

Abstract
Aims/hypothesis
This study aimed to examine changes in the insulin secretory response in early pregnancy, while accounting for changes in insulin sensitivity.
Methods
This is a secondary analysis of a previously conducted longitudinal physiological study. In 34 women, insulin secretory response (by intravenous glucose tolerance test) and insulin sensitivity (by euglycaemic clamp) were assessed prior to pregnancy, in early pregnancy (12-14 weeks gestation) and in late pregnancy (34-36 weeks gestation). Using mixed-effects models, we compared insulin secretory response and sensitivity in early pregnancy to the same variables prior to pregnancy and in late pregnancy, with adjustment for age, obesity status and gestational diabetes mellitus (GDM). We examined changes in insulin secretory response after adjustment for insulin sensitivity using both multivariate modelling and the disposition index (DI). We explored the relationship between insulin secretory response and circulating hormones.
Results
The insulin secretory response increased from prior to pregnancy to early pregnancy (unadjusted mean [SD] first-phase insulin response 465.1 [268.5] to 720 [358.2], p<0.0001) and from early pregnancy to late pregnancy (to 924 [494.6], p=0.01). Insulin sensitivity increased from prior to pregnancy to early pregnancy (insulin sensitivity index 0.10 [0.04] to 0.12 [0.05], p=0.001) and decreased in late pregnancy (to 0.06 [0.03], p<0.0001). Accounting for changes in insulin sensitivity, using either multivariate modelling or the DI, did not attenuate the early-pregnancy augmentation of insulin secretory response. Leptin was positively associated with insulin secretory response, independent of insulin sensitivity and adiposity (p=0.004). Adjustment for leptin attenuated the observed augmentation of insulin secretory response in early pregnancy (adjusted mean change 121.5, p=0.13).
Conclusions/interpretation
The insulin secretory response increases markedly in early pregnancy, prior to and independent of changes in insulin sensitivity. Circulating hormones may mediate this metabolic adaptation. Identifying mediators of this physiological effect could have therapeutic implications for treating hyperglycaemia during and outside of pregnancy.
Keywords: Gestational diabetes, Glucose metabolism, Insulin secretion, Physiology, Pregnancy
Introduction
During pregnancy, there are substantial changes in glucose metabolism (1–3). These alterations in maternal physiology are likely mediated through placental hormones and/or cytokines, though the exact mechanisms are unclear (4, 5).
Pregnancy produces a 50–60% decrease in insulin sensitivity by late gestation. Concomitantly, there is a two- to threefold increase in insulin secretory response. Indeed, limited autopsy studies in humans suggest that beta cell mass increases by at least 40% during pregnancy (6, 7). A prevailing theory is that the increase in insulin secretory response is compensatory for the decrease in maternal insulin sensitivity that occurs with advancing gestation. However, in animal models, lactogenic hormones seem to directly stimulate beta cells, independent of insulin resistance (8–12).
Many of the studies examining changes in maternal glucose metabolism during pregnancy have been performed during late pregnancy and postpartum. Thus, the metabolic changes that occur in early pregnancy are not well described (4, 13, 14). We hypothesised that the insulin secretory response increases in early pregnancy, independent of insulin sensitivity. We tested this hypothesis, then explored the relationships between circulating hormones and cytokines and insulin secretory response or insulin sensitivity.
Methods
Participants
This is a secondary analysis of a prospective longitudinal study of the metabolic changes in glucose metabolism during pregnancy (1–3). The study was approved by the hospital’s institutional review board and written informed consent was obtained from each participant. Nineteen women with normal glucose tolerance (NGT) before and during pregnancy and 15 women with NGT prior to pregnancy who developed gestational diabetes mellitus (GDM) during pregnancy enrolled. We defined obesity as percentage body fat ≥ 25% prior to pregnancy. In order to obtain approximately equal numbers of obese and lean participants who did and did not develop GDM, women were pre-selected based on weight and other risk factors, as previously described (1–3). Women who developed GDM were free of diabetes prior to pregnancy but had a history of GDM in a prior pregnancy and a family history of type 2 diabetes. Participants had no medical or obstetrical problems other than GDM. None of the participants was using tobacco, was breastfeeding or was using hormonal contraception prior to conception.
Experimental protocol
Physiological measures were conducted prior to pregnancy and repeated in early (12–14 weeks) and late (34-36 weeks) gestation. Of the 34 participants, 29 were evaluated in the follicular phase of the menstrual cycle for the studies prior to pregnancy. Each participant was instructed in a dietary regimen for 2 weeks before each study period by a research nutritionist. The dietary regimen was designed to standardise nutritional intake, to maintain weight before pregnancy and to allow appropriate increases in weight during pregnancy. The diet was identical to that used for the treatment of GDM at the hospitals where the study was performed.
The following sets of studies were performed on three separate days: (1) an OGTT and routine laboratory studies; (2) hydrodensitometry to estimate body composition and an intravenous glucose tolerance test (IVGTT); and (3) the hyperinsulinaemic–euglycaemic clamp with infusion of a stable isotope of glucose ([6,6- 2H2]glucose) to estimate residual endogenous glucose production during the clamp.
Oral glucose tolerance testing
Prior to pregnancy, each participant had a 75 g OGTT to ensure that pre-gestational diabetes was not present. During pregnancy, women had an 100 g OGTT and GDM was classified according to the Carpenter and Coustan criteria (15). Plasma glucose concentrations were measured with the glucose oxidase method using a Yellow Springs glucose analyser (Yellow Springs Instrument, Yellow Springs, OH, USA).
Intravenous glucose tolerance testing and insulin secretory response
For participants who were less than 120% ideal body weight, we infused 0.5 g/kg glucose as a bolus over 3 min. Samples of glucose and insulin were obtained at baseline (0 min) and at 1, 3, 5, 10, 15, 30, 45 and 60 min. For those participants whose weight was greater than 120% ideal body weight, the glucose bolus was 19 g/m2 body surface area. Using the trapezoidal rule, we estimated the first-phase insulin response as the AUC from 0 to 10 min. Insulin concentrations were measured with a double-antibody method (16). Insulin was bound to an anti-insulin primary antibody (guinea pig, ICN 65-101) and the complex was precipitated by addition of an anti-guinea pig IgG (Antibodies Incorporated, Davis, CA, USA) and a precipitating agent. Percentage binding was compared against insulin standards. The intra-assay CV was 6% and the inter-assay CV was 8%.
Body composition
We estimated each participant’s body composition by underwater weighing with adjustment for residual lung volume by nitrogen dilution. Percentage of body fat was calculated according to the Keys and Brozek and Siri equations to estimate fat-free mass (17, 18).
Hyperinsulinaemic–euglycaemic clamp and insulin sensitivity
We performed hyperinsulinaemic–euglycaemic clamps to estimate peripheral insulin sensitivity after an 11 h fast, as described by DeFronzo et al (3, 19) An intravenous catheter was placed for infusion of insulin, glucose and [6,6- 2H2]glucose. A catheter was placed retrograde into the contralateral hand for blood withdrawal and warmed to 70°C using a warming box. The infusion of [6,6- 2H2]glucose was continued for 2 h to estimate basal endogenous glucose production. At the completion of the 2 h infusion, the clamp procedure was initiated with a primed constant infusion of insulin, 40 mU m−2 min−1, to achieve a plasma insulin concentration of approximately 600 pmol m−2 ml−1. Plasma glucose was maintained at 5.0 mmol/l for 120 min by sampling the plasma glucose every 5 min and varying the 20% glucose infusion rate. The glucose and insulin concentrations collected every 10 min during the last 40 min of the clamp were averaged and used to estimate glucose disposal. Residual endogenous glucose production during the clamp was estimated by adding a calculated amount of the [6,6- 2H2]glucose to the 20% glucose infusion as described by Finegood et al (20) A penta-acetate derivative of glucose was prepared as described by Tsemg and Kalhan (21) Plasma enrichment was determined with a gas chromatograph mass spectrometer (model 5985B; Hewlett-Packard, Pal Alto, CA, USA).
We estimated insulin sensitivity as the glucose infusion rate required to maintain plasma glucose at 5.0 mmol/l during the clamp. Because there was not complete suppression of endogenous glucose production during insulin infusion and plasma insulin concentrations varied during the clamp across time and between groups, we estimated an insulin sensitivity index to account for these variations. We defined insulin sensitivity index as the glucose infusion rate plus any residual endogenous glucose production per kg of fat-free mass during the clamp divided by the mean insulin concentration achieved during the clamp.
Disposition index
To describe the degree of beta cell compensation for decrements in insulin sensitivity without multivariate modelling, we used the disposition index (DI). The DI is the product of the insulin secretory response (IVGTT first-phase insulin response) and the insulin sensitivity (insulin sensitivity index from hyperinsulinaemic–euglycaemic clamp) (22, 23).
Hormone and cytokine analysis
Measurements of fasting TNF-α, leptin, cortisol and reproductive hormones were run in duplicate. Plasma TNF-α concentrations were measured by an ELISA (Quantikine HS; R&D Systems, Minneapolis, MN, USA). Plasma cortisol, oestradiol, human placental lactogen (hPL) and progesterone were determined using RIA (Diagnostic Products, Los Angeles, CA, USA). Plasma leptin samples were also measured using RIA (Linco Research, St Charles, MO, USA). Plasma human chorionic gonadotropin (hCG) was determined by immunoradiometric assay (Diagnostic Products). Plasma prolactin was measured by RIA (Nichols Institute Diagnostics, San Juan Capistrano, CA, USA). Fifteen participants had levels of leptin and TNF-α measured; reproductive hormone measures were performed for all participants. Assays were performed near the time of the original study and not specifically for the present analysis.
Statistical analysis
We employed mixed-effects repeated-measures ANCOVA models, analysing insulin secretory response for change across study time points (prior to pregnancy, early pregnancy and late pregnancy). We also tested for an effect of study time point on insulin sensitivity (insulin sensitivity index) and DI (product of first-phase insulin response and insulin sensitivity index). We included insulin sensitivity as a covariate in models with insulin secretory response as the dependent variable, a-priori, where noted. Additional covariates considered for initial models, subject to backward elimination, included obesity status, GDM status, their interaction, the interaction of each of these terms with study time point, and also age. Backward elimination was conducted to remove a limited number of non-significant terms to pool error and increase precision and power for remaining terms.
We added individual time-varying cytokines and hormones to models to explore whether they were potential mediators of early-pregnancy changes in insulin secretory response and sensitivity. Fat mass was also included as a covariate in models where the cytokine/hormone of interest was significantly associated with insulin secretory response.
In all models, an unstructured correlation matrix was assumed for the random study time point variable. First-phase insulin response, insulin sensitivity index, DI, fat mass and cytokine/hormone levels were natural log-transformed prior to analysis to reduce their otherwise positively skewed distributions. In some cases, natural log-transformed data were analysed to establish the statistical significance of relations, but descriptively, statistics were reported at the raw data level for ease of interpretation. Residuals from models were checked for conformance to assumptions of normality and homoscedasticity. Statistical analyses were performed with SAS version 9.4 (SAS Institute, Cary, NC, USA). A level of p<0.05 was considered significant.
Results
The characteristics of the participants prior to pregnancy are given in Table 1. Women were studied at a mean (SD) of 10.8 (12.8) weeks prior to the last menstrual period before conception, at 13.6 (1.2) weeks gestation in early pregnancy and at 34.7 (0.88) weeks gestation in late pregnancy. Fifteen (44%) participants were classified as obese (body fat percentage ≥ 25%) prior to pregnancy. Fifteen participants (44%), eight lean and seven obese, developed GDM.
Table 1.
Characteristics of study participants prior to pregnancy
| Characteristic | Mean (SD) |
|---|---|
| n | 34 |
| Age (years) | 30.5 (3.9) |
| Time before LMP (weeks) | 10.8 (12.8) |
| Weight (kg) | 64.8 (14.8) |
| Fat mass (kg) | 16.9 (9.8) |
| Lean body mass (kg) | 47.9 (5.9) |
| BMI (kg/m2) | 23.3 (5.2) |
| Body fat (%) | 24.7 (7.6) |
| First-phase insulin response | 465.1 (268.5) |
| Insulin sensitivity index | 0.10 (0.04) |
| DI | 46.7 (30.9) |
| Leptin (ng/ml)* | 20.7 (17.9) |
| TNF-α (pg/ml)* | 1.8 (1.0) |
n=15
LMP, last menstrual period prior to conception
Longitudinal changes in the first-phase insulin response
Unadjusted changes in first-phase insulin response during pregnancy are presented in Fig. 1a. There was a significant increase in insulin secretory response from pre-pregnancy to early pregnancy (465.1 [268.5] to 720 [358.2] μU/ml, p<0.0001) and early pregnancy to late pregnancy (720 [358.2] to 924 [494.6] μU/ml, p=0.01).
Fig. 1.
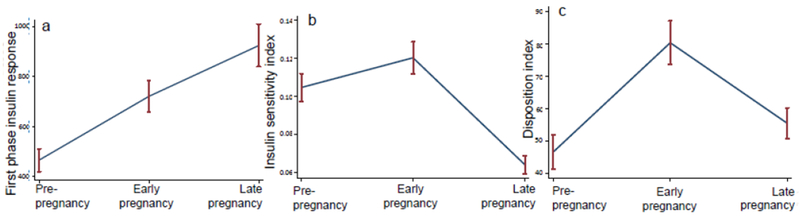
(a–c) Mean first-phase insulin response (a), insulin sensitivity index (b) and DI (c) at the three study time points: pre-pregnancy, early pregnancy and late pregnancy. Error bars denote the SEM for each time point
In the covariate-adjusted models, older age was associated with reduced insulin secretory response across the three study time points (p=0.005). Obese women had higher first-phase insulin response than lean women, independent of age and study time point (adjusted mean difference 197.4, p=0.02), but there was no difference according to obesity status in the changes in insulin secretory response across pregnancy. There were also no significant differences in first-phase insulin response or changes in insulin secretory response across pregnancy between women with and without GDM. Electronic supplementary material (ESM) Fig. 1a shows the changes in insulin secretory response across study time points by obesity and GDM status.
Adjustment for age and obesity status did not attenuate the increase in insulin secretory response observed from pre-pregnancy to early pregnancy (adjusted mean change 260.2 μU/ml, p<0.0001).
Longitudinal changes in insulin sensitivity
Unadjusted changes in insulin sensitivity during pregnancy are presented in Fig. 1b. There was a significant increase in insulin sensitivity from pre-pregnancy to early pregnancy (0.10 [0.04] to 0.12 [0.05], p=0.001) and a significant decrease in insulin sensitivity from early pregnancy to late pregnancy (0.12 [0.05] to 0.06 [0.03], p<0.0001).
As previously reported,(1) GDM was associated with lower insulin sensitivity, independent of study time point (adjusted mean difference −0.04, p=0.0003). There were no significant differences in change in insulin sensitivity across gestation according to obesity or GDM status. ESM Fig. 1b shows the changes in insulin sensitivity across study time points by obesity and GDM status.
Adjustment for GDM status did not attenuate the significant increase in insulin sensitivity observed from pre-pregnancy to early pregnancy (adjusted mean change 0.015, p=0.001).
Longitudinal changes in the disposition index
Unadjusted changes in the DI during pregnancy are presented in Fig. 1c. There was a significant increase in DI from pre-pregnancy to early pregnancy (46.7 [30.9] to 80.4 [39.1], p<0.0001) and a significant decrease from early pregnancy to late pregnancy (to 55.5 [28.0], p=0.0001). Consistent with these findings, adjustment for insulin sensitivity in the model for insulin secretory response did not attenuate the observed augmentation in insulin secretory response in early pregnancy (adjusted mean change 278.5, p<0.0001), but adjustment did eliminate the apparent augmentation of insulin secretory response between early pregnancy and late pregnancy (adjusted mean change 92.4, p=0.30).
Women with GDM had lower DI across the time points studied (adjusted mean difference 22.3, p=0.007). There were no statistically significant differences in change in DI according to obesity or GDM status. ESM Fig. 1c shows the change in DI across study time points by obesity and GDM status.
Adjustment for GDM status did not attenuate the increase in DI observed from pre-pregnancy to early pregnancy (adjusted mean change 33.4, p<0.0001).
Associations between hormones/cytokines and insulin secretory response
Leptin was positively associated with insulin secretory response, independent of insulin sensitivity, age, time point and fat mass (β=0.47, p=0.004, log-transformed values). Changes in leptin across study time points are presented in Fig. 2a. Leptin levels increased from pre-pregnancy to early pregnancy (20.7 [17.9] ng/ml to 30.2 [24.0] ng/ml, p=0.004); leptin remained stable from early to late pregnancy (p=0.80). Adjustment for leptin attenuated the augmentation of insulin secretory response from pre-pregnancy to early pregnancy (adjusted mean change 121.5 μU/ml, p=0.13).
Fig. 2.
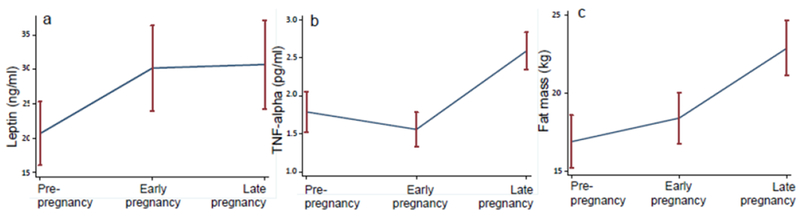
(a–c) Mean leptin levels (a), TNF-α levels (b) and fat mass (c) at the three study time points: pre-pregnancy, early pregnancy and late pregnancy. Error bars denote the SEM for each time point
As expected, fat mass and leptin levels were strongly correlated (r=0,86, p<0.0001, log scale), but the changes in leptin from pre-pregnancy to early pregnancy were independent of fat mass (Fig. 2c) (adjusted mean difference 7.5 ng/ml, p=0.005). Adjustment for fat mass did not attenuate the association between insulin secretory response and leptin (negligible change in effect size, p=0.02 after fat mass adjustment); in fact, fat mass was no longer significantly associated with first-phase insulin response after leptin was introduced into the model.
There was no association between oestradiol, progesterone, cortisol, hPL or TNF-α and insulin secretory response independent of insulin sensitivity, age and study time point.
Associations between hormones/cytokines and insulin sensitivity
As previously reported,(5) TNF-α was inversely associated with insulin sensitivity, independent of study time point and GDM status (β=−0.25, p=0.002). Changes in TNF-α across study time points are presented in Fig. 2b. Adjustment for TNF-α attenuated the changes in insulin sensitivity from pre-pregnancy to early pregnancy (adjusted mean change 0.014, p=0.10).
Leptin was inversely associated with insulin sensitivity, independent of study time point and GDM status (p=0.018). This effect was not independent of fat mass (effect size decreased by 67% with adjustment for fat mass, p=0.49). Adjustment for leptin did not attenuate the significant increase in insulin sensitivity that occurred in early pregnancy; in fact, the apparent positive effect of early pregnancy on insulin sensitivity was enhanced when leptin was included in the model (adjusted mean change 0.024, p=0.004).
There was no association between oestradiol, progesterone, cortisol or hPL and insulin sensitivity independent of study time point and GDM status.
Discussion
Here, using well-validated methods, we demonstrate that the insulin secretory response is markedly augmented in early pregnancy, independent of, and prior to, any decrement in insulin sensitivity. In fact, insulin sensitivity increases in early pregnancy. The observed increases in both insulin sensitivity and insulin secretory response may represent metabolic adaptations for accrual of adipose tissue, consistent with the theory that early pregnancy is an anabolic state. In contrast, the decreases in insulin sensitivity that occur in later gestation may allow amino acids, glucose and lipids to become available for fetal growth. These changes in maternal metabolism are probably mediated through placental hormones and/or cytokines, though our study was not designed to definitively identify the mediators of this effect.
Our findings of increased insulin secretory response in pregnancy are consistent with a small body of literature in humans and a larger body of literature in rodent models. Two studies previously reported an increase of C-peptide in women with a longstanding history of type 1 diabetes in pregnancy, even in women with previously undetectable levels (24, 25). Studies in animal models suggest a robust pregnancy-associated increase in glucose-stimulated insulin release and beta cell mass, with a role for placental lactogens and serotonin in these changes (8–12). Prior to the present study, it was still an open question as to whether the observed increase in insulin secretion in pregnant women without type 1 diabetes was a response to decreased insulin sensitivity. Our data suggest that this augmented insulin secretory response is independent of changes in insulin sensitivity, and thus it may be directly mediated by circulating factors released from the placenta.
Our findings of increased insulin sensitivity in early pregnancy are consistent with well-recognised clinical observations of an increased risk of hypoglycaemia in early pregnancy in women with pre-existing diabetes (26). Much of the increased risk has been ascribed to vigorous attempts to achieve euglycaemia to lower the risk of congenital anomalies and spontaneous abortion. However, even in women with well-controlled pre-existing type 1 diabetes there are often decreases in insulin requirements during gestational weeks 10-20 (27). Previous studies have attributed this to either decreased maternal nutrient intake (because of nausea/vomiting) or the fetal–placental unit functioning as a glucose sink (28, 29). Our study, alternatively, suggests that these clinical observations are due to a physiological increase in insulin sensitivity in early gestation.
We did not observe any differences in the change in insulin secretory response or insulin sensitivity according to GDM status or obesity status across gestation. Thus, these data support the understanding of GDM as a chronic pre-existing condition rather than a disease of pregnancy, with clinical manifestations of hyperglycaemia only becoming apparent with the physiological increases in insulin resistance. However, the women with GDM in the study were chosen because they had risk factors for GDM, so we cannot comment on the pathophysiology of GDM in women without risk factors.
Levels of leptin, a hormone produced by adipose tissue with multiple metabolic effects, are elevated during pregnancy. In non-pregnant individuals, elevated leptin is associated with increased fat mass and reduced insulin sensitivity. The gestational increases in maternal leptin concentration occur before any significant increases in maternal body fat, likely from placental sources (30). We found that maternal leptin was associated with maternal insulin secretory response, across gestation, independent of both insulin sensitivity and fat mass. Adjustment for leptin attenuated the observed increase in insulin secretory response in early pregnancy, identifying it as a potential mediator of this effect. Prior studies suggest that leptin has bidirectional effects on beta cell function and glucose-stimulated insulin secretion (31–33). Using our data, it is difficult to rule out reverse causality, namely that the association between leptin level and insulin secretory response is due to an effect of greater insulin secretory response on fat accrual, leading to a greater total amount of leptin secretion from adipose; yet, adjusting for leptin in our multivariate models eliminated any association between fat mass and insulin secretory response. Future research is needed to definitively identify hormonal mediators of the increased insulin secretory response in early pregnancy. If identified, these mediators would have important implications for the development of novel pharmacological treatments for diabetes outside of pregnancy.
We have previously reported that circulating TNF-α concentrations are inversely associated with insulin sensitivity in pregnancy (5). Although correlation does not prove causation, we have also reported that post-receptor insulin signalling defects in skeletal muscle during pregnancy are consistent with an inflammatory mechanism for decreased insulin sensitivity (34, 35). TNF-α decreased along with the increase in insulin sensitivity in early pregnancy. Adjustment for TNF-α attenuated the association of early pregnancy with increased insulin sensitivity, making the decrease in TNF-α a potential mediator of this physiological effect. Leptin has also been reported to improve insulin sensitivity by attenuating the effects of TNF-α, but our analysis did not suggest leptin as a potential mediator (36).
The strengths of our study include its longitudinal nature and the inclusion of pre-pregnancy measurements from the same participants, which allows us to make definitive statements about the effect of pregnancy on glucose metabolism. In addition, insulin sensitivity (by clamp) and insulin secretory response (by IVGTT) were measured independently using well-validated methods; in most other studies, both variables have been estimated from the same GTT.
Limitations include a small sample size and the measurement of a limited number of reproductive hormones and cytokines. Future research using unbiased approaches to assess for correlations between circulating factors and insulin secretory response or insulin sensitivity may more definitively identify the mediators of the physiological effects we observed. We did not measure C-peptide levels, so we cannot definitively say that the patterns observed are due to endogenous insulin release from beta cells. In addition, the insulin assay used may have had cross-reactivity with proinsulin based on the available technology at the time of the original study. Finally, the women who developed GDM were selected based on risk factors, so we are unable to generalise our findings to women with GDM who do not have pre-pregnancy risk factors.
In conclusion, there is a significant increase in both insulin secretory response and insulin sensitivity in early pregnancy. This suggests that augmented insulin secretory response in pregnancy is independent of insulin sensitivity, as demonstrated by the marked increase in DI in early pregnancy. To our knowledge, this is a unique demonstration of a physiological increase in beta cell function of this magnitude in humans. The changes in insulin secretory response and insulin sensitivity in early pregnancy may be related to effects of placental hormonal and cytokine factors, such as leptin and TNF-α. The alterations in maternal insulin sensitivity provide a mechanism for the clinical observations of increased hypoglycaemia and decreased insulin requirement among women with pre-existing diabetes in early gestation. Identifying mediators of the physiological increase in insulin secretory response has potential therapeutic implications for treating hyperglycaemia in and outside of pregnancy.
Supplementary Material
Research in context.
What is already known about this subject?
During pregnancy, there are profound changes in glucose metabolism: insulin sensitivity declines and the insulin secretory response increases
The physiological changes in glucose metabolism are well characterised in late pregnancy, but poorly understood in early pregnancy
What is the key question?
Is the pregnancy-associated augmentation in insulin secretory response compensatory for insulin resistance or is it an independent phenomenon?
What are the new findings?
We found that insulin secretory response increases early in pregnancy, prior to and independent of any decrement in insulin sensitivity
How might this impact on clinical practice in the foreseeable future?
If the mediators of the pregnancy-associated augmentation of insulin secretory response are identified, this biology could be harnessed for diabetes therapeutics
Acknowledgements
The authors thank the clinical staff of the Clinical Research Unit of the Clinical Translational Science Center at MetroHealth Medical Center for their support in performing the laboratory assays and for research, nutritional and nursing support. We also thank S. Kalhan (Cleveland Clinic Foundation, Cleveland, OH, USA) for performing the stable isotopes assays of glucose.
Funding This work was supported in part by NICHD HD22965 (PMC) and NCRR CTSA Ul1 RR 024989. CEP is supported by career development awards from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (K23DK113218) and the Robert Wood Johnson Foundation’s Harold Amos Medical Faculty Development Program (74256). The data analysis was supported by Harvard Catalyst/Harvard University’s Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL 1TR002541) and financial contributions from Harvard University and its affiliated academic healthcare centres.
Abbreviations
- DI
Disposition index
- GDM
Gestational diabetes mellitus
- hCG
Human chorionic gonadotropin
- hPL
Human placental lactogen
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Data availability The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request after approval is granted by the relevant institutional review boards.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
References
- 1.Catalano PM, Tyzbir ED, Wolfe RR et al. Carbohydrate metabolism during pregnancy incontrol subjects and women with gestational diabetes. Am J Physiol. 1993;264:E60–67. [DOI] [PubMed] [Google Scholar]
- 2.Catalano PM, Tyzbir ED, Roman NM, Amini SB, Sims EA. Longitudinal changes ininsulin release and insulin resistance in nonobese pregnant women. Am J Obstet Gynecol 1991;165:1667–1672. [DOI] [PubMed] [Google Scholar]
- 3.Catalano PM, Huston L, Amini SB, Kalhan SC. Longitudinal changes in glucosemetabolism during pregnancy in obese women with normal glucose tolerance andgestational diabetes mellitus. Am J Obstet Gynecol 1999;180:903–916. [DOI] [PubMed] [Google Scholar]
- 4.Ryan EA, O’Sullivan MJ, Skyler JS. Insulin action during pregnancy. Studies with theeuglycemic clamp technique. Diabetes 1985;34:380–389. [DOI] [PubMed] [Google Scholar]
- 5.Kirwan JP, Hauguel-De Mouzon S, Lepercq J et al. TNF-α is a predictor of insulinresistance in human pregnancy. Diabetes 2002;51:2207–2213. [DOI] [PubMed] [Google Scholar]
- 6.Butler AE, Cao-Minh L, Galasso R et al. Adaptive changes in pancreatic beta cellfractional area and beta cell turnover in human pregnancy. Diabetologia 2010;53:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas inhuman pregnancy. Br J Obstet Gynaecol 1978;85:818–820. [DOI] [PubMed] [Google Scholar]
- 8.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. TrendsEndocrinol Metab 2010;21:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy:increased islet cell proliferation and insulin secretion correlates with the onset ofplacental lactogen secretion. Endocrinology 1992;130:1459–1466. [DOI] [PubMed] [Google Scholar]
- 10.Rieck S, White P, Schug J et al. The transcriptional response of the islet to pregnancy inmice. Mol Endocrinol 2009;23:1702–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H, Toyofuku Y, Lynn FC et al. Serotonin regulates pancreatic beta cell mass duringpregnancy. Nat Med 2010;16:804–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karnik SK, Chen H, McLean GW et al. Menin controls growth of pancreatic beta-cells inpregnant mice and promotes gestational diabetes mellitus. Science 2007;318:806–809. [DOI] [PubMed] [Google Scholar]
- 13.Buchanan TA, Metzger BE, Freinkel N, Bergman RN. Insulin sensitivity and B cellresponsiveness to glucose during late pregnancy in lean and moderately obese womenwith normal glucose tolerance or mild gestational diabetes. Am J Obstet Gynecol 1990;162:1008–1014. [DOI] [PubMed] [Google Scholar]
- 14.Homko C, Sivan E, Chen X, Reece EA, Boden G. Insulin secretion during and afterpregnancy in patients with gestational diabetes mellitus. J Clin Endocrinol Metab 2001;86:568–573. [DOI] [PubMed] [Google Scholar]
- 15.Carpenter MW, Coustan DR. Criteria for screening tests for gestational diabetes. Am JObstet Gynecol 1982;144:768–773. [DOI] [PubMed] [Google Scholar]
- 16.Jaffe B (1979) Methods of hormone radioimmunoassay (2nd ed). Academic Press, New York. [Google Scholar]
- 17.Keys A, Brozek J. Body fat in adult man. Physiol Rev 1953;33:245–325. [DOI] [PubMed] [Google Scholar]
- 18.Siri WE. Body composition from fluid spaces and density: analysis of methods. 1961.Nutrition 1993;9:480–491 [PubMed] [Google Scholar]
- 19.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifyinginsulin secretion and resistance. Am J Physiol 1979;237:E214–223. [DOI] [PubMed] [Google Scholar]
- 20.Finegood DT, Bergman RN, Vranic M. Estimation of endogenous glucose productionduring hyperinsulinemic-euglycemic glucose clamps. Comparison of unlabeled andlabeled exogenous glucose infusates. Diabetes 1987;36:914–924. [DOI] [PubMed] [Google Scholar]
- 21.Tserng KY, Kalhan SC. Calculation of substrate turnover rate in stable isotope tracerstudies. Am J Physiol 1983;245:E308–311. [DOI] [PubMed] [Google Scholar]
- 22.Bergman RN, Ader M, Huecking K, Van Citters G. Accurate assessment of beta-cellfunction: the hyperbolic correction. Diabetes 2002;51(Suppl 1):S212–220. [DOI] [PubMed] [Google Scholar]
- 23.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controllingglucose tolerance in man: measurement of insulin sensitivity and beta-cell glucosesensitivity from the response to intravenous glucose. J Clin Invest 1981;68:1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilic S, Jovanovic L, Wollitzer AO. Is the paradoxical first trimester drop in insulinrequirement due to an increase in C-peptide concentration in pregnant type I diabeticwomen? Diabetologia 2000;43:1329–1330. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen LR, Rehfeld JF, Pedersen-Bjergaard U, Damm P, Mathiesen ER. Pregnancy-induced rise in serum C-peptide concentrations in women with type 1 diabetes. DiabetesCare 2009;32:1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimmerle R, Heinemann L, Delecki A, Berger M. Severe hypoglycemia incidence andpredisposing factors in 85 pregnancies of type I diabetic women. Diabetes Care 1992;15:1034–1037. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Patterson A, Gich I, Amini SB, Catalano PM, de Leiva A, Corcoy R. Insulinrequirements throughout pregnancy in women with type 1 diabetes mellitus: threechanges of direction. Diabetologia 2010;53:446–451. [DOI] [PubMed] [Google Scholar]
- 28.Desoye G, Nolan CJ. The fetal glucose steal: an underappreciated phenomenon indiabetic pregnancy. Diabetologia 2016;59:1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gadsby R, Barnie-Adshead AM, Jagger C. A prospective study of nausea and vomitingduring pregnancy. Br J Gen Pract 1993;43:245–248. [PMC free article] [PubMed] [Google Scholar]
- 30.Highman TJ, Friedman JE, Huston LP, Wong WW, Catalano PM. Longitudinal changesin maternal serum leptin concentrations, body composition, and resting metabolic rate inpregnancy. Am J Obstet Gynecol 1998;178:1010–1015. [DOI] [PubMed] [Google Scholar]
- 31.Covey SD, Wideman RD, McDonald C et al. The pancreatic beta cell is a key site formediating the effects of leptin on glucose homeostasis. Cell Metab 2006;4:291–302. [DOI] [PubMed] [Google Scholar]
- 32.Kulkarni RN, Wang ZL, Wang RM et al. Leptin rapidly suppresses insulin release frominsulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest 1997;100:2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morioka T, Asilmaz E, Hu J et al. Disruption of leptin receptor expression in the pancreasdirectly affects beta cell growth and function in mice. J Clin Invest 2007;117:2860–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedman JE, Ishizuka T, Shao J, Huston L, Highman T, Catalano P. Impaired glucosetransport and insulin receptor tyrosine phosphorylation in skeletal muscle from obesewomen with gestational diabetes. Diabetes 1999;48:1807–1814. [DOI] [PubMed] [Google Scholar]
- 35.Friedman JE, Kirwan JP, Jing M, Presley L, Catalano PM. Increased skeletal muscletumor necrosis factor-alpha and impaired insulin signaling persist in obese women withgestational diabetes mellitus 1 year postpartum. Diabetes 2008;57:606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaspelkis BB 3rd, Davis JR, Saberi M et al. Leptin administration improves skeletalmuscle insulin responsiveness in diet-induced insulin-resistant rats. Am J PhysiolEndocrinol Metab 2001;280:E130–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.