

Abstract
Voltage-gated sodium ion channel subtype 1.7 (NaV1.7) is a high interest target for the discovery of non-opioid analgesics. Compelling evidence from human genetic data, particularly the finding that persons lacking functional NaV1.7 are insensitive to pain, has spurred considerable effort to develop selective inhibitors of this Na+ ion channel target as analgesic medicines. Recent clinical setbacks and disappointing performance of preclinical compounds in animal pain models, however, have led to skepticism around the potential of selective NaV1.7 inhibitors as human therapeutics. In this Perspective, we discuss the attributes and limitations of recently disclosed investigational drugs targeting NaV1.7, and review evidence that, by better understanding the requirements for selectivity and target engagement, the opportunity to deliver effective analgesic medicines targeting NaV1.7 endures.
Graphical Abstract
A. Introduction
Voltage-gated sodium ion channels (NaVs) are integral membrane proteins comprising a pore-forming α-subunit and two accessory β-subunits.1 These ~260 kD transmembrane proteins undergo conformational changes in response to membrane depolarization and open transiently to allow passage of Na+ ions down an electrochemical gradient into the cell. The fast depolarization events produced by NaV channel opening result in generator potentials, action potentials, and pacemaker potentials depending on cell localization and tissue distribution. Ten mammalian isoforms of the NaV α-subunit have been identified (NaV1.1–1.9, Nax), which differ in primary sequence, tissue distribution and gating characteristics.2,3 A number of clinically prescribed small molecule therapeutics modulate NaV activity such as local anesthetics, anticonvulsants and antiarrhythmics.4 Most drugs exhibit limited NaV subtype-selectivity but display remarkably distinct pharmacology, a property that has been attributed to differences in drug affinities for closed, open and inactivated conformations of the channel (“state-dependence”). Drug efficacy may also reflect the influence of ligand binding on the distribution of protein conformations, for instance, by shifting the voltage dependence of activation or inactivation.5 Design of therapeutics targeting NaVs is complicated by the difficulty of understanding the biophysical and pharmacodynamic (PD) consequences of ligand binding to different conformational states of the channel.
The finding that certain NaV isoforms, particularly NaV1.7, NaV1.8 and NaV1.9, are expressed predominantly in unmyelinated and small diameter myelinated afferents that transmit nociceptive signals has led to widespread efforts to discover selective inhibitors of these particular subtypes.6 Selectivity is necessary for drug safety given the obligatory role of off-target NaV isoforms in central and peripheral neuronal conduction (NaV1.1–1.3, 1.6), skeletal muscle contraction (NaV1.4) and the cardiac action potential (NaV1.5).2 Interest in the discovery of selective inhibitors of NaV1.7 was further kindled by the findings that a rare genetic condition in which the channel is nonfunctional leads to insensitivity to pain, and that gain-of-function mutations to the same subtype are associated with episodic extreme pain syndromes such as erythromelalgia and paroxysmal extreme pain disorder.7–9 Based on the biophysical properties of NaV1.7, localization in small diameter peripheral afferents, human genetic findings and studies in transgenic mice, NaV1.7 has been vaunted as one the best validated targets for analgesic drug discovery.10,11 The availability of safe and effective NaV1.7 inhibitors would potentially reduce dependence on opioids for pain treatment.12 Despite this excitement, clinical proof-of-concept has remained elusive with investigation of early lead compounds in acute and chronic pain trials resulting in limited analgesic benefit. Such clinical setbacks have led some to question the obligatory role of NaV1.7 in nociception and to suggest alternative hypotheses to explain the insensitivity to pain observed in humans with NaV1.7 loss-of-function mutations and rodent knockouts.13
In this Perspective, we highlight recent advances in the discovery of small molecule and peptide selective inhibitors of NaV1.7 and discuss a series of pressing questions regarding the physicochemical and pharmacological requirements for a safe and effective antagonist of this target. We review evidence that modest efficacy of early NaV1.7 inhibitors in preclinical and clinical studies stems from insufficient target engagement and discuss strategies to overcome the limitations of disclosed lead compounds in order to realize the therapeutic promise of analgesic medicines targeting NaV1.7.
B. Contribution of Sodium Channel Subtypes to Pain Signaling
Multiple NaV isoforms including NaV1.3, NaV1.6, NaV1.7, NaV1.8 and NaV1.9 are involved in the transmission of pain signals by primary afferent neurons and have been proposed as targets for subtype-selective analgesics.14 NaV1.3 is not normally detected in adult primary afferents but is re-expressed and accumulates at the end of the transected fiber following nerve injury.15 This result led to interest in selective inhibitors of NaV1.3 as therapeutics for neuropathic pain. However, the finding that mechanical allodynia develops normally in NaV1.3 knockout mice calls into question whether selective inhibition of NaV1.3 would be sufficient for analgesia.16 NaV1.6 is expressed throughout both the central and peripheral nervous systems and concentrated at nodes of Ranvier in myelinated neurons where it is necessary for saltatory conduction of action potentials.14 Increased density of NaV1.6-positive nodes of Ranvier is observed in the spared nerve injury (SNI) model of neuropathic pain in which two of the three branches of the sciatic nerve are cut.17 Further, adult-onset knockout of NaV1.6 in primary sensory neurons attenuates development of mechanical allodynia, suggesting a role in peripheral sensitization.17 Despite these findings, achieving safe and effective analgesia by inhibition of NaV1.6 is likely to be challenging due to the broad expression of this isoform.
Three sodium channel isoforms, NaV1.7, NaV1.8 and NaV1.9, are preferentially expressed in peripheral afferents and have garnered attention as targets for novel analgesics.14 NaV1.7 is expressed in different types of primary sensory neurons, including myelinated Aβ- and Aδ-fibers, and unmyelinated C-fibers. Evidence from immunocytochemistry and electrophysiology recordings indicates that NaV1.7 expression is inversely correlated with neuronal size and conduction velocity, suggesting preferential expression in C- and Aδ-fibers that convey nociceptive information.18 Staining with a NaV1.7-selective antibody has demonstrated that the channel is present throughout the length of the neuron, from the free nerve endings to the central terminals within the dorsal horn.19 Despite its association with the peripheral nervous system, NaV1.7 is also expressed in olfactory sensory neurons and discrete regions of the CNS including hypothalamic neurons.20,21 The gating properties of NaV1.7 are characterized by a voltage of half-maximal activation and fast activation kinetics similar to other tetrodotoxin-sensitive (TTX-s) subtypes, but significantly slower closed-state inactivation (~150 ms for NaV1.7 compared to 20 ms for NaV1.6).22 The slow kinetics of closed-state inactivation allow the channel to remain in the closed state during subthreshold depolarizations and generate substantial ramp currents.23 The observations of localization at peripheral terminals, the ability to generate ramp currents and a low threshold for activation have led to a hypothesis that, in addition to contributing to axonal conduction, NaV1.7 serves as a threshold channel in nociceptors, amplifying small depolarizations caused by other channels to generate the action potential (Figure 1).11,14
Figure 1.

Contribution of NaV1.7, NaV1.8 and NaV1.9 to the action potential in peripheral nociceptive neurons, adapted from Bennett et al.14 NaV1.7 and NaV1.9 function as threshold channels, amplifying small depolarizations driven by upstream ion channels.11,14,24 NaV1.7 also contributes to the rising phase of the action potential. NaV1.8 is activated at more depolarized potentials near 0 mV, contributes the majority of current to the rising phase of the action potential, and is capable of high frequency firing due to rapid recovery from inactivation.25–27
The functional role of NaV1.7 as a threshold channel and significant contributor to the upstroke of the action potential in nociceptors may explain the profound insensitivity to acute thermal and mechanical pain observed in NaV1.7 knockout, conditional knockout and inducible knockout mice.28–30 Whereas global NaV1.7 knockout mice exhibit intact intra-epidermal C-fibers, certain human patients with NaV1.7 loss-of-function are reported to lack intra-epidermal C-fiber nociceptors as measured anatomically by skin biopsy and functionally by microneurography recordings.28,31 This finding suggests an important anatomic difference between mice and humans, although the functional consequence of NaV1.7 loss-of-function, insensitivity to acute pain, is similar in both species. The role of NaV1.7 in chronic, and particularly neuropathic pain states is less clear, with evidence that certain types of pain may be independent of NaV1.7.32 One case report describes a woman with genetic NaV1.7 loss-of-function who developed certain symptoms of neuropathic pain including reduced mechanical withdrawal thresholds, tingling and numbness following a pelvic fracture that resulted in an epidural hematoma impinging on the L5 nerve root.33 In adult onset inducible NaV1.7 knockout mice, mechanical allodynia developed following SNI, but was attenuated compared to control animals.30 In contrast, acetone-induced cold allodynia did not develop in the inducible knockout model.30 In a conditional mouse knockout in which NaV1.7 is not present in dorsal root ganglia (DRG) or sympathetic neurons, neither mechanical nor cold allodynia developed in chronic constriction injury (CCI) or spinal nerve transection (SNT) models but both were observed following treatment with the chemotherapeutic oxaliplatin.32 Viewed together, studies involving human patients with NaV1.7 loss-of-function and rodent knockout models confirm the obligatory role of NaV1.7 in acute nociception, but suggest a more nuanced function in the development and maintenance of allodynia in nerve injury models.
NaV1.8, similar to NaV1.7, has generated significant interest as a target for the identification of isoform-selective analgesics. Early reports found that NaV1.8 is preferentially expressed in small diameter primary afferents with the functional characteristics of nociceptors.34 More recent studies demonstrated that NaV1.8 is also present in a population of low threshold mechanoreceptors and hypothalamic neurons.35,36 In contrast to NaV1.7, which exhibits a voltage of half maximal activation near –30 mV in multiple expression systems, NaV1.8 is half-maximally activated at more depolarized membrane potentials near 0 mV.25,26 This difference in the threshold for activation, taken in the context of other biophysical properties of the isoforms, has led to a model for action potential generation in nociceptive sensory neurons in which NaV1.7 sets the threshold for firing and NaV1.8 contributes the majority of current to the upstroke the action potential, setting the amplitude (Figure 1).31 NaV1.8 is also likely to be the main NaV isoform involved in repetitive firing, made possible by fast recovery from inactivation.27 Isoform selective NaV1.8 inhibitors have been reported by Abbott, Pfizer, Vertex and others, and characterized in rodent pain models.37–39 Recently Vertex reported promising clinical results with a selective NaV1.8 inhibitor, VX-150, in three phase 2 studies in patients with osteoarthritis, small fiber neuropathy, and following bunionectomy.40 Collectively, the available evidence suggest that NaV1.8 functions in concert with NaV1.7 and plays a major role in establishing the excitability of nociceptive sensory neurons.
Characterization of the role of NaV1.9 in pain signaling has proven challenging due to instability of current in native neurons and difficulty expressing the isoform in heterologous systems. Recently, interest has been piqued by the identification of genetically-defined human pain disorders involving NaV1.9 and certain technical challenges to studying its functional properties have been overcome.41,42 Subcellular localization at the peripheral and central terminals of small diameter primary afferents and biophysical evidence that NaV1.9 is active at voltages near the resting membrane potential suggest that it serves as a threshold channel, albeit with slower activation/inactivation kinetics than the TTX-s isoforms NaV1.3 and NaV1.7.24 Behavioral studies in rodent knockouts indicate that NaV1.9 has a subtle role in setting thresholds for acute thermal and mechanical pain, but a more profound effect on hyperalgesia following inflammation and cold allodynia upon nerve injury.43,44 Like NaV1.7, mutations in NaV1.9 are associated with rare pain disorders in humans, with examples of both episodic extreme pain and insensitivity to pain.41,42 Paradoxically, one mutation in NaV1.9 found in patients with insensitivity to pain, L811P, results in a hyperpolarized voltage-dependence of activation, a property that would be expected to render neurons hyperexcitable.41 Functional characterization of L811P and a related mutation, L1302F, expressed heterologously in rat DRG neurons demonstrated persistent current and depolarization of the resting membrane potential to voltages that inactivated other NaV isoforms, effectively silencing a subpopulation of neurons.45 These results imply an important functional difference between insensitivity to pain associated with NaV1.7 and NaV1.9 mutations in humans.
C. Sodium Channel Conformational Changes and Binding Sites
The voltage-gated sodium channel α-subunit is composed of four homologous domains, each containing six transmembrane helices (S1–S6), arranged symmetrically around the ion-conducting pore. Each of the four domains undergoes conformational changes in response to variation in the electrical potential across the cell membrane. Cationic “voltage sensors” partially traverse the membrane and drive conformational changes that affect Na+ ion conductance (Figure 2). This mechanism was suggested in prescient work by Hodgkin and Huxley, and confirmed by experimental measurement of gating currents.46,47 Recent X-ray and cryoelectron microscopy (cryo-EM) studies of bacterial NaV homologs (NaVAb, NaVMs, NaVRh), cockroach sodium channel (NaVPaS), electric eel channel (NaV1.4-β1), and human channels (hNaV1.4-β1, hNaV1.7-β1-β2) reveal the structure and trajectory of the voltage sensors in exquisite detail.48–55 At hyperpolarized membrane potentials the S4 helices, which contain arginine residues, are drawn inward to favor the closed, “resting” conformation of the channel. Depolarization of the membrane shifts the equilibrium toward alternate conformations in which one or more S4 helices move ~5–10Å toward extracellular space. Movement of the S4 helices is coupled to “activation” and “inactivation” gating mechanisms that regulate conduction of Na+ ions through the central pore. While significant questions remain regarding gating mechanisms, a compelling body of evidence indicates that activation of the domain I–III voltage sensors is associated with channel opening whereas activation of the domain IV voltage sensor results in movement of a hydrophobic isoleucine/phenylalanine/methionine (IFM) motif and fast inactivation.56–58 Channel closing, recovery from fast inactivation, and onset and recovery of slow inactivation are also voltage-dependent and coupled to changes in the position of voltage sensors, but the molecular details are unclear. An incomplete understanding of the structure, dynamics and relative populations of the different NaV conformers and how small molecule ligands affect the conformational state of the channel present a substantial impediment to drug discovery programs.
Figure 2.

A) The NaV α-subunit consists of four homologous domains arranged symmetrically around an ion-conducting pore. Each domain contains six transmembrane alpha helices, S1–S6. The voltage sensor (S4, green cylinder) partially traverses the membrane in response to voltage changes. B) Activation of domain I–III voltage sensors is coupled to NaV channel opening. Activation of domain IV is coupled to movement of the inactivation motif (IFM) and fast-inactivation.
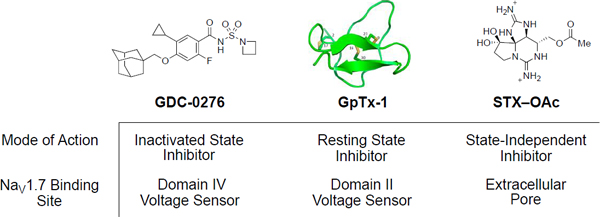
At least seven discrete binding sites have been identified for ligands that modulate NaVs (Figure 3).59 At the voltage sensor domains (VSDs), peptide anemone, cone snail, scorpion and spider toxins bind to extracellular loops, interfering with NaV activation and inactivation.60 Cysteine rich peptides isolated from spider venoms that engage the S3–S4 loop of VSD II and trap the channel in the resting state have attracted particular attention for their selective pharmacology against NaV1.7 (Figure 3, binding site 2).61 Additionally, a class of small molecule sulfonamides that bind to a distinct site on VSD IV of NaV1.7 was disclosed in patent applications filed in 2009 (Figure 3, binding site 4).62,63 Whereas the spider toxins are potent against the resting state of the channel, the sulfonamide ligands preferentially bind the depolarized conformation of VSD IV.64,65 At the pore, cationic guanidinium compounds, saxitoxin (STX) and tetrodotoxin (TTX), associate with the extracellular vestibule and sterically obstruct Na+ ion conductance (Figure 3, binding site 1).66,67 Peptide µ-conotoxins isolated from marine cone snails bind to a proximal site.68 Subtype-selectivity has been profiled for a series of conotoxins across NaV1.1–NaV1.8.69 Classical local anesthetics, such as lidocaine, benzocaine and bupivacaine, bind to a discrete region of NaV that is accessible from the intracellular side of the channel or by diffusion through intramembrane fenestrations (Figure 3, binding site 3).70 Like the sulfonamides, local anesthetics preferentially bind to certain open or inactivated conformations of the channel.71 While both state-dependent, the preference of local anesthetics and sulfonamides for specific conformers of the channel are distinct, and likely influences their pharmacology. Of the known NaV ligand binding sites, three have shown particular promise as targets for selective inhibition of NaV1.7: the sulfonamide binding site at VSD IV, the peptide toxin site at VSD II, and, recently, the extracellular pore.72,73
Figure 3.

Approximate binding sites of compounds targeting NaV1.7 overlaid on cryo-EM structure of hNaV1.7-β1-β2 complex.55 (Left) Extracellular view of hNaV1.7. (Right) Cross section through hNaV1.7. Approximate ligand binding sites: (1) extracellular vestibule, (2) VSD II, (3) local anesthetic binding site, (4) VSD IV.
D. Classes of Selective NaV1.7 Inhibitors
Sulfonamides binding to VSD IV
A series of aryl sulfonamides that bind to VSD IV of NaV1.7 first appeared in patents filed by Pfizer and Icagen in 2009 (Figure 4).62,63 Whereas other compounds characterized as NaV1.7 inhibitors such as XEN-402 (1) and CNV-1014802 (2) exhibit limited isoform selectivity, early sulfonamides were reported as highly selective for NaV1.7 over a subset of off-target isoforms including NaV1.3, NaV1.4, NaV1.5 and NaV1.8; selectivity over NaV1.1, NaV1.2 and NaV1.6 is generally more modest.74 A development candidate PF-05089771 (3) was advanced to the clinic and studied in healthy volunteers with evoked pain endpoints, and in subjects with postoperative dental pain, osteoarthritis of the knee, diabetic peripheral neuropathy and hereditary erythromelalgia.75–77 Results disclosed from these studies have been disappointing (Table 1). Despite an IC50 of 11 nM against hNaV1.7 (measured with a voltage protocol that favors the inactivated state) and maximum plasma exposures > 10,000 ng/mL, a study in healthy volunteers that measured evoked pain and a study in patients with peripheral diabetic neuropathy did not meet efficacy endpoints.75,76 Observations from two studies, one with 235 subjects with postoperative dental pain, and the other with five subjects with inherited erythromelalgia were more promising, showing marginally significant reductions in pain scores at one or more time points.76,77 As of Oct 27, 2015 PF-05089771 (3) was no longer listed in Pfizer’s drug development pipeline.78
Figure 4.

Potency and selectivity of NaV1.7 inhibitors advanced to clinical development. 64,73,79–81
Table 1.
Clinical Studies with PF-05089771
| Indication | Subjects | Arms | Endpoint | Result |
|---|---|---|---|---|
| Evoked pain in healthy volunteers75 | 25 | PF-05089771 (300 mg), PF-05089771 + pregabalin, pregabalin, ibuprofen, placebo | Thermal, UV, pressure, electrical and cold pain thresholds | Not significantly different than placebo |
| Diabetic peripheral neuropathy76 | 141 | PF-05089771 (150 mg), PF-05089711 + pregabalin, pregabalin, placebo | Daily pain numeric rating (NRS) | Trend toward lower NRS, not statistically significant |
| Postoperative dental pain76 | 235 | PF-05089771 (150–1600 mg), ibuprofen, placebo | Total pain relief, 0–6 hours (TOTPAR[6]) | Small statistically-significant effect at 150 mg dose |
| Inherited erythromelalgia77 | 5 | PF-05089771 (1600 mg), placebo, crossover design | Average pain score post dose (PI-NRS) | Lower pain score at 4–5 h and 8–9 h post dose (P < 0.1) |
| Osteoarthritis of the knee | 80 | PF-05089771, placebo | Safety, tolerability, pharmacokinetics | Not disclosed |
Additional characterization of PF-05089771 (3) and related compounds has been disclosed,64,82 and groups from Amgen, Bristol-Myers Squibb, Chromocell, Genentech/Xenon, Merck and other companies have pursued related chemical series (Figure 5).83–87 In several cases, high unbound plasma exposure (i.e. drug concentration corrected for plasma protein binding) relative to in vitro potency against NaV1.7 is required to achieve a pharmacodynamic effect in animal pain models.82,83,88,89 Sulfonamide 5 was studied in a mouse formalin pain model.82 Using an intravenous infusion to maintain unbound plasma concentrations > 60x the mouse NaV1.7 IC50 (< 0.1 nM), no evidence of analgesia was observed.
Figure 5.

NaV1.7 inhibitors targeting VSD IV evaluated in rodent pain behavior models. “+” indicates effective in PD model. “–” indicates not effective. *Kd of GX-585 against mouse TTX-resistant current = 22 µM **Kd of GX-201 against mouse TTX-resistant current = 13 µM. 30,82–84,86–89,93
First-generation sulfonamide inhibitors are largely sequestered by plasma proteins (≥ 99% plasma protein bound). Standard equilibrium dialysis methods of determining plasma protein binding are known to underestimate binding in such instances, possibly explaining the need for high unbound exposures.90 Merck has explored multiple series of aryl sulfonamides bearing basic amine functional groups with an emphasis on improving ligand efficiency and lipophilic ligand efficiency.87,88 Compound 6 exhibits an unbound fraction (fu) of 0.065 in mouse and was efficacious by subcutaneous (SC) administration in a formalin model at an unbound concentration of 1.5 µM, ~50x the mNaV1.7 IC50; however, oral bioavailability was low (F = 2%, 10 mpk in rat).88 A related compound, 7, with improved oral availability (F = 50%) was effective at an unbound concentration ~100x the mNaV1.7 IC50. These exposure multiples are consistent with other reports of the pharmacokinetic/pharmacodynamic (PK/PD) relationship of sulfonamide inhibitors despite the greater unbound fraction and reduced lipophilicity. A more striking result from the team at Merck was achieved with compound 8, which is effective in a mouse formalin model at low multiples of 3.5x the mNaV1.7 IC50.87 This result is a significant improvement from the profile of earlier sulfonamides. Interpretation of these data are complicated, however, by the absence of potency measurements against off-target isoforms such as NaV1.2 and NaV1.6, inhibition of which could contribute to efficacy but also produce undesirable central nervous system, sensory or motor deficits.91,92
An additional hypothesis for why high exposure multiples are required for analgesia with many NaV1.7-selective sulfonamides proposes that free drug concentration in plasma is not representative of drug concentration at the site of action.84 A team at Bristol Myers Squibb identified a series of sulfonamide derivatives bearing amine functional groups that display good selectivity over NaV1.5, reminiscent of compounds disclosed by Merck.84 Selectivity over other off-target NaV isoforms was not reported. Quantification of drug concentrations in dissected mouse DRG one hour after oral (po) dosing showed that total DRG concentrations correlated with apparent permeability (Papp) and ranged from 0.23–6.3x free plasma concentration across a series of four compounds. Two compounds, 9 and 10, were identified with high permeability in a PAMPA assay (Papp pH 7.4 > 20 × 10−6 cm/s), total DRG/free plasma ratios > 5 and similar potency against mNaV1.7. Compound 10 reduced pain behaviors in the mouse formalin assay at total DRG concentrations corresponding to 30x the mNaV1.7 IC50 but 9, at a similar exposure multiple, was ineffective. A measurement of the unbound drug concentrations in DRG would improve interpretation of these findings. Unbound DRG concentrations were measured for a series of indole-acylsulfonamides including 11, however, the results remain confounding.93 11 was effective in the mouse formalin, complete Freund’s adjuvant (CFA), and CCI pain models at doses that achieve unbound DRG exposures of ~3x the mNaV1.7 IC50 (35 nM) and ~9x the mNaV1.7 DRG IC50 (11 nM), whereas a closely related compound that achieved similar unbound DRG exposures was not. One explanation is that measurement of unbound drug concentration in DRG tissue is technically challenging, and may not accurately represent DRG exposure. Alternatively, factors other than poor distribution to DRG neuronal tissue may be responsible for the high exposures required for analgesia with many but not all sulfonamides.
Genentech elucidated the X-ray structure of a NaV1.7/NaVAb chimera with a bound sulfonamide, confirming the association of this ligand with the S4 helix of VSD IV.94 In a collaboration with Xenon Pharmaceuticals, two candidates GDC-0276 (4) and GDC-0310 were advanced for clinical development.81 As of October 2018, development of both compounds has been discontinued for reasons that have not been disclosed. Recent publications have revealed specific in vivo target engagement and PK/PD models that presumably supported these development programs.86 By expressing the I848T hNaV1.7 gain-of-function mutation in mice, an aconitine-induced pain behavior model was established as a preclinical tool to assess NaV1.7 target engagement (IEM model).86 Whereas competitor compounds PF-05089771 (3) and MRL-5 (similar to 8) require free plasma concentrations > 30x NaV1.7 IC50 to reduce flinching in this model, a number of Genentech/Xenon acyl sulfonamide NaV1.7 inhibitors such as 12, 13 and 14 are active in the target engagement assay at concentrations ≤ 5x the NaV1.7 IC50. 30,86,89 Compound 12 is highly potent (IC50 = 0.6 nM) and closely resembles the development candidate GDC-0276. A hypothesis has been put forward that improved target engagement at low exposure multiples compared to competitor compounds is driven by slow ligand-protein dissociation kinetics (koff < 10–4 sec−1).86 Because the kinetics of association are also slow, equilibrium IC50 values for 13 and 14 are difficult to measure.30 The equilibrium dissociation constant (Kd), determined by the kinetics of inhibition (koff/kon) is used as an alternative to IC50 for these compounds in PK/PD assessments. In wild type mice, 13 and 14 were effective in the formalin, CFA, streptozocin (STZ) diabetic neuropathy and SNI models of inflammatory and chronic pain at unbound plasma concentrations 2–12x NaV1.7 Kd. The half maximal effective concentration (EC50) of both compounds in the STZ model decreased by 3–10 fold following 12 days of dosing for reasons that are not understood but may involve a reversal of sensitization driven by suppression of electrical activity. Higher unbound plasma exposures of 13 and 14, 10–30x mNaV1.7 Kd (Figure 6), were required to affect the threshold for response to noxious thermal stimulus (hot plate, tail immersion), noxious mechanical stimulus (Randall-Selitto assay), and thermal stimulus after inflammation (CFA-Hargreaves).30 The availability of an inducible NaV1.7 knockout model makes possible direct comparison of the pharmacodynamic effect of drug treatment to the phenotype of the NaV1.7 knockout. While the effects of compounds 13 and 14 reached statistical significance in acute thermal and mechanical pain behavior assays, animals did not routinely reach the cut-off, as was the case with the knockout. This finding suggests that a larger effect size is possible at higher exposures or with superior target engagement.
Figure 6.

PK/PD relationship of GX-585 (13) and GX-201 (14) in mouse models of thermal, mechanical, chemical and inflammatory pain. “+” indicates effective in PD model. “–” indicates not effective. Ave. Cp = average total plasma concentration.30
One interpretation of the finding that higher unbound plasma concentrations of 13 and 14 are required to demonstrate a PD effect in acute thermal and mechanical pain models than in inflammatory or neuropathic pain models is that the steady-state dynamics between channel conformers in uninjured tissue favors the resting state for which the sulfonamide inhibitors have low affinity. In chronic or inflammatory pain states, depolarization of the resting membrane potential may shift the population of channel conformers toward the inactivated state, improving target engagement in a variable manner that is dependent on the specific pain condition. If this explanation is correct, state-independent inhibitors of NaV1.7 should exhibit a different PK/PD relationship than state-dependent inhibitors. The former may display more consistent pharmacology across a range of pain states because target engagement is not affected by differences in the distribution of NaV1.7 between channel conformers. For certain types of pain, state-dependent inhibition would have the advantage of addressing pathological pain while leaving normal, protective pain reflexes intact. For other types of pain, state-independent inhibition may be necessary to achieve a robust analgesic effect at well-tolerated exposures. While studies with animal PD models provide useful insight into these questions, ultimately the comparative effects of state-dependent and state-independent NaV1.7 inhibitors will need to be explored in human patients to be properly understood.
Amgen has reported a pair of atropisomeric quinolone sulfonamides having different potencies against hNaV1.7.83 The more potent enantiomer, AMG8379 (15), exhibits an IC50 < 10 nM against hNaV1.7 and > 100x selectivity over NaV1.1–1.6. This compound was effective in mouse UVB burn-induced hyperalgesia (UVB), capsaicin-induced nociceptive behavior and histamine-induced itch models at doses of 30–100 mg/kg po with terminal free plasma concentrations in the range of 5–23x mNaV1.7 IC50. The absence of locomotor effects in an open field assay at any dose tested and the lack of efficacy with the less potent atropisomer, AMG8380 (16), support the view that the anti-nociceptive effects are the result of on-target inhibition of mNaV1.7.
Collectively, the available data generated with aryl and acyl sulfonamide inhibitors of NaV1.7 indicate that substantial improvements in NaV subtype selectivity and target engagement have been made since the discovery of first-generation investigational drug candidates such as PF-05089771. Whether these improvements are sufficient to achieve robust analgesia in human pain indications with an acceptable margin of safety remains to be demonstrated.
Cystine knot peptides binding to VSD II
A number of structurally-related, cysteine-rich peptides that bind to the extracellular face of VSD II have been isolated from venomous spiders.61 These cystine knot compounds, named for their three-dimensional loop structures arising from multiple disulfide bridges, preferentially bind to the resting state of the channel and trap the channel in a closed conformation. Certain peptides exhibit selectivity for hNaV1.7 over other human isoforms.95 However, interpretation of in vivo PD results is often complicated by off-target activity against other families of ion channels (e.g. CaV channels).96 GpTx-1, JzTx-V, Huwentoxin IV (HwTx-IV), ProTx-II (17) and Pn3a (18) have been the subject of investigation as therapeutics targeting NaV1.7 and are discussed as representative examples of this class (Figure 7).
Figure 7.

Selective cystine knot toxins that bind to VSD II in NaV1.7. “+” indicates effective in PD model. “–” indicates not effective. r = rat. m = mouse.99–101
An initial report characterized ProTx-II as 100 to 500-fold selective for NaV1.7 (IC50 = 0.3 nM) over other hNaV subtypes (NaV1.2–1.6, 1.8 IC50s = 26–146 nM). 97 This study also found that ProTx-II was not analgesic in a rat CFA model of acute hyperalgesia by intravenous (iv) or intrathecal (it) administration at doses of 0.1 and 0.01 mg/kg, respectively.97 Higher doses were not tolerated and the absence of efficacy was attributed to limited permeability through the peripheral nerve sheath. Subsequent reports have indicated that the potency of ProTx-II against at least one subtype, NaV1.2, is affected by the presence of an accessory β4 subunit and that NaV subtype-selectivity is more modest than originally reported.98,99 Intrathecal administration of 2 µg of toxin in rats produced elevated thermal withdrawal latencies in the Hargreaves test and a significant reduction in phase I and phase II flinching in the formalin test.99 The therapeutic window was narrow; a dose of 0.8 µg had no effect and doses > 2 µg resulted in motor deficits, possibly due to inhibition of NaV1.1 or 1.6. In the absence of published pharmacokinetic (PK) data, it is not possible to understand what level of exposure relative to on- and off-target NaV IC50s was achieved by these dosing regimens.
µ-TRTX-Pn3a (Pn3a, 18), isolated from a tarantula venom, is also highly potent against hNaV1.7 (IC50 = 0.9 nM) with selectivity ranging from 40 to 1000-fold over all other NaV isoforms.100 Pn3a dose-dependently reverses pain behaviors induced by the NaV1.7 activator OD1 at 1 and 3 mg/kg ip, however 3 mg/kg ip produced no analgesic effect in response to inflammatory challenges induced by formalin, carrageenan, or CFA. Interestingly, Pn3a reduces mechanical hypersensitivity in a mouse model of post-surgical pain, an effect that was reversed by co-administration of the opioid antagonist naloxone.102 Pn3a acts on NaV1.7 by reducing peak current and shifting the voltage-dependence of activation to more depolarized membrane potentials; due to this mechanism of action, NaV1.7 current is not fully abolished at all membrane potentials.100 The PK profile of Pn3a has not been reported. In the absence of additional data, these findings suggest that the lack of analgesic efficacy in certain rodent pain models may be due to insufficient inhibition of NaV1.7.
The limited analgesic efficacy and narrow therapeutic window observed in studies with natural cystine knot peptides motivated the design of synthetic peptides with improved NaV subtype selectivity. Amgen and Janssen have advanced synthetic peptides that target VSD II with improved selectivity for NaV1.7 over other hNaV isoforms.95,99,101,103 Using a multi-attribute positional scan approach, Amgen has identified NaV1.7-selective analogues of the tarantula venom peptides GpTx-1 and JzTx-V. A single amino acid variation in GpTX-1, Ile10Glu, confers > 500-fold selectivity over two other peripheral subtypes, NaV1.4 and NaV1.5.101 Similarly, an analogue of JzTx-V AMG6120 (20), was identified that exhibits > 100-fold selectivity over NaV1.4, NaV1.5, NaV1.6 and NaV1.8.103 20 elicited an anti-pruritic effect in a mouse histamine-induced itch model following sc administration but required high plasma exposures, ~100x mNaV1.7 IC50. Interestingly, similar exposures were not effective in a capsaicin induced nociceptive behavior model.
Following a different approach, Janssen prepared a library of over 1500 analogues of ProTx-II, from which JNJ63953918 (21) was identified and found to exhibit 100 to > 1000 fold selectivity over NaV1.1, NaV1.2, NaV1.4, NaV1.5 and NaV1.6 in a manual patch clamp assay.99 Like ProTx-II, 21 is believed to bind the closed, resting state of NaV1.7. Similar potency is observed in electrophysiological assays that favor the resting state (holding potential = –120 mV) and an approximately 1:1 ratio of the resting/inactivated states (V½ inactivation). Species variability, which is often a confounding factor in interpreting behavioral results, was found to be minimal, with similar IC50s measured against human and rat peripheral isoforms NaV1.5, NaV1.6 and NaV1.7. Central and peripheral administration of 21 was studied in rat models of chemical and thermal pain; 0.3–7.5 µg JNJ63953918 (21) it dose-dependently reduced flinching in phase 1 and phase 2 of the rat formalin model and increased withdrawal latencies in the tail flick and hot plate assays. Peri-sciatic (PS) administration of 0.8–2.5 mg increased the thermal withdrawal latency of the ipsilateral paw in a rat Hargreaves test with no evidence of discoordination or motor effects, indicating that the peptide is also effective by local peripheral administration. Interestingly, rats receiving the highest 2.5 mg PS dose exhibited increased thermal withdrawal latencies at both the ipsilateral and contralateral paws with no evidence of gross motor deficits. These findings support the obligatory role of NaV1.7 in the peripheral and spinal transmission of pain and suggest that inhibition in either compartment may be analgesic. Absent PK measurements with 17 or 21, it is not possible to estimate the level of exposure required for analgesia by this mechanism. The finding that peripheral exposures of ~100x mNaV1.7 are required for a pharmacodynamic effect with 20 suggests that at least a subset of cystine knot peptides suffer from similar challenges with target engagement as first-generation aryl sulfonamides (vide supra).
Guanidinium compounds binding to the extracellular vestibule
TTX (22), STX (23) and the gonyautoxins are naturally occurring sodium channel inhibitors that bind to the extracellular vestibule and occlude the ion-conducting pore (Figure 8). These agents show little state-dependence, exhibiting < 3-fold variation in potency between electrophysiology protocols that favor the resting and inactivated conformations of the channel.104 While compounds that block the outer pore of the sodium channel are often dismissed in the pursuit of selectivity given the highly conserved nature of this region in all NaV subtypes, small sequence differences between isoforms exist and markedly affect ligand potency. A single amino acid variation in the domain I pore loop is responsible for the insensitivity of NaV1.5, NaV1.8, and NaV1.9 to the natural product TTX.105,106 TTX has been investigated in a number of rodent models of acute and chronic pain, and was effective at doses of 1–6 µg/kg sc in the formalin, acetic acid induced writhing, and sciatic nerve ligation (SNL) models.107 The finding that, despite relatively low potency against NaV1.5 compared to TTX-s NaV isoforms, TTX exhibits a median lethal dose of 12.5 µg/kg sc in mice is evidence that selectivity over non-cardiac subtypes (e.g., NaV1.4, NaV1.6) is necessary to achieve a high margin of safety with state-independent inhibitors.108 A formulation of TTX is currently in development at Wex Pharmaceuticals for the treatment of chemotherapy-induced peripheral neuropathy and cancer-related pain, and has been evaluated in multiple clinical studies at a doses up to 30 µg administered IM or sc.109
Figure 8.

Natural and synthetic guanidinium compounds that bind to the extracellular pore. 105,107,110–112
STX (23) and certain gonyautoxins are ≥ 100-fold less potent against hNaV1.7 in comparison to other TTX-s hNaV isoforms.113 This difference in potency is primate specific; STX is essentially equipotent against other TTX-s NaV isoforms expressed in other species such as mouse, rat and dog. Mutagenesis experiments have demonstrated that a two amino acid variation in the pore loop of domain III is responsible for the difference in potency between hNaV1.7 and other TTX-s isoforms.111 In principal, this sequence variation could be leveraged to design selective, state-independent inhibitors of hNaV1.7. Modification of the C13-substituent in compound 23 to an acetate (24) improves potency against hNaV1.7 and selectivity over hNaV1.5.111 SiteOne Therapeutics has capitalized on these findings and disclosed a series of compounds that exhibit > 1000x selectivity for NaV1.7 over NaV1.4 and NaV1.6 (e.g., compound 25, Figure 8). Selectivity over other isoforms and characterization in pain behavior models is not reported.112 Future work with this class of compounds will inform our understanding of whether highly selective, state-independent inhibitors of NaV1.7 more fully recapitulate the insensitivity to pain observed in humans with genetic NaV1.7 loss-of-function in comparison to state-dependent inhibitors with low activity against the (closed) resting state of the channel.
E. Discussion
The limited clinical efficacy of certain potent and selective inhibitors of NaV1.7 raises a series of questions regarding the pharmacology of NaV1.7:
Is acute inhibition of NaV1.7 sufficient to produce robust analgesia?
If so, what level of NaV1.7 target occupancy is required for meaningful analgesia?
Do factors such as tissue barriers, underestimated protein binding or state-dependence limit the efficacy of existing chemical series targeting NaV1.7?
What level of selectivity is required to avoid off-target effects mediated by other NaV isoforms?
Aside from analgesia, what other on-target effects may be expected from truly selective NaV1.7 inhibitors?
In the discussion that follows, we review data and hypotheses surrounding each of these questions.
Analgesia by acute inhibition
Genetic and pharmacological evidence from humans and rodents supports the obligatory role of NaV1.7 in the transmission of nociceptive signals. NaV1.7 is the most abundant TTX-s sodium channel in small diameter human and mouse DRG neurons, where it accounts for 75–100% of TTX-s current.64 Inhibitors of NaV1.7 increase the action potential rheobase and elicit action potential failure in human DRG neurons at concentrations ~10x IC50.64 Unfortunately, disappointing outcomes from early clinical studies have generated skepticism that pharmacological inhibition of NaV1.7 is sufficient to produce robust analgesia. These studies were conducted with the nonselective state-dependent NaV inhibitors XEN402 (1) and CNV1014802 (2), and with PF-05089771 (3), an isoform selective, state-dependent inhibitor that targets the inactivated state of the channel and shows very high plasma protein binding, both of which may increase the plasma exposure required for efficacy.
The failure of clinical drug candidates has been ascribed to the expression of putative gene products other than NaV1.7 that are required for loss of pain sensation.13 The observation that an endogenous opioid peptide, met-enkephalin, is upregulated 2–3 fold in conditional NaV1.7 knockout mice has been noted by multiple laboratories and supports the above hypothesis.13,114 However, the finding that an opioid antagonist, naloxone, partially reverses insensitivity to pain in NaV1.7 knockout mice is controversial. Minett et al. showed that naloxone (2 mg/kg ip) reversed insensitivity to thermal (Hargreaves) and mechanical (Randall-Selitto) noxious stimuli in a sensory-neuron specific NaV1.7 KO.13 In contrast, researchers at Genentech found that naloxone did not affect thermal or mechanical nociceptive deficits in an adult onset conditional NaV1.7 knockout and a team from Merck found that naloxone did not affect analgesia mediated by a preclinical NaV1.7 inhibitor.114,115 Moreover, studies with the highly selective gating modifier peptide JNJ63953918 (21) indicate that true NaV1.7 inhibitors are effective in pain models in morphine-tolerant animals and within minutes of administration, faster than can be explained by changes in gene expression.99 While there may be an additive effect of increased expression of endogenous opioids, current evidence supports the conclusion that acute inhibition of NaV1.7 is sufficient to produce analgesia.
NaV1.7 target occupancy
Genetic loss-of-function of NaV1.7 results in essentially complete elimination of Na+ currents mediated by this subtype. In comparison, pharmacological inhibition, especially at concentrations that minimize effects on off-target isoforms, is often incomplete. At this time, the level of NaV1.7 inhibition needed to reduce sensitivity to noxious stimuli remains an outstanding question. Some insight is obtained from ex vivo dose-response studies that determine the relationship between drug concentration and neuronal excitability. TTX is a useful tool compound for performing such measurements because it exhibits similar potency against human, rat, and mouse NaV1.7, very limited state- or use-dependence, and excellent selectivity over the two other principal NaV isoforms expressed in nociceptive afferent neurons, NaV1.8 and NaV1.9. In a study that used live cell imaging of rat DRG cultures to measure calcium release triggered by electrical field stimulation, 100 nM TTX (22) had a maximal effect, reducing the Ca2+ response by ~90%.116 Accordingly, concentrations of TTX ~10x the IC50 against rNaV1.7, which inhibit ~90% of NaV1.7 current, are sufficient to almost completely silence firing of cultured DRGs. Additional insight into the level of NaV1.7 inhibition required to affect pain behavior is gleaned from recent work from Genentech with an adult onset inducible knockout mouse.30 Following treatment with tamoxifen, NaV1.7 protein decreases exponentially with a half-life of ~0.7 weeks. Significant deficits in pain sensitivity are apparent two weeks after treatment (i.e., roughly 10–15% protein remaining) and continue to develop up to 6–8 weeks. These results are consistent with the view that inhibition of 80–90% of NaV1.7 is necessary for robust pharmacodynamic effects on pain behavior.
Factors that may limit efficacy
One explanation for the limited efficacy of investigational drugs targeting NaV1.7 posits that certain factors conspire to prevent target engagement of ostensibly potent and selective chemotypes. Understanding the nature of these factors is an active area of research in NaV1.7 drug discovery programs. Outstanding questions include the importance of central vs peripheral exposure, the level of inhibition required for analgesia (vide supra), the relative availability of the resting vs the inactivated channel conformation, and barriers that may limit drug access or distribution to the target.
Expression of NaV1.7 extends from the peripheral terminals of DRG neurons to the central terminals within the dorsal horn.19 The presence of NaV1.7 in the spinal cord raises the question of whether inhibition of the peripheral population is sufficient for analgesia, or whether distribution within the CNS is required. Preclinical results with aryl and acyl sulfonamides that have low brain/plasma ratios suggest that peripheral inhibition of NaV1.7 is sufficient for analgesia.83 Some caution is necessary in interpreting these data; given the high exposures required, one cannot rule out that efficacy in pain models could be a result of inhibition of multiple peripheral isoforms. The finding that the synthetic peptide JNJ63953918 (21) is analgesic by it or PS administration supports the position that inhibition of either the central or peripheral subpopulation of NaV1.7 is effective. The high selectivity of this compound and absence of gross motor deficits in treated animals increases confidence that the effects are mediated by inhibition of NaV1.7.
Many NaV1.7-selective inhibitors including small molecules from the sulfonamide series and at least one cystine knot peptide (20) require unbound plasma concentrations ≥ 20–100x IC50 for efficacy in rodent PD models. The high exposures needed for efficacy may be partially explained if in fact 80–90% NaV1.7 occupancy is required for meaningful analgesia. In addition, factors such as very high protein binding, tissue barriers including the peripheral nerve sheath or blood brain barrier, rapid protein-ligand dissociation kinetics,86 state-dependent activity, and reduced potency in the presence of accessory subunits may adversely influence target engagement in vivo, giving rise to an apparent PK/PD disconnect. Accurate assessment of plasma protein binding can be difficult for lipophilic compounds, particularly as the bound fraction approaches ≥ 99%. Alternatively, the concentration of unbound drug in plasma may not correlate well with levels of exposure in neuronal tissue.84 Finally, the state-dependent nature of most inhibitors that target NaV1.7 (as measured against cells in culture) may complicate predictions of target engagement in live organisms, as the in vivo distribution of NaV1.7 conformers is not well understood and likely varies between tissues and pain states. The sulfonamide class of inhibitors binds to a site on VSD IV that is exposed upon membrane depolarization. In contrast, cystine knot peptides preferentially bind to the resting conformation of VSD II; guanidinium pore blockers exhibit little if any conformational preference. Understanding which of these factors limit efficacy of certain sulfonamide inhibitors and related investigational drugs is critical for advancing new small molecules that target NaV1.7.
Selectivity
Achieving selectivity over off-target NaV isoforms is a formidable challenge in the development of NaV1.7 inhibitors. Due to the obligatory role of NaVs in the generation and propagation of action potentials, inhibition of off-target isoforms is a profound safety liability. An open question is what level of selectivity over different off-target isoforms, as measured by a functional in vitro assay such as whole-cell electrophysiology, is necessary for a satisfactory therapeutic window in vivo. The requirement for selectivity is compound specific, as differences in tissue distribution, state- and frequency dependent binding will augment or erode the margin of safety in ways that are difficult to predict from in vitro measurements.
The gating modifier toxin JNJ63953918 (21) exhibits > 100x selectivity for hNaV1.7 over hNaV1.1, hNaV1.2, hNaV1.4, hNaV1.5 and hNaV1.6, as measured by whole-cell electrophysiology against recombinant NaV isoforms.99 A more modest 7–16x therapeutic window between the lowest analgesic dose and the onset of muscle weakness was observed in rats following intrathecal administration. This margin of safety is consistent with the view that analgesia requires > 80% block of NaV1.7 and that sensory-motor effects may occur at lower levels of off-target NaV inhibition. Amgen has reported that AMG8379 (15), a state-dependent sulfonamide NaV1.7 inhibitor that exhibits 100–1000x selectivity over other NaV isoforms, is effective in mouse histamine-induced itch and capsaicin-induced nociceptive behavior models at doses of 30 and 100 mg/kg po. The high dose did not cause a decrease in locomotor activity, as measured in an open field assay by automated beam breaks. The finding that a less potent atropisomer AMG8380 (16) is not effective in either behavioral model supports the conclusion that the anti-itch and anti-nociceptive effects of 15 are mediated by NaV1.7. Other reports describing the efficacy of sulfonamides do not discuss the influence of these compounds on locomotion. In addition, absent knowledge of the potency of sulfonamides towards off-target NaVs (preferably with appropriate rodent isoforms), it is not possible to conclude with certainty that analgesia is mediated by NaV1.7 block or by inhibition of more than one NaV isoform. In light of evidence that a high level of NaV1.7 inhibition is required for analgesia by acute dosing and that ostensibly potent and selective NaV1.7 inhibitors exhibit a narrow window between analgesia and adverse pharmacological effects, a high level of selectivity for NaV1.7, perhaps > 100x, may be necessary to attain the former. This level of selectivity may be achieved with compounds that display differential potency against NaV isoforms, functional selectivity driven by differences in NaV isoform tissue distribution, action potential frequency, and state-dependent binding, or a combination of the two. If mild adverse physiological effects such as paresthesias or CNS side effects precede gross motor deficits, selectivity levels greatly exceeding 100x may be required to achieve a robust margin of safety in humans.
Additional on-target effects
While pharmaceutical interest in NaV1.7 has primarily focused on the role of this subtype in pain sensation, recent work has demonstrated that NaV1.7 also mediates olfaction, cough, regulation of glucose sensing, and body weight.21,117–119 NaV1.7 is highly expressed in olfactory sensory neurons and appears to have a non-redundant role in the transmission of action potentials along a section of olfactory sensory neurons within the olfactory glomerulus.20 Mice and humans lacking functional NaV1.7 exhibit a loss of smell (anosmia), and NaV1.7 inhibitors have been shown to impact odor recognition. 30,117 In vagal sensory neurons that arise from the trachea, larynx and the lungs, NaV1.7 is the predominant NaV isoform, thus raising the possibility that NaV1.7 inhibitors could affect the cough reflex or urge to cough. Bilateral treatment of guinea pig nodose ganglia with adeno-associated virus containing NaV1.7 shRNA nearly abolished the cough response to a chemical irritant (0.1 M citric acid).118 NaV1.7 is also expressed in pancreatic β-cells where it is involved in glucose sensing and insulin secretion. NaV1.7 knockout mice exhibit > 3-fold higher insulin content in insulin-producing β cells than wild type controls.119 In mouse hypothalamic neurons, expression of NaV1.7 prolongs excitatory postsynaptic potentials and is involved in integration of synaptic input. NaV1.7 knockout in Agrp+ and Pomc+ subtypes of neurons, respectively, resulted in a decrease and increase in bodyweight.21 These observations suggest additional indications for which selective inhibitors of NaV1.7 may prove useful and draw attention to potential on-target side effects of analgesics targeting NaV1.7 that will need to be monitored during development.
Conclusion
Despite the limited clinical efficacy of first- and second-generation NaV1.7 inhibitors, this channel subtype remains a compelling target for analgesic drug development. Human and rodent genetic validation, and electrophysiological studies in DRG neurons support the view that NaV1.7 has an obligatory role in setting the threshold for action potential generation in nociceptive neurons. The poor showing of early clinical candidates may have resulted from limited selectivity over off-target NaV isoforms, preferential drug binding to the inactivated state, and/or poor drug properties such as high plasma protein binding and high unbound clearance resulting in a level of target engagement that was insufficient to produce robust analgesia. Results from behavioral experiments conducted with compounds that are > 100x selective over other NaV isoforms such as the gating modifier toxin JNJ63953918 (21) and the sulfonamide AMG8379 (15) indicate that analgesia is possible by either central or peripheral inhibition of NaV1.7, but that relatively high levels of target engagement, perhaps 80–95%, are required to achieve an acute pharmacodynamic effect.83,99 Recent studies with the acyl sulfonamides GX-585 (13) and GX-201 (14) suggest that prolonged exposure to these inhibitors through repeat dosing may reverse sensitization in chronic pain models at exposures that do not affect sensitivity to acute thermal and mechanical pain. 30,86 Going forward, efforts that focus on identifying lead compounds with excellent selectivity (e.g. > 100x) over off-target NaV isoforms and evidence of target engagement at low exposure multiples hold promise of yielding safe and effective medicines.
Acknowledgments
Relevant Funding Sources
Authors Mulcahy, Pajouhesh, Beckley and Delwig received funding from National Institutes of Health grant NS081887 and USAMRMC contract W81XWH-17–1-0672.
Abbreviations
- CFA
complete Freund’s adjuvant
- Cryo-EM
cryoelectron microscopy
- DRG
dorsal root ganglia
- fu
unbound fraction
- IEM
inherited erythromelalgia mouse model
- IFM
isoleucine/phenylalanine/methionine motif
- Kd
equilibrium dissociation constant
- NaV
voltage-gated Na+ ion channel
- NRS
numeric rating scale
- Papp
apparent permeability
- PS
peri-sciatic
- SNI
spared nerve injury
- SNL
sciatic nerve ligation
- SNT
spinal nerve transection
- STX
saxitoxin
- STZ
streptozocin
- TTX
tetrodotoxin
- TTX-s
tetrodotoxin-sensitive
- UVB
UVB burn-induced hyperalgesia
- VSD
voltage sensor domain
Biographies
John V. Mulcahy – John Mulcahy is a cofounder and Vice President of Research at SiteOne Therapeutics. He received his PhD from Stanford University, working in the fields of natural product total synthesis and synthetic methodology, and completed a postdoctoral fellowship at the Stanford University School of Medicine in the Department of Anesthesiology.
Hassan Pajouhesh Ph.D. is Director of Chemistry at SiteOne Therapeutics. He has a broad background in organic synthesis and medicinal chemistry and over 20 years of specific training and expertise in the ion channel research area. He has directed successful lead optimization efforts for multiple indications, resulting in clinical candidates and Investigational New Drug (IND) applications. He is author or co-author of more than 22 peer-reviewed publications and a co-inventor on more than 30 issued or pending U.S. patents. Dr. Pajouhesh completed his Ph.D. in the field of organic chemistry at the University of Missouri-Kansas City. He received postdoctoral training at Southern Methodist University where his research contributed to the total synthesis of (+-)-Asparenomycin C.
Jacob T. Beckley is a Principal Scientist at SiteOne Therapeutics and leads the preclinical efficacy testing program. He received his PhD in Neuroscience from the Medical University of South Carolina under the direction of Dr. John J. Woodward. He then completed postdoctoral training in Dr. Dorit Ron’s laboratory at the University of California, San Francisco. His research is focused on assessing the electrophysiological and anti-nociceptive properties of selective sodium channel blockers.
Anton Delwig PhD is a Principal Scientist at SiteOne. He has 12 years of experience in molecular biology, neurophysiology and behavioral studies. Dr. Delwig received his PhD in neurobiology from the University of Vermont and completed his post-doctoral training at UC Berkeley and UCSF. He is a co-Principal Investigator on two small business grants awarded to SiteOne by The National Institutes of Health.
Justin Du Bois joined the Department of Chemistry at Stanford University in 1999 and is currently the Henry Dreyfus Professor. His research interests span chemical design and synthesis, chemical biology, and neuroscience. He is a cofounder and executive board member of SiteOne Therapeutics.
Dr. John Hunter is the Chief Scientific Officer of SiteOne Therapeutics based in South San Francisco, CA. Previously, Dr. Hunter held leadership roles within Merck Research Laboratories, Schering-Plough, Roche Bioscience and the Parke-Davis Neuroscience Research Center. While at Roche he served as Consulting Professor of Anesthesiology at Stanford University School of Medicine. Dr. Hunter received his Bachelor of Science degree with honors and Ph.D. in Pharmacology from the University of Glasgow. MRC Post-Doctoral Fellowships in Neuroscience then followed at the University of Cambridge. His principal areas of research have been neurodegenerative disease and pain. He has authored more than 100 original scientific articles, as well as several reviews and/or book chapters and participated as either editor or reviewer for several journals.
Footnotes
Ancillary Information
Supporting Information – none
References Cited
- (1).Ahern CA; Payandeh J; Bosmans F; Chanda B The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol 2016, 147 (1), 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Catterall WA; Goldin AL; Waxman SG International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev 2005, 57 (4), 397–409. [DOI] [PubMed] [Google Scholar]
- (3).Goldin AL Diversity of mammalian voltage-gated sodium channels. Ann. N. Y. Acad. Sci 1999, 868, 38–50. [DOI] [PubMed] [Google Scholar]
- (4).Lenkey N; Karoly R; Lukacs P; Vizi ES; Sunesen M; Fodor L; Mike A Classification of drugs based on properties of sodium channel inhibition: a comparative automated patch-clamp study. PloS One 2010, 5 (12), e15568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hille B Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol 1977, 69 (4), 497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cummins TR; Sheets PL; Waxman SG The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 2007, 131 (3), 243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cox JJ; Reimann F; Nicholas AK; Thornton G; Roberts E; Springell K; Karbani G; Jafri H; Mannan J; Raashid Y; Al-Gazali L; Hamamy H; Valente EM; Gorman S; Williams R; McHale DP; Wood JN; Gribble FM; Woods CG An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444 (7121), 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nilsen KB; Nicholas AK; Woods CG; Mellgren SI; Nebuchennykh M; Aasly J Two novel SCN9A mutations causing insensitivity to pain. Pain 2009, 143 (1), 155–158. [DOI] [PubMed] [Google Scholar]
- (9).Dib-Hajj SD; Rush AM; Cummins TR; Hisama FM; Novella S; Tyrrell L; Marshall L; Waxman SG Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128 (8), 1847–1854. [DOI] [PubMed] [Google Scholar]
- (10).Clare JJ Targeting voltage-gated sodium channels for pain therapy. Expert Opin. Investig. Drugs 2010, 19 (1), 45–62. [DOI] [PubMed] [Google Scholar]
- (11).Dib-Hajj SD; Yang Y; Black JA; Waxman SG The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci 2013, 14 (1), 49–62. [DOI] [PubMed] [Google Scholar]
- (12).Volkow ND; Collins FS The role of science in addressing the opioid crisis. N. Engl. J. Med 2017, 377 (4), 391–394. [DOI] [PubMed] [Google Scholar]
- (13).Minett MS; Pereira V; Sikandar S; Matsuyama A; Lolignier S; Kanellopoulos AH; Mancini F; Iannetti GD; Bogdanov YD; Santana-Varela S; Millet Q; Baskozos G; MacAllister R; Cox JJ; Zhao J; Wood JN Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel NaV1.7. Nat. Commun 2015, 6, 8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bennett DL; Clark AJ; Huang J; Waxman SG; Dib-Hajj SD The role of voltage-gated sodium channels in pain signaling. Physiol. Rev 2019, 99 (2), 1079–1151. [DOI] [PubMed] [Google Scholar]
- (15).Black JA; Cummins TR; Plumpton C; Chen YH; Hormuzdiar W; Clare JJ; Waxman SG Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J. Neurophysiol 1999, 82 (5), 2776–2785. [DOI] [PubMed] [Google Scholar]
- (16).Nassar MA; Baker MD; Levato A; Ingram R; Mallucci G; McMahon SB; Wood JN Nerve injury induces robust allodynia and ectopic discharges in NaV1.3 null mutant mice. Mol. Pain 2006, 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen L; Huang J; Zhao P; Persson A-K; Dib-Hajj FB; Cheng X; Tan A; Waxman SG; Dib-Hajj SD Conditional knockout of NaV1.6 in adult mice ameliorates neuropathic pain. Sci. Rep 2018, 8 (1), 3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Djouhri L; Newton R; Levinson SR; Berry CM; Carruthers B; Lawson SN Sensory and electrophysiological properties of guinea-pig sensory neurones expressing NaV 1.7 (PN1) Na+ channel α subunit protein. J. Physiol 2003, 546, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Black JA; Frézel N; Dib-Hajj SD; Waxman SG Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol. Pain 2012, 8, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ahn H-S; Black JA; Zhao P; Tyrrell L; Waxman SG; Dib-Hajj SD NaV1.7 is the predominant sodium channel in rodent olfactory sensory neurons. Mol. Pain 2011, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Branco T; Tozer A; Magnus CJ; Sugino K; Tanaka S; Lee AK; Wood JN; Sternson SM Near-perfect synaptic integration by NaV1.7 in hypothalamic neurons regulates body weight. Cell 2016, 165 (7), 1749–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Herzog RI; Cummins TR; Ghassemi F; Dib-Hajj SD; Waxman SG Distinct repriming and closed-state inactivation kinetics of NaV1.6 and NaV1.7 sodium channels in mouse spinal sensory neurons. J. Physiol 2003, 551, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cummins TR; Howe JR; Waxman SG Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the HNE/PN1 sodium channel. J. Neurosci 1998, 18 (23), 9607–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cummins TR; Dib-Hajj SD; Black JA; Akopian AN; Wood JN; Waxman SG A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J. Neurosci 1999, 19 (24), RC43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Akopian AN; Sivilotti L; Wood JN A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996, 379 (6562), 257–262. [DOI] [PubMed] [Google Scholar]
- (26).Sangameswaran L; Delgado SG; Fish LM; Koch BD; Jakeman LB; Stewart GR; Sze P; Hunter JC; Eglen RM; Herman RC Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem 1996, 271 (11), 5953–5956. [DOI] [PubMed] [Google Scholar]
- (27).Renganathan M; Cummins TR; Waxman SG Contribution of NaV1.8 sodium channels to action potential electrogenesis in DRG neurons. J. Neurophysiol 2001, 86 (2), 629–640. [DOI] [PubMed] [Google Scholar]
- (28).Gingras J; Smith S; Matson DJ; Johnson D; Nye K; Couture L; Feric E; Yin R; Moyer BD; Peterson ML; Rottman JB; Beiler RJ; Malmberg AB; McDonough SI Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PloS One 2014, 9 (9), e105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Minett MS; Nassar MA; Clark AK; Passmore G; Dickenson AH; Wang F; Malcangio M; Wood JN Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun 2012, 3, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shields SD; Deng L; Reese RM; Dourado M; Tao J; Foreman O; Chang JH; Hackos DH Insensitivity to pain upon adult-onset deletion of Nav1.7 or its blockade with selective inhibitors. J. Neurosci 2018, 38 (47), 10180–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).McDermott LA; Weir GA; Themistocleous AC; Segerdahl AR; Blesneac I; Baskozos G; Clark AJ; Millar V; Peck LJ; Ebner D; Tracey I; Serra J; Bennett DL Defining the functional role of NaV1.7 in human nociception. Neuron 2019, 101 (5), 905–919.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Minett MS; Falk S; Santana-Varela S; Bogdanov YD; Nassar MA; Heegaard A-M; Wood JN Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep 2014, 6 (2), 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wheeler DW; Lee MCH; Harrison EK; Menon DK; Woods CG Case report: neuropathic pain in a patient with congenital insensitivity to pain. F1000Research 2014, 3, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Djouhri L; Fang X; Okuse K; Wood JN; Berry CM; Lawson SN The TTX-resistant sodium channel NaV1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J. Physiol 2003, 550, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shields SD; Ahn H-S; Yang Y; Han C; Seal RP; Wood JN; Waxman SG; Dib-Hajj SD Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain 2012, 153 (10), 2017–2030. [DOI] [PubMed] [Google Scholar]
- (36).Lu VB; Ikeda SR; Puhl HLA 3.7 Kb fragment of the mouse Scn10a gene promoter directs neural crest but not placodal lineage EGFP expression in a transgenic animal. J. Neurosci 2015, 35 (20), 8021–8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Jarvis MF; Honore P; Shieh C-C; Chapman M; Joshi S; Zhang X-F; Kort M; Carroll W; Marron B; Atkinson R; Thomas J; Liu D; Krambis M; Liu Y; McGaraughty S; Chu K; Roeloffs R; Zhong C; Mikusa JP; Hernandez G; Gauvin D; Wade C; Zhu C; Pai M; Scanio M; Shi L; Drizin I; Gregg R; Matulenko M; Hakeem A; Gross M; Johnson M; Marsh K; Wagoner PK; Sullivan JP; Faltynek CR; Krafte DS A-803467, a potent and selective NaV1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (20), 8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bagal SK; Bungay PJ; Denton SM; Gibson KR; Glossop MS; Hay TL; Kemp MI; Lane CAL; Lewis ML; Maw GN; Million WA; Payne CE; Poinsard C; Rawson DJ; Stammen BL; Stevens EB; Thompson LR Discovery and optimization of selective NaV1.8 modulator series that demonstrate efficacy in preclinical models of pain. ACS Med. Chem. Lett 2015, 6 (6), 650–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Anderson C; Hadida-Ruah SS; Golec JMC; Zhang B; Littler BJ; Keshavarz-Shokri A; Alcacio TE; Belmont DT Prodrugs of pyridone amides useful as modulators of sodium channels Patent WO2015089361, 2015.
- (40).Vertex announces positive phase 2 data in third proof-of concept study with the NaV1.8 inhibitor VX-150 2018, https://investors.vrtx.com/news-releases/news-release-details/vertex-announces-positive-phase-2-data-third-proof-concept-study (accessed Mar 26, 2019).
- (41).Leipold E; Liebmann L; Korenke GC; Heinrich T; Giesselmann S; Baets J; Ebbinghaus M; Goral RO; Stödberg T; Hennings JC; Bergmann M; Altmüller J; Thiele H; Wetzel A; Nürnberg P; Timmerman V; De Jonghe P; Blum R; Schaible HG; Weis J; Heinemann SH; Hübner CA; Kurth I A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat. Genet 2013, 45 (11), 1399–1404. [DOI] [PubMed] [Google Scholar]
- (42).Zhang XY; Wen J; Yang W; Wang C; Gao L; Zheng LH; Wang T; Ran K; Li Y; Li X; Xu M; Luo J; Feng S; Ma X; Ma H; Chai Z; Zhou Z; Yao J; Zhang X; Liu JY Gain-of-function mutations in SCN11A cause familial episodic pain. Am. J. Hum. Genet 2013, 93 (5), 957–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Priest BT; Murphy BA; Lindia JA; Diaz C; Abbadie C; Ritter AM; Liberator P; Iyer LM; Kash SF; Kohler MG; Kaczorowski GJ; MacIntyre DE; Martin WJ Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (26), 9382–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Minett MS; Eijkelkamp N; Wood JN Significant determinants of mouse pain behaviour. PloS One 2014, 9 (8), e104458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Huang J; Vanoye CG; Cutts A; Goldberg YP; Dib-Hajj SD; Cohen CJ; Waxman SG; George AL Sodium channel NaV1.9 mutations associated with insensitivity to pain dampen neuronal excitability. J. Clin. Invest 2017, 127 (7), 2805–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hodgkin AL; Huxley AF A Quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol 1952, 117 (4), 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Armstrong CM; Bezanilla F Currents related to movement of the gating particles of the sodium channels. Nature 1973, 242, 459–461. [DOI] [PubMed] [Google Scholar]
- (48).Payandeh J; Scheuer T; Zheng N; Catterall WA The crystal structure of a voltage-gated sodium channel. Nature 2011, 475 (7356), 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zhang X; Ren W; DeCaen P; Yan C; Tao X; Tang L; Wang J; Hasegawa K; Kumasaka T; He J; Wang J; Clapham DE; Yan N Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 2012, 486 (7401), 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shen H; Zhou Q; Pan X; Li Z; Wu J; Yan N Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355 (6328), eaal4326. [DOI] [PubMed] [Google Scholar]
- (51).Yan Z; Zhou Q; Wang L; Wu J; Zhao Y; Huang G; Peng W; Shen H; Lei J; Yan N Structure of the NaV1.4-β1 complex from electric eel. Cell 2017, 170 (3), 470–482.e11. [DOI] [PubMed] [Google Scholar]
- (52).Sula A; Booker J; Ng LCT; Naylor CE; DeCaen PG; Wallace BA The complete structure of an activated open sodium channel. Nat. Commun 2017, 8, 14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Lenaeus MJ; Gamal El-Din TM; Ing C; Ramanadane K; Pomès R; Zheng N; Catterall WA Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (15), E3051–E3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Pan X; Li Z; Zhou Q; Shen H; Wu K; Huang X; Chen J; Zhang J; Zhu X; Lei J; Xiong W; Gong H; Xiao B; Yan N Structure of the human voltage-gated sodium channel NaV1.4 in Complex with β1. Science 2018, 362 (6412), eaau2486. [DOI] [PubMed] [Google Scholar]
- (55).Shen H; Liu D; Wu K; Lei J; Yan N Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363 (6433), 1303–1308. [DOI] [PubMed] [Google Scholar]
- (56).Chanda B; Bezanilla F Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol 2002, 120 (5), 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Capes DL; Goldschen-Ohm MP; Arcisio-Miranda M; Bezanilla F; Chanda B Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol 2013, 142 (2), 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Goldschen-Ohm MP; Capes DL; Oelstrom KM; Chanda B Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat. Commun 2013, 4, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Stevens M; Peigneur S; Tytgat J Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol 2011, 2, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Israel MR; Tay B; Deuis JR; Vetter I Sodium channels and venom peptide pharmacology. In Advances in Pharmacology; Geraghty DP, Rash LD, Eds.; Elsevier: Cambridge, MA, 2017; Vol. 79, pp 67–116. [DOI] [PubMed] [Google Scholar]
- (61).Bosmans F; Swartz KJ Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci 2010, 31 (4), 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Beaudoin S; Laufersweiler MC; Markworth CJ; Millan DS; Rawson DJ; Reister SM; Sasaki K; Storer RI; Stupple PA; Swain NA; West CW; Zhou S Sulfonamide Derivatives Patent WO2010079443, 2010.
- (63).McCormack K; Santos S; Chapman ML; Krafte DS; Marron BE; West CW; Krambis MJ; Antonio BM; Zellmer SG; Printzenhoff D; Padilla KM; Lin Z; Wagoner PK; Swain NA; de Groot M; Butt RP; Castle NA Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (29), E2724–E2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Alexandrou AJ; Brown AR; Chapman ML; Estacion M; Turner J; Mis MA; Wilbrey A; Payne EC; Gutteridge A; Cox PJ; Doyle R; Printzenhoff D; Lin Z; Marron BE; West C; Swain NA; Storer RI; Stupple PA; Castle NA; Hounshell JA; Rivara M; Randall A; Dib-Hajj SD; Krafte D; Waxman SG; Patel MK; Butt RP; Stevens EB Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS One 2016, 11 (4), e0152405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Theile JW; Fuller MD; Chapman ML The selective NaV1.7 inhibitor, PF-05089771, interacts equivalently with fast and slow inactivated NaV1.7 channels. Mol. Pharmacol 2016, 90 (5), 540–548. [DOI] [PubMed] [Google Scholar]
- (66).Tetrodotoxin, Saxitoxin, and the Molecular Biology of the Sodium Channel; Kao CY, Levinson SR, Eds.; Annals of the New York Academy of Science: New York, 1986. [PubMed] [Google Scholar]
- (67).Thottumkara AP; Parsons WH; Du Bois J Saxitoxin. Angew. Chem. Int. Ed 2014, 53 (23), 5760–5784. [DOI] [PubMed] [Google Scholar]
- (68).Zhang M-M; McArthur JR; Azam L; Bulaj G; Olivera BM; French RJ; Yoshikami D Unexpected synergism between tetrodotoxin and µ-conotoxin in blocking voltage-gated sodium channels. Channels 2009, 3 (1), 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wilson MJ; Yoshikami D; Azam L; Gajewiak J; Olivera BM; Bulaj G; Zhang M-M μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (25), 10302–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Martin LJ; Corry B Locating the route of entry and binding sites of benzocaine and phenytoin in a bacterial voltage gated sodium channel. PLoS Comput. Biol 2014, 10 (7), e1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wang G-K; Strichartz GR State-dependent inhibition of sodium channels by local anesthetics: a 40-year evolution. Biochem. Mosc. Suppl. Ser. Membr. Cell Biol 2012, 6 (2), 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Payandeh J; Hackos DH Selective ligands and drug discovery targeting the voltage-gated sodium channel Nav1.7. Handb. Exp. Pharmacol 2018, 246, 271–306. [DOI] [PubMed] [Google Scholar]
- (73).McKerrall SJ; Sutherlin DP NaV1.7 inhibitors for the treatment of chronic pain. Bioorg. Med. Chem. Lett 2018, 28 (19), 3141–3149. [DOI] [PubMed] [Google Scholar]
- (74).Swain NA; Batchelor D; Beaudoin S; Bechle BM; Bradley PA; Brown AD; Brown B; Butcher KJ; Butt RP; Chapman ML; Denton S; Ellis D; Galan SRG; Gaulier SM; Greener BS; de Groot MJ; Glossop MS; Gurrell IK; Hannam J; Johnson MS; Lin Z; Markworth CJ; Marron BE; Millan DS; Nakagawa S; Pike A; Printzenhoff D; Rawson DJ; Ransley SJ; Reister SM; Sasaki K; Storer RI; Stupple PA; West CW Discovery of clinical candidate 4-[2-(5-Amino-1H-pyrazol-4-yl)-4-chlorophenoxy]-5-chloro-2-fluoro-N-1,3-thiazol-4-ylbenzenesulfonamide (PF-05089771): design and optimization of diaryl ether aryl sulfonamides as selective inhibitors of NaV1.7. J. Med. Chem 2017, 60 (16), 7029–7042. [DOI] [PubMed] [Google Scholar]
- (75).Groeneveld GJ The use of a multi-modal pain test battery in early phase clinical drug development. Presented at The Annual Pain & Migraine Therapeutics Summit, San Diego, 2017. [Google Scholar]
- (76).McDonnell A; Collins S; Ali Z; Iavarone L; Surujbally R; Kirby S; Butt RP Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. PAIN 2018, 159 (8), 1465–1476. [DOI] [PubMed] [Google Scholar]
- (77).Cao L; McDonnell A; Nitzsche A; Alexandrou A; Saintot P-P; Loucif AJC; Brown AR; Young G; Mis M; Randall A; Waxman SG; Stanley P; Kirby S; Tarabar S; Gutteridge A; Butt R; McKernan RM; Whiting P; Ali Z; Bilsland J; Stevens EB Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci. Transl. Med 2016, 8 (335), 335ra56. [DOI] [PubMed] [Google Scholar]
- (78).Pfizer Pipeline As of October 27, 2015, https://www.pfizer.com/sites/default/files/product-pipeline/PfizerPipeline_0.pdf (accessed Mar 26, 2019).
- (79).Goldberg YP; Cohen CJ; Namdari R; Price N; Cadieux JA; Young C; Sherrington R; Pimstone SN Letter to the editor: Pain 2014, 155 (4), 837–838. [DOI] [PubMed] [Google Scholar]
- (80).Deuis JR; Wingerd JS; Winter Z; Durek T; Dekan Z; Sousa SR; Zimmermann K; Hoffmann T; Weidner C; Nassar MA; Alewood PF; Lewis RJ; Vetter I Analgesic effects of GpTx-1, PF-04856264 and CNV1014802 in a mouse model of NaV1.7-mediated pain. Toxins 2016, 8 (3), 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Varney M Roche: at the forefront of R&D innovation and breakthrough treatments, 2016, https://www.roche.com/dam/jcr:5c999124-c278-4549-8e94-4475cc741de1/en/2016_roche_cowen_presentation.pdf (accessed Feb 1, 2019).
- (82).Storer RI; Pike A; Swain NA; Alexandrou AJ; Bechle BM; Blakemore DC; Brown AD; Castle NA; Corbett MS; Flanagan NJ; Fengas D; Johnson MS; Jones LH; Marron BE; Payne CE; Printzenhoff D; Rawson DJ; Rose CR; Ryckmans T; Sun J; Theile JW; Torella R; Tseng E; Warmus JS Highly potent and selective NaV1.7 inhibitors for use as intravenous agents and chemical probes. Bioorg. Med. Chem. Lett 2017, 27 (21), 4805–4811. [DOI] [PubMed] [Google Scholar]
- (83).Kornecook TJ; Yin R; Altmann S; Be X; Berry V; Ilch CP; Jarosh M; Johnson D; Lee JH; Lehto SG; Ligutti J; Liu D; Luther J; Matson D; Ortuno D; Roberts J; Taborn K; Wang J; Weiss MM; Yu V; Zhu DXD; Fremeau RT Jr.; Moyer BD Pharmacologic characterization of AMG8379, a potent and selective small molecule sulfonamide antagonist of the voltage-gated sodium channel NaV1.7. J. Pharmacol. Exp. Ther 2017, 362 (1), 146–160. [DOI] [PubMed] [Google Scholar]
- (84).Wu Y-J; Guernon J; Shi J; Ditta J; Robbins KJ; Rajamani R; Easton A; Newton A; Bourin C; Mosure K; Soars MG; Knox RJ; Matchett M; Pieschl RL; Post-Munson DJ; Wang S; Herrington J; Graef J; Newberry K; Bristow LJ; Meanwell NA; Olson R; Thompson LA; Dzierba C Development of new benzenesulfonamides as potent and selective Nav1.7 inhibitors for the treatment of pain. J. Med. Chem 2017, 60 (6), 2513–2525. [DOI] [PubMed] [Google Scholar]
- (85).Babich O; Luo RZ; Wang-Fisher Y; Palling DJ; Venjatachalan SP Sodium channel modulators for the treatment of pain Patent WO2014151472, 2014.
- (86).Bankar G; Goodchild SJ; Howard S; Nelkenbrecher K; Waldbrook M; Dourado M; Shuart NG; Lin S; Young C; Xie Z; Khakh K; Chang E; Sojo LE; Lindgren A; Chowdhury S; Decker S; Grimwood M; Andrez JC; Dehnhardt CM; Pang J; Chang JH; Safina BS; Sutherlin DP; Johnson JP Jr.; Hackos DH; Robinette CL; Cohen CJ Selective NaV1.7 antagonists with long residence time show improved efficacy against inflammatory and neuropathic pain. Cell Rep 2018, 24 (12), 3133–3145. [DOI] [PubMed] [Google Scholar]
- (87).Roecker AJ; Egbertson M; Jones KLG; Gomez R; Kraus RL; Li Y; Koser AJ; Urban MO; Klein R; Clements M; Panigel J; Daley C; Wang J; Finger EN; Majercak J; Santarelli V; Gregan I; Cato M; Filzen T; Jovanovska A; Wang YH; Wang D; Joyce LA; Sherer EC; Peng X; Wang X; Sun H; Coleman PJ; Houghton AK; Layton ME Discovery of selective, orally bioavailable, N-linked arylsulfonamide NaV1.7 inhibitors with pain efficacy in mice. Bioorg. Med. Chem. Lett 2017, 27 (10), 2087–2093. [DOI] [PubMed] [Google Scholar]
- (88).Pero JE; Rossi MA; Lehman HDGF; Kelly MJ; Mulhearn JJ; Wolkenberg SE; Cato MJ; Clements MK; Daley CJ; Filzen T; Finger EN; Gregan Y; Henze DA; Jovanovska A; Klein R; Kraus RL; Li Y; Liang A; Majercak JM; Panigel J; Urban MO; Wang J; Wang YH; Houghton AK; Layton ME Benzoxazolinone aryl sulfonamides as potent, selective NaV1.7 inhibitors with in vivo efficacy in a preclinical pain model. Bioorg. Med. Chem. Lett 2017, 27 (12), 2683–2688. [DOI] [PubMed] [Google Scholar]
- (89).Sun S; Jia Q; Zenova AY; Wilson MS; Chowdhury S; Focken T; Li J; Decker S; Grimwood ME; Andrez J-C; Hemeon I; Sheng T; Chien-An C; White A; Hackos DH; Deng L; Bankar G; Khakh K; Chang E; Kwan R; Lin S; Nelkenbrecher K; Sellers BD; DiPasquale AG; Chang J; Pang J; Sojo L; Lindgren A; Waldbrook M; Xie Z; Young C; Johnson JP; Robinette CL; Cohen CJ; Safina BS; Sutherlin DP; Ortwine DF; Dehnhardt CM Identification of selective acyl sulfonamide-cycloalkylether inhibitors of the voltage gated sodium channel (NaV) 1.7 with potent analgesic activity. J. Med. Chem 2019, 62 (2), 908–927. [DOI] [PubMed] [Google Scholar]
- (90).Focken T; Chowdhury S; Zenova A; Grimwood ME; Chabot C; Sheng T; Hemeon I; Decker SM; Wilson M; Bichler P; Jia Q; Sun S; Young C; Lin S; Goodchild SJ; Shuart NG; Chang E; Xie Z; Li B; Khakh K; Bankar G; Waldbrook M; Kwan R; Nelkenbrecher K; Tari PK; Chahal N; Sojo L; Robinette CL; White AD; Chen C; Zhang Y; Pang J; Chang JH; Hackos DH; Johnson JP; Cohen CJ; Ortwine DF; Sutherlin DP; Dehnhardt CM; Safina BS Design of conformationally constrained acyl sulfonamide isosteres: identification of N-([1,2,4]triazolo[4,3-a ]pyridin-3-yl)methane-sulfonamides as potent and selective hNaV1.7 inhibitors for the treatment of pain. J. Med. Chem 2018, 61 (11), 4810–4831. [DOI] [PubMed] [Google Scholar]
- (91).O’Brien JE; Meisler MH Sodium channel SCN8A (NaV1.6): Properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front. Genet 2013, 4, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Sanders SJ; Campbell AJ; Cottrell JR; Moller RS; Wagner FF; Auldridge AL; Bernier RA; Catterall WA; Chung WK; Empfield JR; George AL Jr.; Hipp JF; Khwaja O; Kiskinis E; Lal D; Malhotra D; Millichap JJ; Otis TS; Petrou S; Pitt G; Schust LF; Taylor CM; Tjernagel J; Spiro JE; Bender KJ Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci 2018, 41 (7), 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Luo G; Chen L; Easton A; Newton A; Bourin C; Shields E; Mosure K; Soars MG; Knox RJ; Matchett M; Pieschl RL; Post-Munson D; Wang S; Herrington J; Graef J; Newberry K; Sivarao DV; Senapati A; Bristow L; Meanwell NA; Thompson LA; Dzierba CD Discovery of indole- and indazole-acylsulfonamides as potent and selective NaV1.7 inhibitors for the treatment of pain. J. Med. Chem 2019, 62 (2), 831–856. [DOI] [PubMed] [Google Scholar]
- (94).Ahuja S; Mukund S; Deng L; Khakh K; Chang E; Ho H; Shriver S; Young C; Lin S; Johnson JP Jr.; Wu P; Li J; Coons M; Tam C; Brillantes B; Sampang H; Mortara K; Bowman KK; Clark KR; Estevez A; Xie Z; Verschoof H; Grimwood M; Dehnhardt C; Andrez JC; Focken T; Sutherlin DP; Safina BS; Starovasnik MA; Ortwine DF; Franke Y; Cohen CJ; Hackos DH; Koth CM; Payandeh J Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350 (6267), aac5464. [DOI] [PubMed] [Google Scholar]
- (95).Wu Y; Ma H; Zhang F; Zhang C; Zou X; Cao Z Selective voltage-gated sodium channel peptide toxins from animal venom: pharmacological probes and analgesic drug development. ACS Chem. Neurosci 2018, 9 (2), 187–197. [DOI] [PubMed] [Google Scholar]
- (96).Bladen C; Hamid J; Souza IA; Zamponi GW Block of T-type calcium channels by protoxins I and II. Mol. Brain 2014, 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Schmalhofer WA; Calhoun J; Burrows R; Bailey T; Kohler MG; Weinglass AB; Kaczorowski GJ; Garcia ML; Koltzenburg M; Priest BT ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol 2008, 74 (5), 1476–1484. [DOI] [PubMed] [Google Scholar]
- (98).Gilchrist J; Das S; Van Petegem F; Bosmans F Crystallographic insights into sodium-channel modulation by the β4 subunit. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (51), E5016–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Flinspach M; Xu Q; Piekarz AD; Fellows R; Hagan R; Gibbs A; Liu Y; Neff RA; Freedman J; Eckert WA; Zhou M; Bonesteel R; Pennington MW; Eddinger KA; Yaksh TL; Hunter M; Swanson RV; Wickenden AD Insensitivity to pain induced by a potent selective closed-state Nav1.7 inhibitor. Sci. Rep 2017, 7, 39662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Deuis JR; Dekan Z; Wingerd JS; Smith JJ; Munasinghe NR; Bhola RF; Imlach WL; Herzig V; Armstrong DA; Rosengren KJ; Bosmans F; Waxman SG; Dib-Hajj SD; Escoubas P; Minett MS; Christie MJ; King GF; Alewood PF; Lewis RJ; Wood JN; Vetter I Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide Pn3a. Sci. Rep 2017, 7, 40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101) (a).Murray JK; Ligutti J; Liu D; Zou A; Poppe L; Li H; Andrews KL; Moyer BD; McDonough SI; Favreau P; Stöcklin R; Miranda LP; Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the NaV1.7 sodium channel. J. Med. Chem 2015, 58 (5), 2299–2314. [DOI] [PubMed] [Google Scholar]; (b) Murray JK; Long J; Zou A; Ligutti J; Andrews KL; Poppe L; Biswas K; Moyer BD; McDonough SI; Miranda LP Single residue substitutions that confer voltage-gated sodium ion channel subtype selectivity in the NaV1.7 inhibitory peptide GpTx-1. J. Med. Chem 2016, 59 (6), 2704–2717. [DOI] [PubMed] [Google Scholar]
- (102).Mueller A; Starobova H; Morgan M; Dekan Z; Cheneval O; Schroeder CI; Alewood PF; Deuis JR; Vetter I Anti-allodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute post-surgical pain: evidence for analgesic synergy with opioids and baclofen. Pain 2019, DOI: 10.1097/j.pain.0000000000001567. [DOI] [PubMed] [Google Scholar]
- (103).Wu B; Murray JK; Andrews KL; Sham K; Long J; Aral J; Ligutti J; Amagasu S; Liu D; Zou A; Min X; Wang Z; Ilch CP; Kornecook TJ; Lin MJ; Be X; Miranda LP; Moyer BD; Biswas K Discovery of tarantula venom-derived NaV1.7-inhibitory JzTx-V peptide 5-Br-Trp24 analogue AM-6120 with systemic block of histamine-induced pruritis. J. Med. Chem 2018, 61 (21), 9500–9512. [DOI] [PubMed] [Google Scholar]
- (104).Huang C-J; Schild L; Moczydlowski EG Use-dependent block of the voltage-gated Na+ channel by tetrodotoxin and saxitoxin: effect of pore mutations that change ionic selectivity. J. Gen. Physiol 2012, 140 (4), 435–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Tsukamoto T; Chiba Y; Wakamori M; Yamada T; Tsunogae S; Cho Y; Sakakibara R; Imazu T; Tokoro S; Satake Y; Masaatsu A; Nishikawa T, Yotsu-Yamashita M; Konoki K Differential binding of tetrodotoxin and its derivatives to voltage-sensitive sodium channel subtypes (Nav1.1 to Nav1.7). Br. J. Pharmacol 2017, 174 (21), 3881–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Satin J; Kyle JW; Chen M; Bell P; Cribbs LL; Fozzard HA; Rogart RB A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science 1992, 256 (5060), 1202–1205. [DOI] [PubMed] [Google Scholar]
- (107).Marcil J; Walczak J-S; Guindon J; Ngoc AH; Lu S; Beaulieu P Antinociceptive effects of tetrodotoxin (TTX) in rodents. Br. J. Anaesth 2006, 96 (6), 761–768. [DOI] [PubMed] [Google Scholar]
- (108).Lago J; Rodríguez LP; Blanco L; Vieites JM; Cabado AG Tetrodotoxin, an extremely potent marine neurotoxin: distribution, toxicity, origin and therapeutical uses. Mar. Drugs 2015, 13 (10), 6384–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hagen NA; Cantin L; Constant J; Haller T; Blaise G; Ong-Lam M; du Souich P; Korz W; Lapointe B Tetrodotoxin for moderate to severe cancer-related pain: a multicentre, randomized, double-blind, placebo-controlled, parallel-design trial. Pain Res. Manag 2017, 7212713. [DOI] [PMC free article] [PubMed]
- (110).Alonso E; Alfonso A; Vieytes MR; Botana LM Evaluation of toxicity equivalent factors of paralytic shellfish poisoning toxins in seven human sodium channels types by an automated high throughput electrophysiology system. Arch. Toxicol 2016, 90 (2), 479–488. [DOI] [PubMed] [Google Scholar]
- (111).Thomas-Tran R; Du Bois J Mutant cycle analysis with modified saxitoxins reveals specific interactions critical to attaining high-affinity inhibition of hNaV1.7. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (21), 5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Mulcahy J; Pajouhesh H; Delwig A; Beckley J; Shibuya GM; Du Bois J; Miljanich G 11,13-modified saxitoxins for the treatment of pain Patent WO2018183781, 2018.
- (113).Walker JR; Novick PA; Parsons WH; McGregor M; Zablocki J; Pande VS; Du Bois J Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (NaV1.7). Proc. Natl. Acad. Sci. U. S. A 2012, 109 (44), 18102–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Deng L; Shields S; Reese R; Kaminker J; Hackos D Examination of the role of endogenous opioids in the insensitivity-to-pain phenotype of an adult-onset Nav1.7 knockout mouse. Presented at the 16th World Congress on Pain, Yokohama, Japan, Sept 29, 2016; Poster PTH446. [Google Scholar]
- (115).Houghton A; Ballard J; Burgey C; John C; Kim R; Jochnowitz N; Kraus R; Klein B; Layton M A selective Nav1.7 inhibitor is antinociceptive, and potentiates opioid antinociception, in a mouse tail flick assay. Presented at the 16th World Congress on Pain, Yokohama, Japan, Sept 27, 2016; Poster PT0290. [Google Scholar]
- (116).Fouillet A; Watson JF; Piekarz AD; Huang X; Li B; Priest B; Nisenbaum E; Sher E; Ursu D Characterisation of Nav1.7 functional expression in rat dorsal root ganglia neurons by using an electrical field stimulation assay. Mol. Pain 2017, 13, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Weiss J; Pyrski M; Jacobi E; Bufe B; Willnecker V; Schick B; Zizzari P; Gossage SJ; Greer CA; Leinders-Zufall T; Woods CG; Wood JN; Zufall F Loss-of-function mutations in sodium channel NaV1.7 cause anosmia. Nature 2011, 472 (7342), 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Muroi Y; Ru F; Kollarik M; Canning BJ; Hughes SA; Walsh S; Sigg M; Carr MJ; Undem BJ Selective silencing of NaV1.7 decreases excitability and conduction in vagal sensory neurons. J. Physiol 2011, 589 (23), 5663–5676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Szabat M; Modi H; Ramracheya R; Girbinger V; Chan F; Lee JTC; Piske M; Kamal S; Carol Yang YH; Welling A; Rorsman P; Johnson JD High-content screening identifies a role for Na+ channels in insulin production. R. Soc. Open Sci 2015, 2 (12), 150306. [DOI] [PMC free article] [PubMed] [Google Scholar]