I1S. As shown in Fig 1, the exposure to Cs-137 irradiation or PCNA-I1S alone led to the growth inhibition in a dose-dependent fashion in all cells. Moreover, the dose-dependent additive effects on cell growth inhibition were observed in cells exposed to combinations of Cs-137 irradiation and PCNA-I1S (Fig 1). Furthermore, we found that PCNA-I1S at 0.5 uM produced greatest growth inhibition either alone or in combination with Cs-137 irradiation.
Fig 1. The combinatorial effects of PCNA-I1S and Cs-137 irradiation on cell growth inhibition.

After irradiation with Cs-137 at 2 or 4 Gy, the cells were plated into 96-well plates (1,000 to 3,000 cells/well, 4 to 6 wells/treatment group) and allowed to attach for 3 hours. The cells were then treated with increasing concentrations of PCNA-I1S for 24 hours. After removing PCNA-I1S containing medium, the cells were then cultured in fresh medium for additional 72 hours and stained with MTT. Data shown are from one representative experiment of three. *, p<0.05 (Cs-137 irradiation alone vs. the combinations of Cs-137 irradiation and PCNA-I1S).
The combinatorial effects of PCNA-I1S and UV-C irradiation on cell growth inhibition
UV-C irradiation leads to the formation of three major types of DNA lesions, including CPDs, pyrimidine 6–4 pyrimidone photoproducts, and their Dewar isomers [37]. CDP in human cells is repaired mainly by NER [37, 38]. We determined the combinatorial effects of PCNA-I1S and UV-C irradiation on cell growth. PC-3 and A549 cells were irradiated with UV-C at 3, 10, or 20 mJ/cm2, followed by incubation in medium without or with 0.5 or 1 uM PCNA-I1S for 24 hours. The cells were then cultured for additional 72 hours in the absence of PCNA-I1S. As shown in Fig 2, UV-C irradiation induced growth inhibition in both PC-3 and A549 cells in a dose-dependent manner. Targeting PCNA with PCNA-I1S alone led to growth inhibition and produced additive effects on cell growth inhibition with UV-C irradiation (Fig 2).
Fig 2. The combinatorial effects of UV-C irradiation and PCNA-I1S on cell growth inhibition.

PC-3 (2,000 cells/well, 4 wells/treatment group) (A) and A549 (1,000 cells/well, 4 wells/treatment group) (B) in phenol red-free medium were plated into 96-well plates. After an overnight incubation, the cells were exposed to UV-C, followed by incubation for 24 hours in medium without or with either 0.5 or 1 uM PCNA-I1S. The cells were then cultured for 72 hours in fresh medium, followed by MTT assay. Data shown are from one representative experiment of three. *, p<0.05 (UV-C alone vs. the combinations of UV-C plus PCNA-I1S).
The combinatorial effects of PCNA-I1S and cisPt on cell growth inhibition
CisPt, a DNA damaging compound, is the first line chemotherapeutic agent used with another drug for treatment of advanced NSCLC [39]. We determined the combinatorial effects of PCNA-I1S with cisPt on growth inhibition in lung cancer cells. NSCLC A549 cells were treated with increasing concentrations of cisPt (1.5 to 12 uM) for 24 hours to induce DNA damages. After the removal of cisPt, the cells treated with PCNA-I1S (0.5 or 1 uM) for 24 hours. After incubation for additional 48 hours in the absence of the drugs, the cultures were stained with MTT. The treatment with cisPt or PCNA-I1S alone induced cell growth inhibition in a dose-dependent fashion (Fig 3A). Additive effects on cell growth inhibition were observed in cells exposed to combinations of cisPt and PCNA-I1S, most significantly at the low concentration of 1.5 uM cisPt (Fig 3A).
Fig 3. The combinatorial effects of cisPt and PCNA-I1S on cell growth inhibition.

A549 cells (1,000 cells/well, 4 wells/treatment group) were plated into 96-well plates and incubated overnight. The cells were treated with cisPt (A) or PCNA-I1S (B) for 24 hours. After removal of cisPt or PCNA-I1S, the cells were treated with PCNA-I1S (A) or cisPt (B) for 24 hours. After an incubation for additional 48 hours in the absence of the drugs, cells in the cultures were subjected to MTT assay. Data shown are from one representative experiment of three. *, p<0.05 (Fig 3A, cisPt alone vs. the combinations of cisPt plus PCNA-I1S).
To explore the potential mechanisms underlying the additive effects of cisPt and PCNA-I1S on cell growth inhibition, A549 cells were treated with PCNA-I1S and cisPt in a reverse sequence as that in the experiment shown in Fig 3A, i.e., first with PCNA-I1S (0.5 uM) for 24 hours, followed by cisPt (1.5 uM) for 24 hours. The treatment with either PCNA-I1S or cisPt alone induced growth inhibition in A549 cells to the similar extent as those shown in Fig 3A (Fig 3B). However, the additive effects of PCNA-I1S and cisPt on cell growth inhibition were not observed in this reverse treatment setting (Fig 3B), suggesting that the additive effects are due to attenuation of DNA repair by PCNA-I1S.
The effects of PCNA-I1S on PCNA association with chromatin and DNA damage
PCNA is a nucleoplasmic protein and must be relocalized to execute its functions in response to various stimuli. In replicating cells, growth-stimulating signals induce PCNA relocalization to DNA replication forks to serve as platforms on DNA templates for partner proteins involved in DNA synthesis [2, 16, 40]. In quiescent cells, on the other hand, PCNA relocalizes to DNA repair foci upon DNA damage and is a critical component in multiple DNA repair pathways [41, 42]. Oxidative stress, spontaneous hydrolysis, and chemotherapeutic alkylating drugs cause DNA base damage, which is repaired by NER or BER, two processes requiring the participation of PCNA at DNA repair foci [42, 43]. PCNA relocalization to DNA repair foci induced by H2O2 is a reliable model for investigating oxidative stress-induced DNA damage and repair [42, 43]. We determined the effects of PCNA-I1S on oxidative stress-induced PCNA association to chromatin. To preferentially evaluate the association of PCNA with DNA repair foci, not the replication forks, we determined the effects of PCNA-I1S in PC-3 cells that were arrested at G1 phase of the cell cycle after starvation in serum-free medium for 72 hours [29]. The serum-starved PC-3 cells were incubated for 30 minutes in fresh medium or the medium with PCNA-I1S (1 uM) and then treated with H2O2 (100 uM) for 1 hour in the absence or presence of PCNA-I1S. After treatment with the hypotonic buffer containing NP-40 to release the free-form PCNA, chromatin-bound PCNA was revealed by immunofluorescent staining. As shown in Fig 4A, a low level of chromatin-bound PCNA was detected in the quiescent PC-3 cells. The chromatin-bound PCNA was greatly elevated in cells treated with H2O2 (Fig 4A). Treatment with PCNA-I1S significantly reduced H2O2-induced PCNA association with chromatin (Fig 4A). For validating the observation in immunocytochemistry, we performed immunoblotting. Data in Fig 4B shows that treatment with H2O2 and PCNA-I1S alone, or in combination did not significantly alter the level of free-form PCNA in the cells (NP-E fraction). In contrast, H2O2 enhanced the level of chromatin-bound PCNA (NP-R) by approximately 5 folds in the serum-starved PC-3 cells and the effect of H2O2 was significantly attenuated by PCNA-I1S.
Fig 4. The effects of H2O2 and PCNA-I1S on PCNA association with chromatin.

A. PC-3 cells (2 x 104/well) were plated into chamber slides and incubated overnight. After starvation in serum-free medium for 3 days, the cells were treated with PCNA-I1S (1 uM) for 1.5 hours and/or H2O2 (100 uM) for 1 hour. The cells were lysed in a hypotonic buffer containing 0.5% NP-40, fixed in cold methanol, stained with PCNA antibody (PC10) and fluorescent secondary antibody. The cells were counterstained with DAPI to reveal cell density and nucleus. B. The cells were starved and treated as described in A. The NP-E and NP-R proteins were analyzed by immunoblotting to detect the free-form and chromatin-bound PCNA with α-tubulin and histone H1 levels as loading controls for NP-E and NP-R PCNA, respectively. The optical density of chromatin-bound PCNA was normalized to histone H1. The raw images of immunoblotting are provided as the supplementary data (S1 Raw Images).
CisPt binds to and induces DNA damages by forming monoadducts, intrastrand crosslinks, interstrand crosslinks (ICLs), and eventually DSBs [9, 44]. The accumulation of γH2AX is a reliable biomarker for DNA DSBs [45–47]. We determined the accumulation of γH2AX in A549 cells treated with PCNA-I1S and/or cisPt. As shown in Fig 5A, treatment with PCNA-I1S (1 uM) or cisPt (5 uM) alone for 24 hours increased γH2AX level in the cells. The expression of γH2AX was significantly elevated in cells treated with cisPt plus PCNA-I1S, suggesting an additive effect of PCNA-I1S and cisPt on DNA damage. We further determined the effects of cisPt and PCNA-I1S on DNA damage in single cells using the alkaline Comet assay that detects DNA SSBs and DSBs [34, 35]. Fig 5B shows that treatment with cisPt or PCNA-I1S alone induced DNA damage in A549 cells and the extent of DNA damage was further increased in the cells treated with cisPt plus PCNA-I1S. The images were quantitatively analyzed with OpenComet software to derive Tail DNA% and Extent Tail moment, two parameters recommended for monitoring the extent of DNA damage in mammalian cells [48]. Both Tail DNA% (Fig 5C) and Extent Tail Moment (Fig 5D) were significantly increased in cells treated with cisPt or PCNA-I1S alone. An additive effect of cisPt and PCNA-I1S was observed in the induction of Tail DNA% and Extent Tail Moment (Fig 5C and 5D).
Fig 5. The effects of cisPt and PCNA-I1S on DNA damage.

A. A549 cells (1 x 104/well) were plated into chambered slides. After an overnight incubation. the cells were treated with PCNA-I1S and/or cisPt and immunofluorescently stained with antibody to γH2AX. The cells were counterstained with DAPI to reveal cell density and nucleus as controls. B. A549 cells (1 x 106/plate) were plated into 60-mm plates and incubated overnight. After treatment with PCNA-I1S and/or cisPt, the cells were gently detached with a rubber scraper and analyzed with the Comet assay kits following the manufacture’s instruction. The images of Comet assay with 60–100 cells each treatment group were analyzed with OpenComet software to derive Tail DNA% and Extent Tail Moment, which were showed in C and D, respectively. *, p < 0.05 for all the comparisons as indicated in Fig 5C and Fig 5D.
The effects of PCNA-I1S on DNA repair
PCNA is a critical component in multiple DNA repair pathways [49, 50], such as BER [42, 51], NER [50, 52], and HRR [50, 53]. The interference of DNA repair could be a major mechanism responsible for DNA damage (Fig 5) and growth inhibition (Figs 1–3) observed in PCNA-I1S-treated cells. We have shown that PCNA-I1S interferes with PCNA recruitment to chromatin induced by H2O2 (Fig 4). We further determined the effects of PCNA-I1S on DNA repair in two well-characterized model systems.
DNA DSBs in cells are repaired mainly through HRR and the nonhomologous end joining (NHEJ) [50, 53, 54]. HRR requires the participation of PCNA [50, 53, 54], whereas NHEJ is largely PCNA-independent [55]. We determined the effects of PCNA-I1S on HRR, a pathway mainly responsible for repairing DSBs at the S and G2 phases of the cell cycle in replicating cells [56]. PC-3 and A549 cells were transiently transfected for 4 hours with linearized pDR-GFP or control plasmid pDsRed. The transfected cells were then treated for 48 hours with PCNA-I1S (0.5 uM). Expression of GFP, the indicator of HRR activity, in control and treated cells was examined. Data in Fig 6 showed that the numbers of GFP-expressing PC-3 cells (Fig 6A and 6C) and A549 cells (Fig 6B and 6C), but not those of pDsRed expressing cells, were significantly reduced by PCNA-I1S, indicating that PCNA-I1S attenuated HRR in the cells.
Fig 6. The effects of PCNA-I1S on HRR.
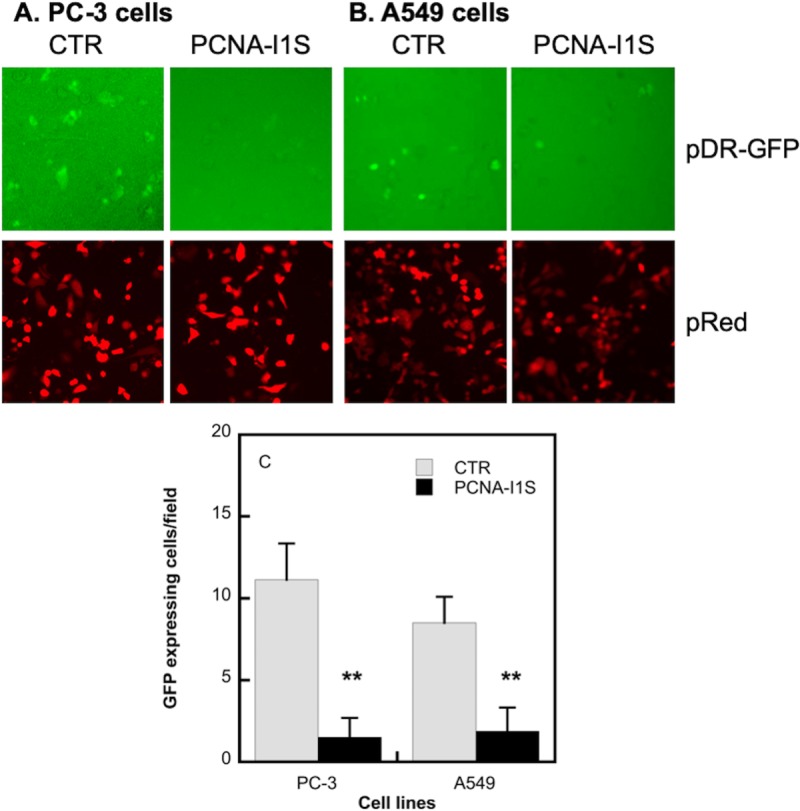
PC-3 cells (2 x 104cells/well, 6 well/treatment group) (A and C) and A549 cells (2 x 104cells/well, 6 well/treatment group) (B and C) were plated into 96-well plates and incubated overnight. The cells were transfected for 4 hours with I-SCE1-linearized pDR-GFP or pDsRed using Lipofectamine 3000 and incubated for additional 48 hours in the absence or presence of 0.5 uM PCNA-I1S. GFP-expressing cells in 10 fields were counted and calculated (C). **, p<0.01.
UV irradiation induces two types of DNA damage: CPD and pyrimidine (6–4) pyrimidone photoproducts [50, 52]. CDP in cells is mainly repaired by NER. NER is also responsible for repair DNA damage induced by some chemotherapeutic agents, such as cisPt [57, 58]. Indeed, platinum drug resistance was shown to be associated with tumor cells with an elevated NER capacity [58]. We have observed the additive effects of PCNA-I1S and UV-C irradiation in the growth inhibition experiments (Fig 2). Using the UV-induced CPD as a model system [38], we further determined the effects of PCNA-I1S on repair of CPD. A strong nuclear CPD staining was detected in 30 minutes in all PC-3 and A549 cells upon exposure to UV-C at 10 mJ/cm2 (Fig 7, UV-C; CPD vs. DAPI), but not in the control cells (Fig 7, CTR). The nuclear CPD staining was not observed in the cells cultured in medium or treated with PCNA-I1S (0.5 uM) for 24 hours (S1 Supplementary Data). The number of CPD-positive cells, as well as CDP staining intensity, was significantly reduced in the cells exposed to 10 mJ/cm2 UV-C, followed by incubation for 24 hours in medium (Fig 7, UV-C + medium for 24 h), suggesting that CPD was repaired in the cells. In contrast, almost all cells (CPD vs. DAPI), that were exposed to UV-C, followed by incubation with PCNA-I1S (0.5 uM) for 24 hours, remained positive with nuclear CPD staining (Fig 7, UV-C+PCNA-I1S for 24 h). However, the intensity of nuclear CPD staining in these cells was significantly reduced in comparison with those stained in 30 minutes after UV-C irradiation (Fig 7, UV-C vs. UV-C+PCNA-I1S for 24 h). These data implicate that treatment with PCNA-I1S attenuated NER-mediated removal of CPD.
Fig 7. The effects of PCNA-I1S on repair of UV-C-induced CPD.

PC-3 and A549 cells (2 x 104cells/well) in phenol red-free medium were plated into 96-well plates and incubated overnight. The cells were exposed to 10 mJ/cm2 UV-, followed by incubation for 24 hours in the absence or presence of 0.5 uM PCNA-I1S. CPD in the cells were immunofluorescently stained in 30 minutes after UV-C irradiation or at end of the 24-hour incubation with kits following the manufacture’s instruction. The cells were counterstained with DAPI to reveal cell density and nucleus as a control.
Discussion
Previously we reported the identification and characterization of a novel class of small-molecule PCNA inhibitors that bind directly to PCNA trimers, stabilize the trimer structure, reduce PCNA association with chromatin, inhibit DNA replication, and selectively inhibit tumor cell growth [29]. Moreover, we found that treatment with the lead compound PCNA-I1 induces DNA damage and apoptosis, and suppresses tumor growth in mice [31]. Through the structure-activity relationship analysis and functional validation, we identified another lead compound PCNA-I1S that is more potent than PCNA-I1 in suppressing cell growth and stabilizing PCNA trimer structure [30]. The objective of the present study was to further investigate the effects of PCNA-I1S alone or in combination with several types of DNA damaging agents on cell growth inhibition, DNA damage, and to evaluate the effects of PCNA-I1S on DNA repair in prostate and lung cancer cells. Our data show that the combination of PCNA-I1S with DNA damage agents Cs-137 irradiation, UV-C irradiation, or cisPt produced significant additive effects on cell growth inhibition and DNA damage, and PCNA-I1S attenuated DNA repair in human prostate and lung cancer cells.
PCNA is indispensable for DNA replication and multiple DNA repair pathways [2, 11, 15, 16, 54]. Targeting PCNA with PCNA-I1S may inhibit cell growth through inhibition of DNA replication as well as attenuation of DNA repair. Suppression of PCNA relocalization to DNA replication forks will lead to stalling of DNA replication and increase susceptibility of DNA damage, resulting in the formation of DNA DSBs and cell growth inhibition [59]. Indeed, we found that targeting PCNA with PCNA-I1 inhibits DNA replication, induces S-G2-M phase arrest, results in the accumulation of DNA DSB marker γH2AX and activation of Chk2, increases expression of p53 and phosphorylation of p53, leading to growth inhibition [30, 31]. The similar effects of PCNA-I1 on cell growth and DNA damage were observed in cells treated with PCNA-I1S in the present study (Figs 1–4).
PCNA participates in multiple DNA repair pathways, such as BER, NER, HRR, and mismatch repair (MMR), which require DNA re-synthesis and involve PCNA interaction with DNA polymerase δ or polymerase ε [2, 16, 54]. In NER that repairs DNA damage induced by UV-C irradiation and some chemotherapeutic agents, such as cisPt [57, 58], PCNA facilitates the recruitment of the essential proteins endonuclease XP-G and XP-A for DNA repair [60, 61]. In BER that repairs DNA base damage induced by oxidative stress, spontaneous hydrolysis, and chemotherapeutic alkylating drugs [42, 43], PCNA interacts with polymerase β, and AP endonucleases Apn and Apn2, as well as other essential protein components [16]. For HRR that repairs DNA DSBs at S and G2 phases of the cell cycle in replicating cells [56], PCNA interacts with WRN helicase at the N-terminus domain [62]. Targeting PCNA with PCNA-I1S attenuated PCNA relocalization to chromatin (Fig 4), suppressed HRR- and NER-mediated DNA repair (Figs 6 and 7), and produced an additive growth inhibition with three types of DNA damaging agents (ionizing radiation with Cs-137, chemotherapeutic drug cisPt, and UV-C irradiation). Moreover, the combinatorial treatment of PCNA-I1S and cisPt produced additive effects on DNA damage (Fig 5). These observations confirmed and extended the findings reported by others on the additive effects of cisPt, bleomycin, or taxanes with small molecule PCNA inhibitors targeting PIP box [25, 63] or caPCNA [26], or a peptide inhibitor targeting APIM sequence [21, 28] on DNA damage, DNA repair, and cytotoxicity. Interestingly, we noted that the additive effects of PCNA-I1S with cisPt were observed only in the cells exposed to cisPt first, followed by PCNA-I1S, but not in those treated with the two agents in a reverse sequence. Given the significant inhibitory effects of PCNA-I1S on HRR- and NER-mediated DNA repair observed in this study, this data suggests that attenuation of DNA repair by PCNA-I1S is very likely the main mechanism responsible for the additive effects of PCNA-I1S with DNA damaging agents on tumor cell growth inhibition.
In conclusion, the data in this study provide strong evidence that targeting PCNA with PCNA-I1S suppresses PCNA association with chromatin in quiescent cells, inhibits DNA repair pathways, and produces the additive growth inhibitory effects with DNA damage agents. Moreover, the data suggest that targeting PCNA with these PCNA inhibitors may provide a novel approach for enhancing the efficacy of chemotherapy and radiation therapy in treatment of human prostate and lung cancers.
Supporting information
(TIFF)
PC-3 and A549 cells (2 x 104cells/well) in phenol red-free medium were plated into 96-well plates and incubated overnight. The cells were incubated for additional 24 hours in the absence or presence of 0.5 uM PCNA-I1S. CPD in the cells were immunofluorescently stained with kits following the manufacture’s instruction. The cells were also counterstained with DAPI to reveal cell density and nucleus as a control.
(TIFF)
Acknowledgments
The authors would like to thank Drs. EL Mustapha Bahassi and Peter Stambrook for generously providing us with the homologous recombination repair reporter system, Dr. Harold Davis for assistance in irradiating cells with Cs-137 source, Mrs. Sue Chen and Mr. Zongqin Tan for technical assistance in carrying out some experiments.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported in part by a 1R21CA223049-01 grant (ZD) from the National Institutes of Health, National Cancer Institute, Millennium Scholar Funds from the University of Cincinnati Cancer Center (ZD), and the Dean’s Bridge Funding of College of Medicine, University of Cincinnati (ZD). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Almendral JM, Huebsch D, Blundell PA, Macdonald-Bravo H, Bravo R. Cloning and sequence of the human nuclear protein cyclin: homology with DNA-binding proteins. Proc Natl Acad Sci U S A. 1987;84(6):1575–9. 10.1073/pnas.84.6.1575 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stoimenov I, Helleday T. PCNA on the crossroad of cancer. Biochemical Society transactions. 2009;37(Pt 3):605–13. 10.1042/BST0370605 . [DOI] [PubMed] [Google Scholar]
- 3.Lu S, Lee J, Revelo M, Wang X, Lu S, Dong Z. Smad3 is overexpressed in advanced human prostate cancer and necessary for progressive growth of prostate cancer cells in nude mice. Clin Cancer Res. 2007;13(19):5692–702. 10.1158/1078-0432.CCR-07-1078 . [DOI] [PubMed] [Google Scholar]
- 4.Harper ME, Glynne-Jones E, Goddard L, Wilson DW, Matenhelia SS, Conn IG, et al. Relationship of proliferating cell nuclear antigen (PCNA) in prostatic carcinomas to various clinical parameters. Prostate. 1992;20(3):243–53. 10.1002/pros.2990200309 . [DOI] [PubMed] [Google Scholar]
- 5.Zdunek M, Korobowicz E. Expression of PCNA in non-small cell lung cancer before and after treatment with cisplatin and vepeside. Polish journal of pathology: official journal of the Polish Society of Pathologists. 2000;51(2):77–81. Epub 2000/09/07. . [PubMed] [Google Scholar]
- 6.Kimos MC, Wang S, Borkowski A, Yang GY, Yang CS, Perry K, et al. Esophagin and proliferating cell nuclear antigen (PCNA) are biomarkers of human esophageal neoplastic progression. Int J Cancer. 2004;111(3):415–7. 10.1002/ijc.20267 . [DOI] [PubMed] [Google Scholar]
- 7.Cappello F, Ribbene A, Campanella C, Czarnecka AM, Anzalone R, Bucchieri F, et al. The value of immunohistochemical research on PCNA, p53 and heat shock proteins in prostate cancer management: a review. Eur J Histochem. 2006;50(1):25–34. . [PubMed] [Google Scholar]
- 8.Malkas LH, Herbert BS, Abdel-Aziz W, Dobrolecki LE, Liu Y, Agarwal B, et al. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc Natl Acad Sci U S A. 2006;103(51):19472–7. 10.1073/pnas.0604614103 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nature Reviews: Cancer. 2008;8(3):193–204. Epub 2008/02/08. 10.1038/nrc2342 . [DOI] [PubMed] [Google Scholar]
- 10.Hosoya N, Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014;105(4):370–88. 10.1111/cas.12366 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naryzhny SN. Proliferating cell nuclear antigen: a proteomics view. Cell Mol Life Sci. 2008;65(23):3789–808. 10.1007/s00018-008-8305-x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79(7):1233–43. 10.1016/0092-8674(94)90014-0 . [DOI] [PubMed] [Google Scholar]
- 13.Bowman GD, O'Donnell M, Kuriyan J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature. 2004;429(6993):724–30. 10.1038/nature02585 . [DOI] [PubMed] [Google Scholar]
- 14.Fukuda K, Morioka H, Imajou S, Ikeda S, Ohtsuka E, Tsurimoto T. Structure-function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J Biol Chem. 1995;270(38):22527–34. Epub 1995/09/22. 10.1074/jbc.270.38.22527 . [DOI] [PubMed] [Google Scholar]
- 15.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129(4):665–79. 10.1016/j.cell.2007.05.003 . [DOI] [PubMed] [Google Scholar]
- 16.Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116(Pt 15):3051–60. 10.1242/jcs.00653 [DOI] [PubMed] [Google Scholar]
- 17.Witko-Sarsat V, Mocek J, Bouayad D, Tamassia N, Ribeil JA, Candalh C, et al. Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J Exp Med. 2010;207(12):2631–45. 10.1084/jem.20092241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohayon D, De Chiara A, Chapuis N, Candalh C, Mocek J, Ribeil JA, et al. Cytoplasmic proliferating cell nuclear antigen connects glycolysis and cell survival in acute myeloid leukemia. Sci Rep. 2016;6:35561 Epub 2016/10/21. 10.1038/srep35561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilljam KM, Feyzi E, Aas PA, Sousa MM, Muller R, Vagbo CB, et al. Identification of a novel, widespread, and functionally important PCNA-binding motif. The Journal of cell biology. 2009;186(5):645–54. Epub 2009/09/09. 10.1083/jcb.200903138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horton NC, Mathew SO, Mathew PA. Novel interaction between proliferating cell nuclear antigen and HLA I on the surface of tumor cells inhibits NK cell function through NKp44. PloS one. 2013;8(3):e59552 10.1371/journal.pone.0059552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller R, Misund K, Holien T, Bachke S, Gilljam KM, Vatsveen TK, et al. Targeting proliferating cell nuclear antigen and its protein interactions induces apoptosis in multiple myeloma cells. PloS one. 2013;8(7):e70430 Epub 2013/08/13. 10.1371/journal.pone.0070430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sogaard CK, Moestue SA, Rye MB, Kim J, Nepal A, Liabakk NB, et al. APIM-peptide targeting PCNA improves the efficacy of docetaxel treatment in the TRAMP mouse model of prostate cancer. Oncotarget. 2018;9(14):11752–66. 10.18632/oncotarget.24357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith SJ, Gu L, Phipps EA, Dobrolecki L, Mabrey KS, Gulley P, et al. A peptide mimicking a region in proliferating cell nuclear antigen specific to key protein interactions Is cytotoxic to breast cancer. Mol Pharmacol. 2015;87:263–76. 10.1124/mol.114.093211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Actis M, Inoue A, Evison B, Perry S, Punchihewa C, Fujii N. Small molecule inhibitors of PCNA/PIP-box interaction suppress translesion DNA synthesis. Bioorganic & medicinal chemistry. 2013;21(7):1972–7. Epub 2013/02/12. 10.1016/j.bmc.2013.01.022 . [DOI] [PubMed] [Google Scholar]
- 25.Punchihewa C, Inoue A, Hishiki A, Fujikawa Y, Connelly M, Evison B, et al. Identification of a small molecule PCNA inhibitor that disrupts interactions with PIP-Box proteins and inhibits DNA replication. Journal of Biological Chemistry. 2012;287(17):14289–300. Epub 2012/03/03. 10.1074/jbc.M112.353201 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu L, Lingeman R, Yakushijin F, Sun E, Cui Q, Chao J, et al. The Anticancer Activity of a First-in-class Small-molecule Targeting PCNA. Clin Cancer Res. 2018;24(23):6053–65. Epub 2018/07/04. 10.1158/1078-0432.CCR-18-0592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desplancq D, Freund G, Conic S, Sibler AP, Didier P, Stoessel A, et al. Targeting the replisome with transduced monoclonal antibodies triggers lethal DNA replication stress in cancer cells. Exp Cell Res. 2016;342(2):145–58. 10.1016/j.yexcr.2016.03.003 . [DOI] [PubMed] [Google Scholar]
- 28.Gederaas OA, Sogaard CD, Viset T, Bachke S, Bruheim P, Arum CJ, et al. Increased Anticancer Efficacy of Intravesical Mitomycin C Therapy when Combined with a PCNA Targeting Peptide. Transl Oncol. 2014;7(6):812–23. 10.1016/j.tranon.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan Z, Wortman M, Dillehay KL, Seibel WL, Evelyn CR, Smith SJ, et al. Small-molecule targeting of proliferating cell nuclear antigen chromatin association inhibits tumor cell growth. Molecular pharmacology. 2012;81(6):811–9. Epub 2012/03/09. 10.1124/mol.112.077735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dillehay KL, Seibel WL, Zhao D, Lu S, Dong Z. Target validation and structure-activity analysis of a series of novel PCNA inhibitors. Pharmacol Res Perspect. 2015;3(2):e00115 10.1002/prp2.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dillehay K L., Lu S, Dong Z. Antitumor effects of a novel small molecule targeting PCNA chromatin association in prostate cancer. Mol Cancer Ther. 2014;13:2817–26. 10.1158/1535-7163.MCT-14-0522 [DOI] [PubMed] [Google Scholar]
- 32.Tichy ED, Pillai R, Deng L, Liang L, Tischfield J, Schwemberger SJ, et al. Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem cells and development. 2010;19(11):1699–711. 10.1089/scd.2010.0058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong ZY, Ward NE, Fan D, Gupta KP, O'Brian CA. In vitro model for intrinsic drug resistance: effects of protein kinase C activators on the chemosensitivity of cultured human colon cancer cells. Mol Pharmacol. 1991;39(4):563–9. Epub 1991/04/01. . [PubMed] [Google Scholar]
- 34.Shibata Y, Nakamura T. Defective flap endonuclease 1 activity in mammalian cells is associated with impaired DNA repair and prolonged S phase delay. J Biol Chem. 2002;277(1):746–54. 10.1074/jbc.M109461200 . [DOI] [PubMed] [Google Scholar]
- 35.Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Molecular biotechnology. 2004;26(3):249–61. 10.1385/MB:26:3:249 . [DOI] [PubMed] [Google Scholar]
- 36.Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, Clement MV. OpenComet: an automated tool for comet assay image analysis. Redox Biol. 2014;2:457–65. Epub 2014/03/14. 10.1016/j.redox.2013.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rastogi RP, Richa, Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids. 2010;2010:592980 Epub 2011/01/07. 10.4061/2010/592980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soehnge H, Ouhtit A, Ananthaswamy ON. Mechanisms of induction of skin cancer by UV radiation. Front Biosci. 1997;2:d538–51. Epub 1997/10/31. 10.2741/a211 . [DOI] [PubMed] [Google Scholar]
- 39.Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382(9893):709–19. 10.1016/S0140-6736(13)61502-0 . [DOI] [PubMed] [Google Scholar]
- 40.Strzalka W, Ziemienowicz A. Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann Bot. 2011;107(7):1127–40. Epub 2010/12/21. 10.1093/aob/mcq243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karmakar P, Balajee AS, Natarajan AT. Analysis of repair and PCNA complex formation induced by ionizing radiation in human fibroblast cell lines. Mutagenesis. 2001;16(3):225–32. 10.1093/mutage/16.3.225 . [DOI] [PubMed] [Google Scholar]
- 42.Savio M, Stivala LA, Bianchi L, Vannini V, Prosperi E. Involvement of the proliferating cell nuclear antigen (PCNA) in DNA repair induced by alkylating agents and oxidative damage in human fibroblasts. Carcinogenesis. 1998;19(4):591–6. 10.1093/carcin/19.4.591 . [DOI] [PubMed] [Google Scholar]
- 43.Balajee AS, Dianova I, Bohr VA. Oxidative damage-induced PCNA complex formation is efficient in xeroderma pigmentosum group A but reduced in Cockayne syndrome group B cells. Nucleic Acids Res. 1999;27(22):4476–82. Epub 1999/10/28. 10.1093/nar/27.22.4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blommaert FA, van Dijk-Knijnenburg HC, Dijt FJ, den Engelse L, Baan RA, Berends F, et al. Formation of DNA adducts by the anticancer drug carboplatin: different nucleotide sequence preferences in vitro and in cells. Biochemistry. 1995;34(26):8474–80. Epub 1995/07/04. 10.1021/bi00026a031 . [DOI] [PubMed] [Google Scholar]
- 45.Kuo LJ, Yang LX. Gamma-H2AX—a novel biomarker for DNA double-strand breaks. In vivo (Athens, Greece). 2008;22(3):305–9. . [PubMed] [Google Scholar]
- 46.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. Journal of Biological Chemistry. 1998;273(10):5858–68. Epub 1998/04/16. 10.1074/jbc.273.10.5858 . [DOI] [PubMed] [Google Scholar]
- 47.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic acids research. 2008;36(17):5678–94. Epub 2008/09/06. 10.1093/nar/gkn550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee E, Oh E, Lee J, Sul D, Lee J. Use of the tail moment of the lymphocytes to evaluate DNA damage in human biomonitoring studies. Toxicol Sci. 2004;81(1):121–32. Epub 2004/06/05. 10.1093/toxsci/kfh184 . [DOI] [PubMed] [Google Scholar]
- 49.Wit N, Buoninfante OA, van den Berk PC, Jansen JG, Hogenbirk MA, de Wind N, et al. Roles of PCNA ubiquitination and TLS polymerases kappa and eta in the bypass of methyl methanesulfonate-induced DNA damage. Nucleic Acids Res. 2015;43(1):282–94. 10.1093/nar/gku1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iyama T, Wilson DM 3rd., DNA repair mechanisms in dividing and non-dividing cells. DNA Repair. 2013;12(8):620–36. 10.1016/j.dnarep.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scovassi AI, Prosperi E. Analysis of proliferating cell nuclear antigen (PCNA) associated with DNA. Methods in molecular biology. 2006;314:457–75. Epub 2006/05/06. 10.1385/1-59259-973-7:457 . [DOI] [PubMed] [Google Scholar]
- 52.Balajee AS, Geard CR. Chromatin-bound PCNA complex formation triggered by DNA damage occurs independent of the ATM gene product in human cells. Nucleic Acids Res. 2001;29(6):1341–51. 10.1093/nar/29.6.1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maugeri-Sacca M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012;11(8):1627–36. 10.1158/1535-7163.MCT-11-1040 . [DOI] [PubMed] [Google Scholar]
- 54.Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20(5):648–59. 10.1016/j.chembiol.2013.04.007 . [DOI] [PubMed] [Google Scholar]
- 55.Pospiech H, Rytkonen AK, Syvaoja JE. The role of DNA polymerase activity in human non-homologous end joining. Nucleic Acids Res. 2001;29(15):3277–88. 10.1093/nar/29.15.3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan MA, Lawrence TS. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin Cancer Res. 2015;21(13):2898–904. Epub 2015/07/03. 10.1158/1078-0432.CCR-13-3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scharer OD. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013;5(10):a012609 Epub 2013/10/03. 10.1101/cshperspect.a012609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res. 2008;14(5):1291–5. 10.1158/1078-0432.CCR-07-2238 . [DOI] [PubMed] [Google Scholar]
- 59.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. Epub 2013/12/25. 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gary R, Ludwig DL, Cornelius HL, MacInnes MA, Park MS. The DNA repair endonuclease XPG binds to proliferating cell nuclear antigen (PCNA) and shares sequence elements with the PCNA-binding regions of FEN-1 and cyclin-dependent kinase inhibitor p21. J Biol Chem. 1997;272(39):24522–9. Epub 1997/09/26. 10.1074/jbc.272.39.24522 . [DOI] [PubMed] [Google Scholar]
- 61.Miura M, Sasaki T. Effect of XPA gene mutations on UV-induced immunostaining of PCNA in fibroblasts from xeroderma pigmentosum group A patients. Mutat Res. 1996;364(1):51–6. Epub 1996/09/02. 10.1016/0921-8777(96)00021-3 . [DOI] [PubMed] [Google Scholar]
- 62.Lebel M, Spillare EA, Harris CC, Leder P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J Biol Chem. 1999;274(53):37795–9. Epub 1999/12/23. 10.1074/jbc.274.53.37795 . [DOI] [PubMed] [Google Scholar]
- 63.Inoue A, Kikuchi S, Hishiki A, Shao Y, Heath R, Evison BJ, et al. A small molecule inhibitor of monoubiquitinated Proliferating Cell Nuclear Antigen (PCNA) inhibits repair of interstrand DNA cross-link, enhances DNA double strand break, and sensitizes cancer cells to cisplatin. J Biol Chem. 2014;289(10):7109–20. 10.1074/jbc.M113.520429 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIFF)
PC-3 and A549 cells (2 x 104cells/well) in phenol red-free medium were plated into 96-well plates and incubated overnight. The cells were incubated for additional 24 hours in the absence or presence of 0.5 uM PCNA-I1S. CPD in the cells were immunofluorescently stained with kits following the manufacture’s instruction. The cells were also counterstained with DAPI to reveal cell density and nucleus as a control.
(TIFF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.