

Abstract
The best global seller among oncology drugs in 2018 is lenalidomide, an analog of thalidomide. It took 53 years and a circuitous route from the discovery of thalidomide to approval of an analog for use in treatment of cancer. We understand now a lot more about the genetic and molecular basis of diseases than we did in 1953 when thalidomide was discovered. We have also no shortage of chemical libraries with hundreds of thousands of compounds, both synthetic and natural. What is needed are better ways to search among these rich resources for compounds with the potential to do what we want them to do. This review summarizes examples from the literature that make Drosophila melanogaster a good model for screen for drugs, and discusses knowledge gaps and technical challenges that make Drosophila models not as widely used as they could or should be.
Keywords: Drosophila, drug-screening, cancer, neurodegeneration
Graphical Abstract:

Drosophila melanogaster is ideal for modeling complex genotypes and phenotypes for drug screening.
Introduction
Oncology remains the largest disease area in the pharmaceutical business (IgeaHub, 2018). Among oncology drugs, the best global seller in 2018 in terms of revenues was Revlimid (lenalidomide), an analog of (infamous) thalidomide. The story of thalidomide is a lesson in drug development (Rehman, Arfons, & Lazarus, 2011). Thalidomide was first discovered in or about 1953, inadvertently by German scientists who were searching for new antibiotics after World War II. It was a by-product in a chemical reaction and was recognized with cautious excitement as a structural analog of glutethimide, a popular sedative. (Kessel, 2013). Glutethimide was being marketed then as a safe alternative to barbiturates but was turning out to have its own addiction problems and severe withdrawal symptoms. This motivated the study of thalidomide as a potentially safer alternative to glutethimide. We are now only too familiar with thalidomide’s short tenure as ‘the’ morning-sickness drug, followed by its horrible legacy of birth defects. One outcome of this period was the recognition that teratogenic properties of thalidomide, which included suppression of blood vessel formation, are potentially anti-tumorigenic because tumors require blood supply for growth. Indeed, thalidomide proved to be a potent anti-cancer agent, with the ability to inhibit not only angiogenesis but also other aspects of cancer cell biology, and its analog, lenalidomide, received FDA-approval in 2006 for the treatment of multiple myeloma.
The year associated with thalidomide discovery, 1953, was also the year that Watson and Crick published the structure of DNA. It took about 53 years and a circuitous route for its analog, lenalidomide, to be approved for cancer. We understand now a lot more about the genetic and molecular basis of diseases. We have also access to large libraries of chemicals, both synthetic and natural. What is needed are better ways to search among these rich resources for chemicals with potential to do what we want them to do. To this end, many screens exist and can be put into two buckets, activity-based and phenotype-based. Activity-based screens rely on a known target such as an enzyme or a pathway whose active state can be detected with a reporter. Inherent in this type of screen is the belief that the target is critical to the disease of interest. The main advantage of activity-based screens is that one has some idea of the molecular target of compounds identified in the screen. The main disadvantage is that modulation of that target by the drug may not be important for the intended use. In phenotype-based screens, the read-out is a biological phenotype that is often complex and many have multiple inputs, both known and unknown. The advantage and disadvantage of phenotype-based screens are complementary to those of activity-based screens; the compounds identified in the former likely matter for the intended use but one does not know the molecular target or the mechanism of action. As seen in examples below, Drosophila can be used for both types of screens, and the reasons to do so are many.
WHY FLIES?
Simpler gene families
As human and Drosophila genome sequences were being completed, it became apparent that while many genes are conserved between the two organisms__ an estimated 60% of human genes associated with disease have fly homologs__ conserved gene families have fewer members in Drosophila (Schneider, 2000; Wangler et al., 2017). To provide disease-relevant examples, there are at least 7 E2F transcription factors in mammals to two in Drosophila, and four notch genes, three hedgehog genes and three members of the Fragile X gene family (with one linked to Fragile X Syndrome) in mammals to one each in Drosophila. The presence of larger gene families in mammals has been attributed to whole genome duplication events in during vertebrate evolution (Dehal & Boore, 2005). Understanding the function of a gene typically requires studying the consequences of inhibiting that gene in loss-of-function studies. Large gene families in which multiple genes provide similar function mean many genes must be inhibited simultaneously to understand their function, a technically challenging feat. Simpler gene families in Drosophila simplified many loss-of-function studies that led to our basic understanding of disease-relevant genes. Smaller gene families of Drosophila are also a potential advantage for drug discovery because fewer genes would need to be modulated to set up a sensitized condition for drug screening. A good example is an activity-based screen designed by Gonsalves, DasGupta and colleagues, to identify inhibitors of Wingless pathway in Drosophila S2 cells in culture (Gonsalves et al., 2011).
Wg is a homolog of mammalian Wnt1 (wingless-type MMTV integration site family, member 1). Wg/Wnt1 pathway controls differentiation, proliferation, survival, and cell motility during normal development (Clevers, 2006). Abnormal activation of Wnt signaling is associated with and contributes to oncogenesis (Miller, 2002). Wg/Wnt1 signaling pathway has core components as well as positive and negative regulators (Clevers, 2006). One key inhibitor is Axin, a member of a protein complex that targets β-catenin for degradation by the Anaphase Promoting Complex (APC) in the absence of Wg/Wnt ligand (Fig. 1). When Wg/Wnt1 ligand is present, Axin is sequestered, β-catenin is stabilized and can activate transcription by complexing with co-activator TCF. Drosophila has a single Axin as opposed to two, Axin 1 and Axin 2, in mammals. Gonsalves, DasGupta and colleagues depleted Axin in Drosophila S2 cells by expressing dsRNA, to generate a condition of constitutively active Wg signaling (Gonsalves et al., 2011). With Axin depleted, β−catenin became stabilized, which was detected using a luciferase reporter under the control of a transcriptional enhancer with TCF-binding sites. This situation mimics constitutive Wnt activation in cancer that occurs by mutations in a different inhibitor, the APC tumor suppressor (Fig. 1). APC mutations are associated with cancer, for example, 80–85% of sporadic colorectal cancer (Zhang & Shay, 2017).
Figure 1.
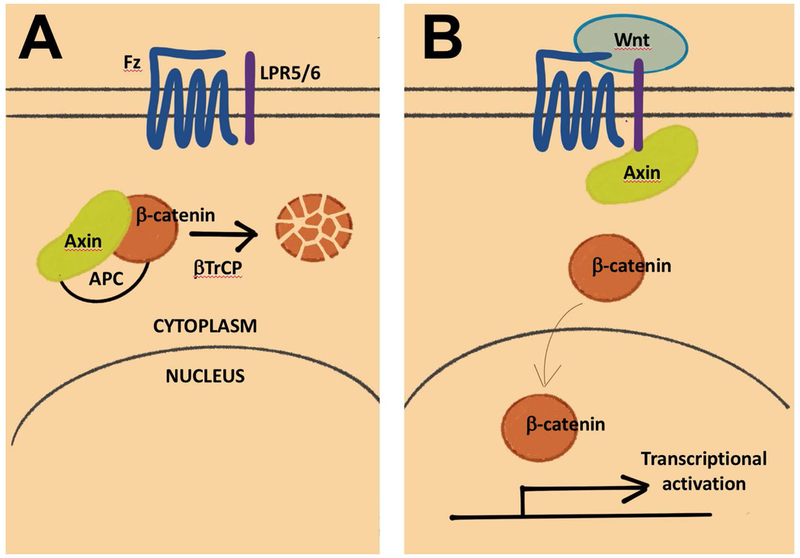
The canonical Wnt pathway, without (A) and with (B) ligand [after (Clevers, 2006)]. Only the components discussed in this review are shown.
Compounds that altered luciferase activity were screened in a high throughput fashion, with non-specific effects on luciferase activity ruled out in a secondary screen. One could argue that a similar screen could be set up in mammalian cells using double RNAi against Axin1 and Axin2. But what Drosophila offered was the ability to take advantage again of simpler gene families, to pinpoint where in the Wg signaling pathway the compounds act. Downstream of Axin are Ubiquitin (Ub) ligase complexes that degrade β-catenin, with Slimb (Supernumerary limbs) providing substrate specificity. Drosophila has one slimb gene but the mammalian homolog, βTrCP (Fig. 1), is encoded by two genes, βTrCP1 and βTrCP2 (Kato et al., 1990). So, a single RNAi knock down of Slimb in S2 cells activated the Wg signaling constitutively similar to Axin but a one step further down in the pathway. A secondary screen using this set up showed that most compounds from the Axin screen (21 of 23 tested) inhibited luciferase in the Slimb screen, indicating they act downstream of the Ub-ligase step. Additional studies of lead compounds including in silico modeling and biochemical analysis identified their activity to be at the level of β-catenin/TCF complex formation or function.
The ability to model complex genotypes; Drosophila ‘avatars’
The screen for Wg inhibitors described above used models in which genes have been knocked out singly. Cancer is a disease of multiple genetic aberrations. Colorectal cancer (CRC) provides a good example. Bangi, Cagan and colleagues analyzed of the data from the Cancer Genome Atlas (TCGA) and found that 90% of patient samples in CRC have mutations in two or more signaling pathways, with recurrent mutations affecting WNT, RAS/MAPK, PI3K, TGF-β and p53 pathways (Bangi, Murgia, Teague, Sansom, & Cagan, 2016). The authors then used a combination of tissue-specific RNAi and transgene expression to model 4 or 5 recurrent mutations in the Drosophila adult hindgut (‘4- or 5-hit models’), which allowed them to test the effect of FDA-approved cancer drugs. In these models, cells of the Drosophila hindgut, a structure analogous to the mammalian colon which is the site of most intestinal cancers in human, show characteristics of transformation that include overgrowth, extra cell layers, mesenchymal appearance, disruption of the basement membrane, and dissemination of GFP-marked transformed cells into the body cavity. Relevant to this review, drugs that were developed based on their effect in a single mutation model work poorly in the context of other mutations to rescue dissemination. For example, of 12 targeted agents effective in an oncogenic RasG12V model, none were effective when this oncogene was expressed in the context of PTEN, p53 and APC depletion (4-hit model). Testing various combinations of genetic manipulations suggested that increasing mTORC1 activity could restore sensitivity a targeted agent against PI3K, a finding that was confirmed by pretreatment with drugs that activate mTORC1. Such pre-treatment increased the sensitivity to PI3K inhibitors in the Drosophila model, human colorectal cancer cells and a mouse model of colorectal cancer (Bangi et al., 2016).
The clinical utility of multi-hit Drosophila models is being tested at the Center for Personalized Cancer Therapeutics, Icahn School of Medicine at Mount Sinai (Misiukiewicz, 2018). Here, genomics data from patients are used to build Drosophila ‘avatars’ with multiple genomic alterations that mimic the complex genotype of the patient’s tumor. The avatars are then used to systematically screen FDA approved drugs, singly and in combination, in a 96-well format. The results, along with independent considerations of toxicity, dosing, etc. are incorporated into a recommended personalized treatment. This approach is being applied to patients with colorectal and Medullary Thyroid cancer patients (Misiukiewicz, 2018). This approach offers exciting possibilities, but it will be a while before the data is in.
The ability to model complex phenotypes
As a multicellular organism, Drosophila allows the modeling of complex traits that are relevant to human. Many complex traits share common genetic determinants in fly and human, such that we may reasonably expect what is discovered in fly to apply to human. Examples of such conservation abound. The first to come to mind is circadian rhythm. As seen in the Nobel prize-winning work by Hall, Rosbash, and Young, the biological clock that dictates when we sleep, when we eat, relies on a Ubiquitin-based self-timing molecular clock comprised of proteins such as Time, Clock, and Period (Bargiello, Jackson, & Young, 1984; Hardin, Hall, & Rosbash, 1990; Konopka & Benzer, 1971; Zehring et al., 1984). All components are conserved in human not only in DNA and protein sequence but also in function. In another example, embryonic development was dissected using Drosophila genetics by Lewis, Nusslein-Volhard and Wieschaus (Lewis, 1978; Nusslein-Volhard & Wieschaus, 1980). Their Nobel prize-winning work led to our understanding of genes such as hedgehog and Polycomb, which are also conserved in human. The following sections describe examples of complex disease phenotypes that have lent themselves to Drosophila models, some of which were used in drug screens.
Cancer; modeling cellular heterogeneity
The study of Drosophila avatars illustrates the need to model not one but multiple genetic alterations in cancer. A natural assumption is that these mutations occur in the same cell. A remarkable study showed that this is not necessarily the case (Wu, Pastor-Pareja, & Xu, 2010). Drosophila larvae may be induced to form epithelial tumors by mutations in at least two genes. Commonly used tumor models combine in the same cell one oncogenic mutation (for example, RasG12V or constitutive activated Notch) and homozygous loss of one tumor suppressor such as Scribble, homolog of human tumor suppressor hScrib. Clones of cells in the Drosophila neuroepithelium that are scrib−/− and express RasG12V grow into metastatic tumors. Wu, Xu and colleagues showed that these two genetic aberrations need not be in the same cell. Generation of RasG12V cells and scrib−/− cells next to each other in the epithelium also induced metastatic tumors. Metastatic behavior was achieved by JNK-mediated activation of JAK/STAT signaling. This could happen cell-autonomously when Ras and Scrib mutations are in the same cell, or cell non-autonomously when the mutations were in adjacent cells (Wu et al., 2010).
These findings in Drosophila were followed by similar findings in mammalian tumor models (e.g. (Cleary, Leonard, Gestl, & Gunther, 2014)). We have known for a long time that cell types within a tumor are not homogeneous. Heterogeneity may actually be a requirement for certain tumors as there is mounting evidence for cells of different genotypes within a tumor cooperating to drive growth and invasion (reviewed in (Caswell & Swanton, 2017)). Tumor cell heterogeneity is not represented in human cancer cell-based models currently used for drug screening. The closest we come to approximating cell-cell interactions may be in Drosophila tumor models where tumors are grown in situ as in the example below.
Cancer; modeling cell-microenvironment interaction
The concept of cancer stem cells has both supporters and critics, but there is experimental evidence that within a tumor, some cells are better than others at initiating new tumors (Marjanovic, Weinberg, & Chaffer, 2013; Ye & Weinberg, 2015). Such tumor initiating cells or cancer stem-like cells (CSCs) are thought to represent a sub-population that resist treatment to initiate regrowth and recurrence. Drugs that can inhibit CSCs would be valuable, hence the modeling of Drosophila to identify such drugs. To identify compounds that inhibit ‘stem-ness’, Markstein, Perrimon and colleagues took a naturally occurring somatic stem cell population in the adult gut and turned them cancerous by cell-type specific expression of a gain-of-function allele of Raf kinase (Markstein et al., 2014) (amino acid 2–431 deleted; (Brand & Perrimon, 1994)). The resulting over growth is heterogenous, comprised of mitotic and polyploid cells, and can initiate new tumors when transplanted into wild type adult hosts. Co-expression of luciferase in the same cells allowed them to use luciferase activity as a read out in 96-well screens for compounds that reduced tumor growth (Figure 2). Screening through ~6000 molecules produced some unexpected results, that some chemicals that inhibited the growth of cancerous stem cells actually stimulated the growth of their normal counterparts, stem cells in the gut. Stimulation of normal stem cell proliferation was through increased production of Upd3, a IL-6-like cytokine, in the daughter of the stem cells, followed by activation of the cognate JAK/STAT signaling in the stem cells. The authors suggest that stimulation of proliferation of normal stem cells could have deleterious consequences such as promoting new tumors.
Figure 2.
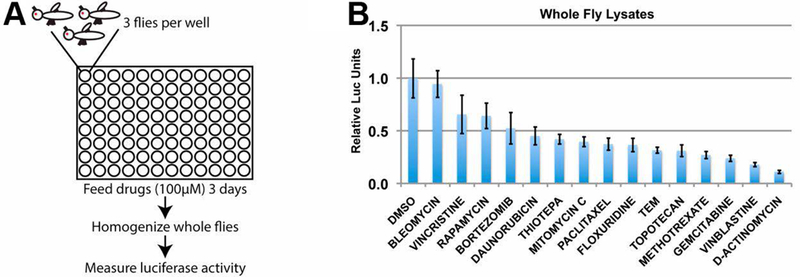
A screen for drugs that inhibit the growth of intestinal stem cell-derived tumors (Markstein et al., 2014). (A) Flies harboring luciferase-expressing tumors were treated with drugs in 96-well plates. Flies were homogenized and luciferase activity measured to quantify drug effect. (B) A sample dataset with 15 compounds.
Cancer cell-microenvironment interaction through conserved signaling molecules is proving to be highly relevant to human cancers. In breast cancer, although chemotherapeutic agents kill cancer cells, these treatments stimulate the production of the cytokine TNF-α in endothelial and other stromal cells, which leads to chemo-resistance in cancer cells and metastasis (Acharyya et al., 2012). In another example, after treatment with DNA damaging chemotherapy agents, thymus cells release cytokines that promote survival of Burkitt’s lymphoma cells (Gilbert & Hemann, 2010). Thus, similar to the case in Drosophila, IL-6/Upd-3 production by normal cells in response to treatment promotes therapy-induced proliferation of abnormal cancer cells. In other words, the Drosophila gut tumor model system has the potential to tease apart effect on cancer vs. normal cells and to find inhibitors of the cell-cell interaction that may result in new tumors (Markstein et al., 2014).
The differential behavior of transformed and normal cells to therapy was apparent also in a study by Sonoshita, Cagan and colleagues of a Drosophila RET-driven tumor model (Sonoshita et al., 2018). Mutations in the proto-oncogene that encodes RET kinase is associated with several types of human cancer including medullary thyroid carcinoma (MTC). Tissue-specific expression of a cancer-relevant mutant form of Drosophila Ret, dRetMEN2B, in a subset of larval epithelial cells result in partial lethality between embryo and pupa stages. Drosophila RET models have been used to characterize inhibitors of RET signaling including ZD6474 (Vandetanib), which is now approved for the treatment of MTC (Dar, Das, Shokat, & Cagan, 2012; Vidal, Wells, Ryan, & Cagan, 2005). Lethality in these models allows screens for drugs that inhibit RET signaling using the rescue of lethality as a read-out. The authors screened through 31 kinase inhibitors approved for cancer use. Of the three strongest inhibitors that rescued lethality, multi-kinase inhibitor sorafenib was studied further because of its known efficacy in human MTC models. The authors identified chemicals and mutations that modified the efficacy of sorafenib or its chemical derivatives. One striking finding concerned Lk6, the fly homolog of mammalian MKNKs (MAP kinase interacting serine/threonine kinases). Knocking down Lk6 in the same cells that express dRetMEN2B rescued viability (that is, enhanced the efficacy of the sorafenib derivative) but knocking down Lk6 in the whole animal had the opposite effect (that is, reduced the efficacy of the same sorafenib derivative) (Sonoshita et al., 2018). These results illustrate the utility of using whole animals with complex cell types.
Cancer; modeling resistance to treatment
Ionizing radiation or IR, alone or in combination with drugs, is used to treat more than half of cancer patients (www.cancer.org). Regrowth of irradiated tumors, however, reduces the success of radiation therapy. We modeled the regrowth of tissues after irradiation in Drosophila larval imaginal discs, which are precursors of adult organs. Doses of IR that kills about half of the cells still allow regeneration to produce viable adults (Bryant, 1970; Haynie & Bryant, 1977; James & Bryant, 1981). Chemicals that, when added to larval food after irradiation, reduced survival to adulthood were screened (Figure 3; (Gladstone et al., 2012; Gladstone & Su, 2011)). Such chemicals have the potential to enhance radiation therapy of human cancers. Larvae mutants in p53 or Checkpoint Kinase 1 (Chk1) were used in the primary screen and the hits were counter-screened against wild type. Screens through two libraries from the National Cancer Institute (total ~2000 molecules) produced 16 hits that fit the criteria, representing a variety of molecular mechanisms. One of these, Bouvardin, was chosen for further study because it inhibits the elongation step of translation by a mechanism previously unknown in human (Gladstone et al., 2012; Stickel, Gomes, Frederick, Raben, & Su, 2015). In an independent study, regulation of elongation was critical for recovery from radiation exposure in human osteosarcoma cells (Kruiswijk et al., 2012), indicating a commonality between Drosophila and human. Bouvardin enhanced the effect of radiation in many preclinical models of human cancer (Gladstone et al., 2012; Stickel et al., 2015). Proprietary derivatives of Bouvardin are being developed for clinical use (US Patent 9452215).
Figure 3.
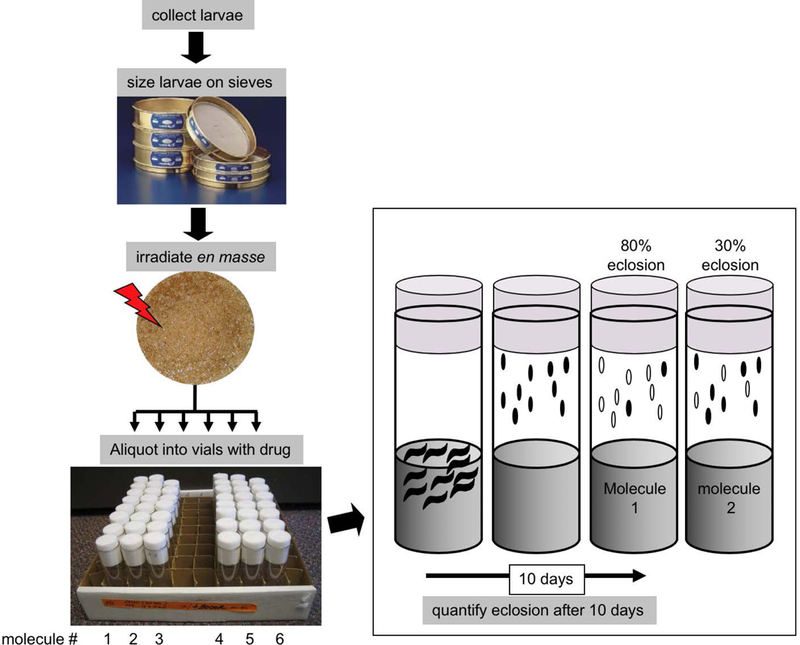
Screen for modulators of radiation (Gladstone & Su, 2011). Third instar larvae were ‘sized’ by passing through sieves and irradiated en mass. Irradiated larvae were aliquoted into culture vials that contained one drug per vial mixed with the food. Survival to adulthood (‘eclosion’) was quantified 10 days later by counting empty (live) and full (dead) pupa cases.
We also have been using the Drosophila irradiation model to address a recently recognized un-wanted effect of cancer therapy, namely therapy-induced stem-ness. As described in a preceding section, ‘Cancer Stem-like Cells’ (CSCs) are recognized by specific protein markers and by their greater ability, relative to cells without such markers, to form tumor spheres in culture or new tumors in mice (Marjanovic et al., 2013; Ye & Weinberg, 2015). However, not only do CSCs generate non-stem cancer cells, it is now recognized that non-stem cancer cells are capable of converting to CSCs. The plasticity that allows non-stem cancer cells and CSCs to interconvert presents a major challenge to any therapy that targets CSCs. Even more concerning, cancer treatments themselves promote the conversion of non-stem cancer cells into CSCs, including chemotherapy (Debeb et al., 2012; Pisco & Huang, 2015) and radiation (Lagadec, Vlashi, Della Donna, Dekmezian, & Pajonk, 2012; Lee et al., 2017; Vlashi et al., 2016). We found that ionizing radiation also induces stem-like behavior in columnar epithelial cells of the larval tissues (Verghese & Su, 2016, 2017, 2018). These cells are resistant to IR-induced apoptosis, change fate, and change location, to participate in regeneration of parts of the tissues that suffered more apoptosis. We have performed genetic screens in this model, but the system should be amenable to drug-screening, to identify chemicals that can inhibit therapy-induced stemness.
In a systematic approach to addressing therapy-induced resistance, Das, Cagan and colleagues used the above-described Drosophila RET-driven tumor model wherein expression of a cancer-relevant mutant form of Drosophila Ret, dRetMEN2B, in larval epithelial tissues resulted in partial lethality between embryo and pupa stages (Das, Esernio, & Cagan, 2018). Here, the authors used the Drosophila RET model to ask what OTHER signaling pathways become activated/repressed in response to treatment with a known/approved drug. The authors hypothesized that pathway(s) activated by a given drug but are not the primary target of the drug, called the ‘network’ by the authors, form the basis for toxicity or resistance to that drug. This concept is not new and has been used effectively in human cancer models; for example, non-canonical Wnt signaling is activated and mediates resistance to the MEK½ inhibitor selumetinib in colorectal cancer cells (Spreafico et al., 2013). What is different here is that Drosophila allowed the authors to identify such networks not only in cells expressing the oncogene but also in surrounding normal cells. Further, the effect of modulating network components in the normal cells of the larvae can be tested using Drosophila genetics, thus identifying common and unique cellular responses that could explain toxicity and resistance. Once a functionally relevant network has been identified, the authors used known inhibitors to retrain the network to non-drug levels, so-called ‘network breaks’, with the goal of reducing toxicity or resistance (Das et al., 2018). For example, sorafenib, a poly-kinase inhibitor approved for the treatment of kidney, liver and thyroid cancers, activated a network that consisted of Erk, Src and Akt among others. They identified bortezomib (proteasome inhibitor) and vorinostat (HDAC inhibitor), for example, as network breaks. Interestingly, the combination of bortezomib and vorinostat restrained networks activated not only by sorafenib but also by another clinically relevant kinase inhibitor erlotinib, and delayed the emergence of drug-resistant cells from prolonged treatment of human cancer cells with erlotinib (Das et al., 2018).
Obesity and Feeding Behavior
Obesity is a health problem world-wide and uncontrolled feeding is one basis for obesity. Drosophila larvae are voracious eaters, increasing body weight by more than 200-fold within 4 days (O’. Farrell, 2009). Gasque, Voshall and colleagues designed a 96-well high-throughput screen in Drosophila larvae to identify chemicals that inhibit feeding in larvae (Gasque, Conway, Huang, Rao, & Vosshall, 2013). They mixed a dye (fluorescein) with food so that food uptake could be monitored using gut fluorescence. A screen through 3630 compounds yielded 32 that inhibited feeding and 10 that stimulated feeding by more than 1 standard deviation away from the average of solvent controls. Of these, two showed a robust dose-response but one of these, reserpine, also reduced larval locomotor activity and was therefore discarded. The other, metitepine, did not affect locomotor activity yet reduced the amount of food in the larval gut. Metitepine reduced the frequency of mouth-hook contractions (feeding behavior), indicating that it reduced food uptake as opposed to accelerating excretion. Thus, metitepine fits the properties of a feeding modulator. Metitepine is a known broad-spectrum antagonist of vertebrate serotonin (5-hydroxytryptamine, or 5-HT) receptors, which are G protein-coupled receptors and ligand-gated ion channels found in the central and peripheral nervous systems. Drosophila genome encodes five 5-HT receptors. Of these, mutations in just one, 5-HT2A, suppressed feeding and rendered the larvae insensitive to metitepine, indicating that this receptor is responsible for mediating the effect of the drug. Suppression of feeding by 5-HT2A mutation supports the idea that metitepine modulates feeding behavior as opposed to more pedestrian explanations such as making the food unpalatable to the larvae. The addition of metitepine to food inhibited feeding immediately, but, in unwelcome news for would-be dieters, the effect lasted just 2 hours. Even worse, at 24 hours after treatment, metitepine-treated larvae ate significantly more than solvent-treated controls (Gasque et al., 2013).
Sleep
Sleep is important for well-being; sleep disorders are associated with increased risk of diseases ranging from depression to cardiovascular diseases (e.g. (Hertenstein et al., 2018; Javaheri & Redline, 2017)). Drosophila adults undergo predictable periods of rest and wakefulness that can be quantified, lending itself to genetic and chemical screens that disrupt sleep (Nall & Sehgal, 2013). Nall and Sehgal screened 1280 commercially available compounds by monitoring their effect on sleep-wake patterns of drug-fed flies for 5 days. Four compounds decreased and one compound increased periods of sleep in reproducible and dose-dependent fashion. The single sleep-promoting drug was reserpine, a plant alkaloid previously known to reduce hypertension (Shamon & Perez, 2016), with additional antipsychotic activity (Indave, Minozzi, Pani, & Amato, 2016). It was also identified as a modulator of feeding behavior in Drosophila larvae as described in the preceding section, but was discarded because it reduced larval locomotion overall and not just feeding (Gasque et al., 2013). Its known target is VMAT (vesicular monoamine transporter) that packages monoaminergic neurotransmitters into presynaptic vesicles (Henry & Scherman, 1989). Mutations in VMAT also increased sleep but reserpine did not increase sleep further in VMAT mutants, indicating that the effect of the drug is through VMAT (Nall & Sehgal, 2013). Reserpine or VMAT mutations did not reduce the activity of adult flies generally during the waking periods, so their effects appear specific for sleep. This is in contrast to the effect of reserpine on the larvae, which was to reduce locomotion (Gasque et al., 2013). The use of VMAT mutants to confirm that reserpine acts though this receptor and the above-described use of 5-HT mutants to identify 5-HT2A as the receptor that responds to metitepine illustrate the utility of Drosophila in combining genetics and pharmacology to identify drug targets.
Degeneration and other neurological disorders
Normal neuronal function relies on interactions between multiple cell types, for example, between neurons and dendrites, between neurons and muscle, or between neurons and glia. Because drugs that correct abnormal neuronal function will need to work in a multi-cellular is context, screening for them in in vivo metazoan models makes sense. Genetic models in Drosophila have been used to screen drugs for at least three neurological disorders as summarized here.
Fragile X Syndrome is a heritable mental retardation disorder and a cause for autism (reviewed in (Drozd, Bardoni, & Capovilla, 2018). FXS results from mutations in or inactivation otherwise of the FMR1 gene. Encoded FMRP protein binds mRNAs to regulate translation, and loss of FMR1 activity is hypothesized to result in excess glutamate signaling. Drosophila and human FMRP homolog share >50% amino acid similarity. FMR1 mutant flies display phenotypes that are considered to parallel those of human FXS patients including defective neuronal architecture, increased translation of mRNA targets, and abnormal social (courtship) behavior. The finding that raising Drosophila FMR1 mutant embryos on high-glutamate diet causes lethality before pupariation formed the basis for a screen for chemical molecules that rescued survival when added to the diet. A screen through 2000 molecules identified 9 chemicals with reproducible effect. These include agonists of two other neurotransmitter pathways, GABAergic and muscarinic acetylcholine receptor (mAChR) signaling (Chang et al., 2008). Direct testing showed that raising FMR1 mutants on GABA could rescue the neuroarchitecture and courtship defects.
The second neurological disorder to be modeled in Drosophila was Alexander Disease, which results from mutations in an intermediate filament protein specific to the glia called GFAP (glial fibrillary acidic protein). The symptoms of AD include seizures and movement disorders. Expression of a disease-causing GFAPR79H mutant but not wild type GFAP in Drosophila glia recapitulates several pathological features of AD including GFAP protein aggregates in astrocytes (a type of glia), apoptosis in glia, non-autonomous neuronal cell death, and seizures (Wang, Colodner, & Feany, 2011). Compounds were screened by feeding adult flies and assaying for cleaved caspase 3, an apoptotic marker, in glia in brain slices (Wang, Hagemann, Messing, & Feany, 2016). A screen through 1987 compounds identified 4 hits that showed dose-dependent reduction in glial toxicity in secondary tests. One of these, glycopyrrolate, an antagonist of mAChR, was further studied and found to reduce seizures as well. The role of mAChR was confirmed using RNAi against mAChR and two additional antagonists (drugs). Two lines of data indicate that data from Drosophila are relevant to AD. First, mAChR subtype M1 was found to be over-expressed in astrocytes of human AD patients and in astrocytes of a mouse AD model that is also based on GFAPR79H expression. Second, administration of an mAChR antagonist reduced a pathological feature in the mouse AD model, namely the expression of NRF2 in astrocytes.
The third neurological disorder to be modeled in Drosophila was Friedreich’s Ataxia, a neurodegenerative disorder that results from mutations in the frataxin (FXN) gene (reviewed in (Monnier, Llorens, & Navarro, 2018). Additional pathological features of FA include loss of vision and hearing and heart defects. FXN protein binds iron to form iron-sulphur clusters, but its exact function remains unclear. Frataxin is conserved in yeast and Drosophila. Screening for compounds that rescue the slow-growth phenotype of yeast frataxin mutants identified from 6380 total (Seguin et al., 2015). Twelve showed specificity (they did not rescue another slow-growing strain) and were not toxic at high doses. These compounds were tested in a Drosophila FA model in which ubiquitous expression of fxn RNAi delayed development. Two compounds rescued the developmental delay and one of these rescued also cardiac dilatation defects of Drosophila fxn RNAi adults.
Screens on the horizon
In addition to the examples discussed already, several other complex phenotypes of biomedical relevance have been modeled in Drosophila. In some instances, proof-of-concept data exist to suggest that the models may be used to screen for drugs with potential clinical relevance. Three such examples are presented in the following section.
Aging
Somatic stem cells provide a pool of regenerative cells for repair and regeneration. Exhaustion of somatic stem cell pools is considered both a cause and an effect of aging (for example, (Kosan, Heidel, Godmann, & Bierhoff, 2018; Lopez-Otin, Blasco, Partridge, Serrano, & Kroemer, 2013)). Haller, Jasper and colleagues found that there is a limit to how many times somatic stem cells may be called upon to fulfill tissue repair in Drosophila (Haller et al., 2017). Studying somatic stem cells in Drosophila adult intestine, the authors found that TOR (Target of Rapamycin) is activated for stem cells to proliferate in response to injury such as that from bacterial infection. Repeated infection and activation of TOR, however resulted in stem cell loss. A similar process operates through mTROC1 in the stem cells of the mouse airway epithelium in response to SO2-injury. Inhibition of TOR/mTORC1 with the drug rapamycin prevented stem cell loss after repeated injury in mice. Rapamycin rescued also natural stem cell loss that occurs in aging flies and mice. This system has not been used for drug screening but the data with rapamycin provides proof of concept that it may lend itself to identify drugs that prevent stem cell loss.
Traumatic Brain Injury (TBI)
Mechanical injury, such as that caused by repeatedly hitting the culture vial on a hard surface, elicits phenotypes in Drosophila that are seen in human Traumatic Brain Injury patients, including temporary incapacitation, ataxia, activation of the innate immune response, neurodegeneration, and death (Katzenberger et al., 2013) (Figure 4). Anderson, Pandey and colleagues found that similar injury to Drosophila adult or larvae resulted in the accumulation of Stress Granules (SG) in the brain (Anderson et al., 2018). Feeding rapamycin to injured larvae decreased stress granule accumulation and improved organism survival. Rapamycin in this case is thought to act by inhibiting TOR to promote autophagy, which has been shown to clear SG.
Figure 4.

The TBI machine (Katzenberger et al., 2013), before (A) and after (B) the strike. (C–D) show flies at the corresponding states.
Polycystic Kidney Disease
Drosophila Malpighian Tubules (MT) are functionally equivalent to mammalian kidneys because both provide detoxification through filtration (Reviewed in (Millet-Boureima, Porras Marroquin, & Gamberi, 2018)). MTs have been used to model Polycystic Kidney Disease (PKD), the most common fatal monogenic disease in human for which there is no approved drugs. Two Drosophila genetic models that represent PKD have been described.
One of the genes responsible is polycystin-1; human PKD patients with polycystin-1 mutations show reduced levels of BICC1 protein, a translation regulator implicated in renal cystogenesis (Gamberi, Hipfner, Trudel, & Lubell, 2017). Drosophila mutants in BicC (Bicaudal C) show increased TOR signaling, progressive cystic degeneration of MT, reduced renal function and reduced survival, all of which are phenotypes of human PKD. Feeding Drosophila BicC mutants TOR inhibitor rapamycin reduced the number of size of cysts and prolonged survival, suggesting that this model may be used to screen for drugs with the potential to correct PKD.
The second Drosophila PKD model is based on another of the genes responsible for PKD, polycistin-2 or PKD2, which encodes a calcium channel of the transient receptor potential (TRP) family called TRPP2. PKD2D511V mutants show reduced TRPP2 protein level that could be rescued by blocking lysosomal degradation with the drug chloroquine. D511V mutation was engineered into the fly ortholog, Amo, at the same conserved amino acid (Hofherr, Wagner, Watnick, & Kottgen, 2016). Interestingly, fly AmoDV mutants show male sterility which has also been seen sporadically PKD patients; MT phenotypes in this model were not discussed. Nonetheless, adding chloroquine to the diet rescued the level of Amo protein in the sperm and male sterility of AmoDV flies, suggesting that rescue of male sterility may be used to screen for compounds that benefit human PKD patients. In fact, the human TRP family includes 28 members, 11 of which are implicated in Mendelian (single locus) disorders that affect the central and peripheral nervous system, cardiac tissue, skin, vision or gastrointestinal system (reviewed in (Wangler et al., 2017)). Therefore, it remains possible that generating fly models for the other TRP mutants will likewise facilitate drug discovery.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a fatal neurodegenerative disease that accompanies progressive loss of locomotor function (reviewed in (Lin, Mao, & Bellen, 2017)). TDP-43 is an RNA-binding protein that is normally found in the nucleus, but degenerating neurons in ALS patients accumulate TDP-43 protein in the cytoplasm (Kim & Taylor, 2017). Overexpression in Drosophila larval motor neurons of human TDP-43, either wild type or a disease-associated mutant, resulted in neuronal toxicity, larval locomotion defects and reduced survival (Estes et al., 2013). Metabolomic profiling of TDP-43 overexpressing larvae showed increased fatty acid-carnitine conjugates and decreased beta-hydroxybutyrate, which are signatures of defective mitochondrial lipid metabolism (Manzo et al., 2018). Increasing dietary fatty acids and ketones towards correcting the metabolic defects also restored locomotor function in TDP-43 overexpressing larvae, providing proof of concept that this model may be used to identify drugs that correct the effects of TDP-43 overexpression.
TRANSLATION FROM FLY TO HUMAN; EFFICACY vs. TOXICITY
Drosophila screens may yield compounds with the desired efficacy profiles, but would they behave the same in human cells and disease? Of human genes associated with disease, 60% have Drosophila homologs (Schneider, 2000). This gives hope that drugs discovered in Drosophila screen will behave similarly in human. Top three inhibitors of Wg signaling found in Drosophila S2 cells inhibited the growth of two colorectal cancer cell lines with constitutive Wnt pathway activation but not three cancer cell lines that lacked this signature (Gonsalves et al., 2011). Select radiation enhancers we found in Drosophila also enhanced the effect of radiation in human cancer cells and xenografts (Edwards et al., 2011; Gladstone et al., 2012; Stickel et al., 2015). At least sixty drugs have the same target or mechanism of action in Drosophila and human and include inhibitors of such diverse cellular activity as cytoskeleton function; DNA, RNA and protein synthesis; kinases and phosphatases; ion channels and cell surface receptors; chaperones and proteases (Fernandez-Hernandez, Scheenaard, Pollarolo, & Gonzalez, 2016). But what is still lacking is a systemic survey of hits in a given Drosophila screen, to assess what fraction of these translate to human models. A systemic study in yeast found that 40% of all yeast genes can be replaced with human homologs (Kachroo et al., 2015). Given that Drosophila and human are closer than yeast and human in evolutionary terms, functional conservation of genes, encoding proteins for drug targets, may be greater than 40%. It will be a tremendous amount of work but knowing what fraction of hits in a Drosophila screen that can be expected to translate from fly to human will boost interest in using flies as a drug screening model. The ongoing work with Drosophila avatars described in an earlier section, which is using Drosophila models to identify therapy combinations that will later by tested on cancer patients, should yield some insight into the translational value of Drosophila screens.
Many properties beyond efficacy makes a potential drug a viable clinical candidate. Toxicity, typically due to the effect of the drug on normal cells and issues, is one of these properties. Several screens in Drosophila take toxicity into consideration in the screen design. In our screen for radiation modulators, we screened with p53 and Chk1 mutants and counter-screened with wild type. The goal was to identify chemicals that had greater efficacy on cancer-relevant mutants compared to wild type (Edwards et al., 2011; Gladstone et al., 2012). Likewise, two screens for small molecule inhibitors in Drosophila tumor models aimed to identify drugs that not only promote efficacy but also reduce toxicity. Levine and Cagan modeled non-small cell lung cancer in flies by expressing RASV12 and PTENRNAi in the larval trachea (Levine & Cagan, 2016). The authors assessed not only efficacy in the transgenic tumor model but also toxicity in non-transgenic larvae. 1000 FDA-approved drugs were screened for those that reduced larval lethality due to transgene expression. The screen yielded two hits, trametinib (inhibits MEK½ kinases) and fluvastatin (a member of statin drugs used to treat cardiovascular disease). The two drugs are found to synergize to enhance each other’s efficacy when administered together in Drosophila and in human lung cancer cells. Interestingly, high levels of trametinib was toxic to non-tumor larvae (reducing survival) but this toxicity was reduced by co-administration of Fluvastatin. In another example, Willoughby, Brumby and colleagues targeted the expression of RASV12 and clonal loss of cell polarity regulator scribble to the eye-antennae disc in Drosophila larvae (Willoughby et al., 2013). RASV12 scrib−/− clones grew as hyperplastic tumors marked with GFP. This experimental design up allowed the authors to identify compounds that reduced GFP without being toxic to the larvae, with toxicity measured in terms of larval survival. A screen through ~2000 compounds yielded one, a glutamine analog, that reproducibly fit the criteria.
Drosophila genetics allowed Dar, Cagan and colleagues to address the molecular basis for toxicity in a Drosophila RET tumor model ((Dar et al., 2012) see preceding sections for the description of the model). Expressing a cancer-relevant mutant form of Drosophila Ret, dRetMEN2B, in a subset of larval epithelial cells result in partial larval lethality. Screening for compounds that rescued lethality yield a class of multi-kinase inhibitors with a pyrazolopyrimidine core and a hydrophobic element (called ‘AD’ by the authors). Members of the AD class showed different efficacy (assessed as reducing transformation of dRetMEN2B cells) and toxicity (assessed by organismal lethality). A more toxic member showed a greater inhibition of TOR and a weaker inhibition of RAF in in vitro kinase assays. The contribution of TOR/RAF status to AD toxicity in the Ret model was confirmed using Drosophila genetics to reduce TOR/RAF. This knowledge was used to design additional AD analogs predicted to have greater efficacy/toxicity ratios. One such optimized analog reduced tumor volume in MEN2 xenografts in mice without changing body weight, a measure of toxicity in this model. Thus, a chemical genetics approach in Drosophila enabled the optimization of a polypharmacologic (multi-target) drug. Furthermore, this example illustrates that toxicity, or lack thereof, in Drosophila may predict toxicity in a human cancer model. But as in the case of efficacy, how predictable Drosophila models are for toxicity remains to be assessed systematically. Further, in examples cited here, toxicity in Drosophila was assessed in terms of organism survival. But technologies exist to monitor toxicity at the organ level such as cardiac function (for example, (Monck et al., 2017)), neurological function such as learning and memory (for example, (Pitman et al., 2009)) and locomotor function as in climbing assays (for example, (Willenbrink et al., 2016)). There is potential to exploit these assays that are relatively low in expense compared to similar assays in rodent models. Proof of concept data for the relevance of Drosophila toxicity data to mammals will of course be needed.
Conclusion
Genetic tools, conserved pathways, target-identification with mutants that are no longer responsive to the drug, and the ability to address mechanisms of drug response/resistance make flies a viable drug screening model. Added to these are practical considerations such as fast generation times and low cost, which allows the usage of a large number of animals for statistical significance to increase the robustness of the screen. The use of an intact organism and drug delivery by feeding in many of the screens described above is also argued to select for bioavailable compounds that can cross the gut and reach target organs. But with increasing ability to model complex cellular contexts in organoids or organs-on-chip, it is worth asking what advantages of flies as a drug screening will remain. Of the advantages of using Drosophila listed in this review, simplicity in gene families, low cost, and the richness of genetic tools will likely remain. In addition, it will be hard to recapitulate in organoid and organ-on-chip models the interaction among multiple cell types and systemic input such as hormones. In fact, one drawback to organoids and similar models is that many growth factors are added exogenously and thus may or may not recapitulate systemic inputs found in vivo. Drugs that work by modulating the in vivo microenvironment may be missed in organoid screens (Markstein, 2013).
Despite these advantages, wider uses of Drosophila screens are held back by limitations in automation technology. In our radiation-modulator screen, we hand-counted surviving larvae in each drug vial, a slow and laborious process; it took us about 4 years to screen through a 10,000 molecule commercial library (our unpublished data). While most researchers understand that one must sacrifice speed to be able to screen in vivo, such low speed would discourage anyone who wants to find truly new agents from large compound libraries. To make Drosophila attractive for drug screening, we must think beyond 1–2000-molecule libraries that repeatedly show up in Drosophila drug screening studies. This is not to discount the value of screening through extant drugs and repurposing them; thalidomide/ linolidimide provides a powerful example for repurposing. But the problem of screening through the same libraries is illustrated by the fact that rapamycin was a hit in three diverse screens described in this review, as a modulator of stem cell aging, TBI and PKD! Even reserpine showed up in two different screens, for modulators of sleep and feeding. Indeed, many screens described here could be automated with appropriate hardware and software. Focusing our efforts to automation of existing screens would be a worthwhile goal towards making Drosophila a model of choice for drug-screening.
Acknowledgments
TTS thanks Michele Markstein for critical comments and many helpful suggestions.
Funding Information
TTS is supported by NIH grants (R01 GM106317 and R35 GM130374).
Footnotes
Conflict of interest: none
References
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, … Massague J (2012). A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell, 150(1), 165–178. doi: 10.1016/j.cell.2012.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EN, Gochenaur L, Singh A, Grant R, Patel K, Watkins S, … Pandey UB (2018). Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum Mol Genet, 27(8), 1366–1381. doi: 10.1093/hmg/ddy047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangi E, Murgia C, Teague AG, Sansom OJ, & Cagan RL (2016). Functional exploration of colorectal cancer genomes using Drosophila. Nat Commun, 7, 13615. doi: 10.1038/ncomms13615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiello TA, Jackson FR, & Young MW (1984). Restoration of circadian behavioural rhythms by gene transfer in Drosophila. Nature, 312(5996), 752–754. [DOI] [PubMed] [Google Scholar]
- Brand AH, & Perrimon N (1994). Raf acts downstream of the EGF receptor to determine dorsoventral polarity during Drosophila oogenesis. Genes Dev, 8(5), 629–639. [DOI] [PubMed] [Google Scholar]
- Bryant PJ (1970). Cell lineage relationships in the imaginal wing disc of Drosophila melanogaster. Developmental biology, 22(3), 389–411. [DOI] [PubMed] [Google Scholar]
- Caswell DR, & Swanton C (2017). The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med, 15(1), 133. doi: 10.1186/s12916-017-0900-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, & Warren ST (2008). Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol, 4(4), 256–263. doi: 10.1038/nchembio.78 [DOI] [PubMed] [Google Scholar]
- Cleary AS, Leonard TL, Gestl SA, & Gunther EJ (2014). Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature, 508(7494), 113–117. doi: 10.1038/nature13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta-catenin signaling in development and disease. Cell, 127(3), 469–480. doi: 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- Dar AC, Das TK, Shokat KM, & Cagan RL (2012). Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature, 486(7401), 80–84. doi: 10.1038/nature11127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das TK, Esernio J, & Cagan RL (2018). Restraining Network Response to Targeted Cancer Therapies Improves Efficacy and Reduces Cellular Resistance. Cancer Res, 78(15), 4344–4359. doi: 10.1158/0008-5472.CAN-17-2001 [DOI] [PubMed] [Google Scholar]
- Debeb BG, Lacerda L, Xu W, Larson R, Solley T, Atkinson R, … Woodward WA (2012). Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/beta-catenin signaling. Stem Cells, 30(11), 2366–2377. doi: 10.1002/stem.1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P, & Boore JL (2005). Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol, 3(10), e314. doi: 10.1371/journal.pbio.0030314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozd M, Bardoni B, & Capovilla M (2018). Modeling Fragile X Syndrome in Drosophila. Front Mol Neurosci, 11, 124. doi: 10.3389/fnmol.2018.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards A, Gladstone M, Yoon P, Raben D, Frederick B, & Su TT (2011). Combinatorial effect of maytansinol and radiation in Drosophila and human cancer cells. Dis Model Mech, 4(4), 496–503. doi: 10.1242/dmm.006486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes PS, Daniel SG, McCallum AP, Boehringer AV, Sukhina AS, Zwick RA, & Zarnescu DC (2013). Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis Model Mech, 6(3), 721–733. doi: 10.1242/dmm.010710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Hernandez I, Scheenaard E, Pollarolo G, & Gonzalez C (2016). The translational relevance of Drosophila in drug discovery. EMBO Rep, 17(4), 471–472. doi: 10.15252/embr.201642080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamberi C, Hipfner DR, Trudel M, & Lubell WD (2017). Bicaudal C mutation causes myc and TOR pathway up-regulation and polycystic kidney disease-like phenotypes in Drosophila. PLoS Genet, 13(4), e1006694. doi: 10.1371/journal.pgen.1006694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasque G, Conway S, Huang J, Rao Y, & Vosshall LB (2013). Small molecule drug screening in Drosophila identifies the 5HT2A receptor as a feeding modulation target. Sci Rep, 3, srep02120. doi: 10.1038/srep02120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, & Hemann MT (2010). DNA damage-mediated induction of a chemoresistant niche. Cell, 143(3), 355–366. doi: 10.1016/j.cell.2010.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladstone M, Frederick B, Zheng D, Edwards A, Yoon P, Stickel S, … Su TT (2012). A translation inhibitor identified in a Drosophila screen enhances the effect of ionizing radiation and taxol in mammalian models of cancer. Dis Model Mech, 5(3), 342–350. doi: 10.1242/dmm.008722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladstone M, & Su TT (2011). Screening for radiation sensitizers of Drosophila checkpoint mutants. Methods Mol Biol, 782, 105–117. doi: 10.1007/978-1-61779-273-1_9 [DOI] [PubMed] [Google Scholar]
- Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, … DasGupta R (2011). An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A, 108(15), 5954–5963. doi: 10.1073/pnas.1017496108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller S, Kapuria S, Riley RR, O’Leary MN, Schreiber KH, Andersen JK, … Jasper H (2017). mTORC1 Activation during Repeated Regeneration Impairs Somatic Stem Cell Maintenance. Cell Stem Cell, 21(6), 806–818 e805. doi: 10.1016/j.stem.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin PE, Hall JC, & Rosbash M (1990). Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature, 343(6258), 536–540. doi: 10.1038/343536a0 [DOI] [PubMed] [Google Scholar]
- Haynie JL, & Bryant PJ (1977). The effects of X-rays on the proliferation dynamics of cells in the imaginal wing disc ofDrosophila melanogaster. Wilhelm Roux’s archives of developmental biology, 183(2), 85–100. [DOI] [PubMed] [Google Scholar]
- Henry JP, & Scherman D (1989). Radioligands of the vesicular monoamine transporter and their use as markers of monoamine storage vesicles. Biochem Pharmacol, 38(15), 2395–2404. [DOI] [PubMed] [Google Scholar]
- Hertenstein E, Feige B, Gmeiner T, Kienzler C, Spiegelhalder K, Johann A, … Baglioni C (2018). Insomnia as a predictor of mental disorders: A systematic review and meta-analysis. Sleep Med Rev, 43, 96–105. doi: 10.1016/j.smrv.2018.10.006 [DOI] [PubMed] [Google Scholar]
- Hofherr A, Wagner CJ, Watnick T, & Kottgen M (2016). Targeted rescue of a polycystic kidney disease mutation by lysosomal inhibition. Kidney Int, 89(4), 949–955. doi: 10.1016/j.kint.2015.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IgeaHub. (2018, 5/28/2018). 10 Best Selling Drugs 2018 – Oncology Retrieved from https://www.igeahub.com/2018/05/28/10-best-selling-drugs-2018-oncology/
- Indave BI, Minozzi S, Pani PP, & Amato L (2016). Antipsychotic medications for cocaine dependence. Cochrane Database Syst Rev, 3, CD006306. doi: 10.1002/14651858.CD006306.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AA, & Bryant PJ (1981). A quantitative study of cell death and mitotic inhibition in gamma-irradiated imaginal wing discs of Drosophila melanogaster. Radiation research, 87(3), 552–564. [PubMed] [Google Scholar]
- Javaheri S, & Redline S (2017). Insomnia and Risk of Cardiovascular Disease. Chest, 152(2), 435–444. doi: 10.1016/j.chest.2017.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, & Marcotte EM (2015). Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science, 348(6237), 921–925. doi: 10.1126/science.aaa0769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato I, Kimura S, Furuhashi T, Nakayoshi H, Takayama S, & Uenishi N (1990). Effects of recombinant murine interferon-gamma on pregnant mice and their fetuses. Fundam Appl Toxicol, 14(4), 658–665. [DOI] [PubMed] [Google Scholar]
- Katzenberger RJ, Loewen CA, Wassarman DR, Petersen AJ, Ganetzky B, & Wassarman DA (2013). A Drosophila model of closed head traumatic brain injury. Proc Natl Acad Sci U S A, 110(44), E4152–4159. doi: 10.1073/pnas.1316895110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel N (2013). ‘Doriden von Ciba’: sleeping pills, pharmaceutical marketing, and Thalidomide, 1955–1963. History and Technology, 29(2), 153–168. doi: 10.1080/07341512.2013.828509 [DOI] [Google Scholar]
- Kim HJ, & Taylor JP (2017). Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron, 96(2), 285–297. doi: 10.1016/j.neuron.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka RJ, & Benzer S (1971). Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A, 68(9), 2112–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosan C, Heidel FH, Godmann M, & Bierhoff H (2018). Epigenetic Erosion in Adult Stem Cells: Drivers and Passengers of Aging. Cells, 7(12). doi: 10.3390/cells7120237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruiswijk F, Yuniati L, Magliozzi R, Low TY, Lim R, Bolder R, … Guardavaccaro D (2012). Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci Signal, 5(227), ra40. doi: 10.1126/scisignal.2002718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagadec C, Vlashi E, Della Donna L, Dekmezian C, & Pajonk F (2012). Radiation-induced reprogramming of breast cancer cells. Stem Cells, 30(5), 833–844. doi: 10.1002/stem.1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH, … Kang HS (2017). Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer, 16(1), 10. doi: 10.1186/s12943-016-0577-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine BD, & Cagan RL (2016). Drosophila Lung Cancer Models Identify Trametinib plus Statin as Candidate Therapeutic. Cell Rep, 14(6), 1477–1487. doi: 10.1016/j.celrep.2015.12.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB (1978). A gene complex controlling segmentation in Drosophila. Nature, 276(5688), 565–570. [DOI] [PubMed] [Google Scholar]
- Lin G, Mao D, & Bellen HJ (2017). Amyotrophic Lateral Sclerosis Pathogenesis Converges on Defects in Protein Homeostasis Associated with TDP-43 Mislocalization and Proteasome-Mediated Degradation Overload. Curr Top Dev Biol, 121, 111–171. doi: 10.1016/bs.ctdb.2016.07.004 [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, & Kroemer G (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. doi: 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzo E, O’Conner AG, Barrows JM, Shreiner DD, Birchak GJ, & Zarnescu DC (2018). Medium-Chain Fatty Acids, Beta-Hydroxybutyric Acid and Genetic Modulation of the Carnitine Shuttle Are Protective in a Drosophila Model of ALS Based on TDP-43. Front Mol Neurosci, 11, 182. doi: 10.3389/fnmol.2018.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjanovic ND, Weinberg RA, & Chaffer CL (2013). Cell plasticity and heterogeneity in cancer. Clin Chem, 59(1), 168–179. doi: 10.1373/clinchem.2012.184655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M (2013). Modeling colorectal cancer as a 3-dimensional disease in a dish: the case for drug screening using organoids, zebrafish, and fruit flies. Drug Discov Today Technol, 10(1), e73–81. doi: 10.1016/j.ddtec.2012.12.005 [DOI] [PubMed] [Google Scholar]
- Markstein M, Dettorre S, Cho J, Neumuller RA, Craig-Muller S, & Perrimon N (2014). Systematic screen of chemotherapeutics in Drosophila stem cell tumors. Proc Natl Acad Sci U S A, 111(12), 4530–4535. doi: 10.1073/pnas.1401160111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR (2002). The Wnts. Genome Biol, 3(1), REVIEWS3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet-Boureima C, Porras Marroquin J, & Gamberi C (2018). Modeling Renal Disease “On the Fly”. Biomed Res Int, 2018, 5697436. doi: 10.1155/2018/5697436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misiukiewicz K (2018). The Center for Personalized Cancer Therapeutics Retrieved from https://icahn.mssm.edu/research/personalized-cancer-therapeutics
- Monck H, Toppe D, Michael E, Sigrist S, Richter V, Hilpert D, … Schwarzel M (2017). A new method to characterize function of the Drosophila heart by means of optical flow. J Exp Biol, 220(Pt 24), 4644–4653. doi: 10.1242/jeb.164343 [DOI] [PubMed] [Google Scholar]
- Monnier V, Llorens JV, & Navarro JA (2018). Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia. Int J Mol Sci, 19(7). doi: 10.3390/ijms19071989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nall AH, & Sehgal A (2013). Small-molecule screen in adult Drosophila identifies VMAT as a regulator of sleep. J Neurosci, 33(19), 8534–8540. doi: 10.1523/JNEUROSCI.0253-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, & Wieschaus E (1980). Mutations affecting segment number and polarity in Drosophila. Nature, 287(5785), 795–801. [DOI] [PubMed] [Google Scholar]
- O’. Farrell PH (2009). How Metazoans Reach Their Full Size: The Natural History of Bigness. In Hall MN, Raff M, & Thomas G (Eds.), Cell Growth: Control of Cell Size (pp. 1–22): Cold Spring Harbor Laboratory Press. [Google Scholar]
- Pisco AO, & Huang S (2015). Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br J Cancer, 112(11), 1725–1732. doi: 10.1038/bjc.2015.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman JL, DasGupta S, Krashes MJ, Leung B, Perrat PN, & Waddell S (2009). There are many ways to train a fly. Fly (Austin), 3(1), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman W, Arfons LM, & Lazarus HM (2011). The rise, fall and subsequent triumph of thalidomide: lessons learned in drug development. Ther Adv Hematol, 2(5), 291–308. doi: 10.1177/2040620711413165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D (2000). Using Drosophila as a model insect. Nat Rev Genet, 1(3), 218–226. doi: 10.1038/35042080 [DOI] [PubMed] [Google Scholar]
- Seguin A, Monnier V, Palandri A, Bihel F, Rera M, Schmitt M, … Lesuisse E (2015). A Yeast/Drosophila Screen to Identify New Compounds Overcoming Frataxin Deficiency. Oxid Med Cell Longev, 2015, 565140. doi: 10.1155/2015/565140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamon SD, & Perez MI (2016). Blood pressure-lowering efficacy of reserpine for primary hypertension. Cochrane Database Syst Rev, 12, CD007655. doi: 10.1002/14651858.CD007655.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoshita M, Scopton AP, Ung PMU, Murray MA, Silber L, Maldonado AY, … Dar AC (2018). A whole-animal platform to advance a clinical kinase inhibitor into new disease space. Nat Chem Biol, 14(3), 291–298. doi: 10.1038/nchembio.2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreafico A, Tentler JJ, Pitts TM, Tan AC, Gregory MA, Arcaroli JJ, … Eckhardt SG (2013). Rational combination of a MEK inhibitor, selumetinib, and the Wnt/calcium pathway modulator, cyclosporin A, in preclinical models of colorectal cancer. Clin Cancer Res, 19(15), 4149–4162. doi: 10.1158/1078-0432.CCR-12-3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickel SA, Gomes NP, Frederick B, Raben D, & Su TT (2015). Bouvardin is a Radiation Modulator with a Novel Mechanism of Action. Radiat Res, 184(4), 392–403. doi: 10.1667/RR14068.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese S, & Su TT (2016). Drosophila Wnt and STAT Define Apoptosis-Resistant Epithelial Cells for Tissue Regeneration after Irradiation. PLoS Biol, 14(9), e1002536. doi: 10.1371/journal.pbio.1002536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese S, & Su TT (2017). STAT, Wingless, and Nurf-38 determine the accuracy of regeneration after radiation damage in Drosophila. PLoS Genet, 13(10), e1007055. doi: 10.1371/journal.pgen.1007055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese S, & Su TT (2018). Ionizing radiation induces stem cell-like properties in a caspase-dependent manner in Drosophila. PLoS Genet, 14(11), e1007659. doi: 10.1371/journal.pgen.1007659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Wells S, Ryan A, & Cagan R (2005). ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res, 65(9), 3538–3541. doi: 10.1158/0008-5472.CAN-04-4561 [DOI] [PubMed] [Google Scholar]
- Vlashi E, Chen AM, Boyrie S, Yu G, Nguyen A, Brower PA, … Pajonk F (2016). Radiation-Induced Dedifferentiation of Head and Neck Cancer Cells Into Cancer Stem Cells Depends on Human Papillomavirus Status. Int J Radiat Oncol Biol Phys, 94(5), 1198–1206. doi: 10.1016/j.ijrobp.2016.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Colodner KJ, & Feany MB (2011). Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J Neurosci, 31(8), 2868–2877. doi: 10.1523/JNEUROSCI.3410-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hagemann TL, Messing A, & Feany MB (2016). An In Vivo Pharmacological Screen Identifies Cholinergic Signaling as a Therapeutic Target in Glial-Based Nervous System Disease. J Neurosci, 36(5), 1445–1455. doi: 10.1523/JNEUROSCI.0256-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangler MF, Yamamoto S, Chao HT, Posey JE, Westerfield M, Postlethwait J, … Bellen HJ (2017). Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics, 207(1), 9–27. doi: 10.1534/genetics.117.203067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenbrink AM, Gronauer MK, Toebben LF, Kick DR, Wells M, & Zhang B (2016). The Hillary Climber trumps manual testing: an automatic system for studying Drosophila climbing. J Neurogenet, 30(3–4), 205–211. doi: 10.1080/01677063.2016.1255211 [DOI] [PubMed] [Google Scholar]
- Willoughby LF, Schlosser T, Manning SA, Parisot JP, Street IP, Richardson HE, … Brumby AM (2013). An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis Model Mech, 6(2), 521–529. doi: 10.1242/dmm.009985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Pastor-Pareja JC, & Xu T (2010). Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature, 463(7280), 545–548. doi: 10.1038/nature08702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, & Weinberg RA (2015). Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol, 25(11), 675–686. doi: 10.1016/j.tcb.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehring WA, Wheeler DA, Reddy P, Konopka RJ, Kyriacou CP, Rosbash M, & Hall JC (1984). P-element transformation with period locus DNA restores rhythmicity to mutant, arrhythmic Drosophila melanogaster. Cell, 39(2 Pt 1), 369–376. [DOI] [PubMed] [Google Scholar]
- Zhang L, & Shay JW (2017). Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. J Natl Cancer Inst, 109(8). doi: 10.1093/jnci/djw332 [DOI] [PMC free article] [PubMed] [Google Scholar]