

Abstract
The dynamics of antimicrobial resistance (AMR) in developing countries are poorly understood, especially in community settings, due to a sparsity of data on AMR prevalence and genetics. We used a combination of phenotyping, genomics and antimicrobial usage data to investigate patterns of AMR amongst atypical enteropathogenic Escherichia coli (aEPEC) strains isolated from children younger than five years old in seven developing countries (four in sub-Saharan Africa and three in South Asia) over a three-year period. We detected high rates of AMR, with 65% of isolates displaying resistance to three or more drug classes. Whole-genome sequencing revealed a diversity of known genetic mechanisms for AMR that accounted for >95% of phenotypic resistance, with comparable rates amongst aEPEC strains associated with diarrhoea or asymptomatic carriage. Genetic determinants of AMR were associated with the geographic location of isolates, not E. coli lineage, and AMR genes were frequently co-located, potentially enabling the acquisition of multi-drug resistance in a single step. Comparison of AMR with antimicrobial usage data showed that the prevalence of resistance to fluoroquinolones and third-generation cephalosporins was correlated with usage, which was higher in South Asia than in Africa. This study provides much-needed insights into the frequency and mechanisms of AMR in intestinal E. coli in children living in community settings in developing countries.
Subject terms: Bacterial genomics, Bacterial infection, Antibiotics, Antimicrobial resistance
Genomic analysis of atypical enteropathogenic Escherichia coli in South Asia and sub-Saharan Africa uncovers high antibiotic resistance prevalence, which is correlated with antibiotic use.
Main
Certain pathotypes of Escherichia coli are important causes of diarrhoea in children, especially in the developing countries of sub-Saharan Africa and South Asia1. Intestinal E. coli is also an important source and reservoir of genes that encode antimicrobial resistance (AMR). One pathotype of intestinal E. coli, known as atypical enteropathogenic E. coli (aEPEC), is defined by the presence of the locus of enterocyte effacement pathogenicity island, and the absence of Shiga toxins (denoting enterohaemorrhagic E. coli) and type IV bundle-forming pili (indicating typical EPEC)2. Atypical enteropathogenic E. coli causes a variety of disease symptoms ranging from sporadic and persistent diarrhoea to asymptomatic carriage1,3–5. We recently identified distinct lineages of aEPEC, including ten common clonal groups6.
AMR has been reported in E. coli from various animal species, the environment and in hospitalized patients globally7–12. Many strains exhibit multi-drug resistance (MDR; resistance to one or more agents in at least three different antimicrobial categories13). Strains that are resistant to fluoroquinolones and/or produce extended-spectrum β-lactamases (ESBL) or carbapenemases are of particular concern14. Although several recent studies of pathogenic E. coli from countries in sub-Saharan Africa and South Asia have reported increases in ESBLs15,16, as well as increasing resistance to gentamicin17,18 and ciprofloxacin18–22, these data are mostly derived from E. coli responsible for extra-intestinal infections in hospital settings. Thus, there remain major gaps in knowledge of the global prevalence of AMR in human intestinal E. coli, particularly in developing nations where the burden of infectious diseases is highest and AMR may result in infections that are unresponsive to treatment14.
Enhancing our knowledge of AMR amongst gut-dwelling E. coli is important for two reasons: (1) E. coli is a leading cause of extra-intestinal infections and strains colonising the gastrointestinal tract of patients are the major reservoir of these infections; and (2) most AMR in E. coli is encoded on mobile genetic elements that are transferable between bacteria, thus enabling the rapid dissemination and maintenance of resistance genes between bacteria of different species23,24. Although antimicrobials are not recommended for the treatment of uncomplicated gastroenteritis, they are commonly administered to diarrhoeic children in developing countries to treat dysentery25 and prolonged diarrhoea, of which aEPEC is a major cause4,5.
Here we present AMR data for 185 aEPEC isolates collected during the Global Enteric Multicenter Study (GEMS)1,26. Using phenotypic susceptibility data and whole-genome sequence analysis, we determined the prevalence, mechanisms of resistance and potential drivers of variation in AMR profiles. These isolates, collected from healthy children living in a community setting and children with diarrhoea at seven sites in sub-Saharan Africa and South Asia, provided a unique opportunity to investigate the prevalence of AMR in intestinal bacteria that were not selected on the basis of AMR profile.
Results
Antimicrobial susceptibility profiles
Susceptibility testing of 185 aEPEC isolates (Supplementary Table 1) to 16 antimicrobials (Supplementary Table 2) revealed resistance to 14 of the drugs investigated (Fig. 1a; Supplementary Table 3), with 121 MDR isolates (65%; Fig. 1c). No resistance was detected to the ‘last-line’ drugs, amikacin (an aminoglycoside) or meropenem (a carbapenem). Only 35 isolates (19%) were susceptible to all drugs tested: 17 from cases (18%) and 18 from controls (20%). Resistance to ‘older’ antimicrobials was common, with 121 (65%) isolates resistant to ampicillin, 124 (67%) to trimethoprim, 122 (66%) to trimethoprim/sulphamethoxazole and 104 (56%) to tetracycline (Fig. 1a). Approximately half (n = 96, 52%) the isolates were resistant to three or more of these drugs. Streptomycin resistance was common (43%), although this antibiotic is not used to treat diarrhoea. Resistance to other aminoglycosides tested was rare (3%), with five isolates from India resistant to tobramycin, four of which were also resistant to gentamicin. Fluoroquinolone resistance was relatively infrequent, with 31 isolates (17%) resistant to norfloxacin and 8 (4%) resistant to both norfloxacin and ciprofloxacin. Resistance to chloramphenicol (11%) and azithromycin (7%) was also infrequent. Among the β-lactam antibiotics, ampicillin resistance was common (65%), but resistance to ceftriaxone (3%), ceftazidime (3%) and cefepime (2%) was rare (Fig. 1a,b).
Fig. 1. Prevalence of AMR-associated gene content and AMR phenotypes in 185 aEPEC isolates.

a,c AMR phenotypes of the aEPEC isolates. a, AMR profiles grouped by the drug class to which aEPEC strains were phenotypically resistant. c, Histogram illustrating the number of drug classes to which aEPEC strains were phenotypically resistant. b,d AMR-associated gene content of aEPEC strains. b, Genes detected in the genomes associated with AMR are shown to the left of the graph and are grouped by drug class. Gene that contain point mutations that result in AMR and that are not acquired through horizontal gene transfer are indicated with a cross. d, Histogram illustrating the number of classes to which aEPEC strains were detected as having AMR-associated gene content.
Genetic determinants of AMR
The genomes of the 185 aEPEC isolates were screened for known genetic determinants of AMR, including horizontally acquired genes and point mutations in chromosomal genes associated with resistance to fluoroquinolones and nitrofurantoin (Fig. 1b; Supplementary Fig. 1 and Supplementary Table 4). More than forty different acquired AMR genes were detected, along with four point mutations (two in gyrA, one in parC (both gyrA and parC are associated with quinolone resistance) and one in nfsA (associated with resistance to nitrofurantoin)). Extensive diversity of AMR genotypes was observed, with 104 distinct combinations of AMR determinants across the 185 isolates (Fig. 1b, Supplementary Fig. 1). Nevertheless, four acquired AMR genes were detected in more than half the isolates. These were alleles of blaTEM (ampicillin), strA and strB (streptomycin), and sul2 (sulphonamides). Alleles of dihydrofolate reductase (dfr) genes encoding trimethoprim resistance were detected in 132 (71%) isolates. The most common of these were dfrA1 (12%), dfrA5 (23%), dfrA7 (20%) and dfrA8 (16%).
Investigation of mobile genetic elements associated with transfer of AMR genes
As MDR was common in the bacteria studied (Fig. 1), we hypothesized that this was due to the co-transfer of groups of AMR genes via mobile elements. The pairwise co-occurrence matrix of AMR genes was sparse (Supplementary Fig. 2), with only a few clusters of genes frequently detected together in the same genome. The mean co-occurrence value across all gene pairs was 6.4 strains. Figure 2a shows AMR gene co-occurrence networks, constructed using different thresholds for co-occurrence across the aEPEC collection. The most common gene network comprised sul2, strA and strB, which co-occurred in 112 genomes (61%); the combination of sul2, strA and strB occurred with blaTEM-198 in 46 (25%) and with blaTEM-191 in 20 (11%; and Supplementary Fig. 2 and Fig. 2a) genomes. Using a minimum threshold of co-occurrence in ≥20 strains (mean plus s. d. of all co-occurrence values), we detected a large network of genes comprising sul2, strA, strB and blaTEM-198, as well as sul1, blaTEM-191, dfrA14, dfrA8, dfrA7, tet(A) and tet(B).
Fig. 2. Co-occurrence and characterization of common mobile elements of AMR-associated genes in aEPEC strains.

a, Visualization of AMR gene co-occurrence networks among GEMS aEPEC strains, using different frequency thresholds. Solid lines join genes that occur together on known mobile elements at a high frequency; dashed lines join genes that occur together on known mobile elements at a lower frequency. The mean frequency of co-occurrence in strains was 6.4, with standard deviation 13.1. b, BLAST comparison of pCERC1 and pCERC2 to representative plasmids from two GEMS aEPEC strains (400897 and 402635). Blue shading indicates regions of sequence homology. c, Gene arrangement in composite transposons previously identified in Salmonella plasmid pSRC26 and assembly graph for GEMS aEPEC strain 401596, showing how this composite transposon appears in assemblies inferred from short-read Illumina data. The assembly graph was visualized in Bandage; coloured blocks indicate BLAST hits to AMR genes as labelled.
Resolving the genetic context of AMR genes is generally not possible using short-read sequence data, because repeated sequences (such as insertion sequences) and variable plasmid copy numbers cause uncertainty in the de novo assembly graphs27,28. We therefore sought only to make broad classifications about the potential mobile elements associated with the AMR gene networks present in the aEPEC genomes, through comparison with elements that have previously been found to mobilize these combinations of genes.
The genes sul2, strA and strB frequently move together on small (approximately 6,000 base pairs (kbp)) plasmids related to pCERC229 (Fig. 2b). The combination of the pCERC2 plasmid backbone and the sul2, strA and strB sequences was present in 42 isolates, 40 of which (95%) also carried dfrA gene sequences, including dfrA1, dfrA14 and dfrA8. In some genomes, the plasmid sequences could be resolved completely, showing that the dfr gene was located on the plasmid. For example, GEMS strain 400897 carried dfrA1 gene adjacent to strB on a pCERC2-like plasmid, while GEMS strain 402635 carried dfrA14 inserted within strA as in pCERC129 (Fig. 2b).
The sul2, strA and strB genes also occur together with blaTEM genes (predominately blaTEM-198) in transposon Tn6029, which is commonly found in E. coli in a range of distinct plasmid backbones27,29,30 (Fig. 2c). Tn6029 is mobilized by the flanking copies of IS26. A third copy of IS26 is located between blaTEM and the other AMR genes, resulting in separation of the transposon into two separate contigs in short-read assemblies (Fig. 2c). We detected the presence of both Tn6029 contigs in 33 genomes, which are therefore likely to carry the complete transposon. As the flanking IS26 sequence is present in many different locations, we could not determine the insertion site of Tn6029 within the draft genomes. However, Tn6029 is frequently located within Tn1696, which includes a class I integron that carries variable AMR genes (often including dfr genes) within the cassette, and sul1 downstream of the cassette. For an example of a composite transposon structure from Salmonella plasmid pSRC2631, see Fig. 2c. In total, we identified class I integron sequences in 38 aEPEC genomes, 26 of which included both Tn6029 contigs. An example assembly graph, showing how these contigs are connected to one another in a manner consistent with previously sequenced composite transposons, is shown in Fig. 2c. Overall, we identified five different integron gene cassettes, the most prevalent of which carried dfrA7 (n = 30 genomes, including 25 with Tn6029), whereas the others carried dfrA1 (n = 4), dfrA1 and aadA (n = 1 genome, which also carried Tn6029), dfrA17 and aad5 (n = 2), and dfrA5 (n = 1).
We found two common genes that encode tetracycline resistance efflux pumps. The most prevalent was tet(A), which was found in 63 (34%) aEPEC genomes. Although tet(A) is associated with the Tn1721 transposon, the full transposon was detected in only three genomes. The tet(B) gene was detected in 50 (27%) genomes, five of which also carried tet(A). The tet(B) gene can be mobilized by Tn10, which is flanked by IS10 genes, but the complete transposon was present in only two genomes. The linkage of tet(A) or tet(B) to other AMR-related elements was not resolvable from the draft genome assemblies, however both were found in association with other common AMR genes (Fig. 2a).
Although it is not possible to resolve plasmid sequences from draft short-read assemblies or determine linkage between specific plasmid replicons and AMR genes32, our screening for markers of plasmid replicons revealed several that are often associated with large AMR plasmids (Supplementary Fig. 1d). The most prevalent amongst the aEPEC collection were FII (n = 131, 71%) and FIBA (n = 104, 56%). Notably, F plasmids are also associated with the carriage of genes for virulence determinants, such as adhesins of enteropathogenic E. coli, but our data did not permit the determination of which plasmids were associated with AMR genes versus virulence genes. An IncC plasmid replicon (also known as IncA/C) was detected in four genomes that carried blaTEM-198 but not sul2, strA or strB. It was not possible, however, to determine whether the blaTEM-198 gene was located on this plasmid.
Prediction of AMR phenotypes from genotypes
In vitro resistance was largely explained by the presence of known genetic determinants of AMR (Fig. 1, Table 1). For most drugs, the detection of resistance genes was both sensitive (>95%) and specific (>90%) in predicting AMR phenotype. The frequency of very major errors (failure to detect phenotypic resistance) exceeded the minimum acceptable threshold of 1.5% for five drugs (Table 1). These were: ampicillin (4.9%), streptomycin (2.2%), trimethoprim (2.2%), trimethoprim/sulphamethoxazole (2.2%) and tetracycline (2.2%). Major errors (predicting resistance when none is present) were also detected for these and several other antimicrobials (Table 1). The highest major error rates were observed for streptomycin (26.5%), ampicillin (7.0%), trimethoprim (5.9%), trimethoprim/sulphamethoxazole (5.4%) and tetracycline (4.3%). The sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) for each drug are shown in Supplementary Table 5. Sensitivity and NPV were greater than 90% for all drugs tested with the exception of azithromycin (85% sensitivity) and ampicillin (85% NPV). Specificity and PPV were more variable, reflecting the error rates summarized above (Table 1).
Table 1.
Comparison of phenotypic and genotypic AMR profiles of 185 aEPEC isolates
| Resistant phenotype | Resistant genotype | Very major errora | Susceptible phenotype | Susceptible genotype | Major errorb | |
|---|---|---|---|---|---|---|
| β-lactams | ||||||
| Ampicillin | 121 | 112 | 9 (4.9%) | 64 | 51 | 13 (7.0%) |
| Cefepime | 3 | 3 | 0 | 182 | 178 | 4 (2.2%) |
| Ceftriaxone | 6 | 6 | 0 | 179 | 174 | 5 (2.7%) |
| Ceftazidime | 6 | 6 | 0 | 179 | 174 | 5 (2.7%) |
| Meropenem | 0 | 0 | 0 | 185 | 185 | 0 |
| Aminoglycosides | ||||||
| Streptomycin | 80 | 76 | 4 (2.2%) | 105 | 56 | 49 (26.5%) |
| Gentamicin | 5 | 5 | 0 | 180 | 180 | 0 |
| Tobramycin | 5 | 5 | 0 | 180 | 180 | 0 |
| Amikacin | 0 | 0 | 0 | 185 | 185 | 0 |
| Folate pathway inhibitors | ||||||
| Trimethoprim | 124 | 120 | 4 (2.2%) | 61 | 50 | 11 (5.9%) |
| Trimethoprim/sulphamethoxazolec | 122 | 118 | 4 (2.2%) | 63 | 53 | 10 (5.4%) |
| Nitrofurantoin | ||||||
| Nitrofurantoin | 0 | 0 | 0 | 185 | 182 | 3 (1.6%) |
| Chloramphenicol | ||||||
| Chloramphenicol | 21 | 19 | 2 (1.1%) | 164 | 160 | 4 (2.2%) |
| Macrolides | ||||||
| Azithromycin | 13 | 11 | 2 (1.1%) | 172 | 166 | 6 (3.2%) |
| Tetracyclines | ||||||
| Tetracycline | 104 | 100 | 4 (2.2%) | 81 | 73 | 8 (4.3%) |
| Fluoroquinolones | ||||||
| Ciprofloxacind | 8 | 8 | 0 | 177 | 173 | 4 (2.2%) |
| Norfloxacin | 31 | 30 | 1 (0.5%) | 154 | 149 | 5 (2.7%) |
aVery major errors (resistant isolate genotyped as susceptible) at frequencies >1.5% are shown in bold.
bMajor errors (susceptible isolate genotyped as resistant) at frequencies >3% are shown in bold.
cWhen two genes are required for resistance, both were required for genotypic resistance: strA and strB for streptomycin, and a dfr gene plus a sul gene for resistance to trimethoprim/sulphamethoxazole.
dAt least one point mutation in gyrA and a second in gyrA, gyrB, parC or parE.
Potential sources of variation in AMR profiles
Given the diversity of AMR profiles in our aEPEC strains, we determined whether the distribution of AMR genes was associated with disease status, phylogenetic lineage or the geographic location from which each strain was isolated. First, we compared the frequency of AMR phenotypes and genotypes among aEPEC isolated from diarrhoea cases and asymptomatic controls. Only data from confirmed cases (n = 94) and controls (n = 88) were used for this analysis. For each drug class, neither AMR phenotype nor AMR predicted from genotype, were statistically different between cases and controls (Supplementary Table 6). The frequencies of individual AMR genes were also similar in isolates obtained from cases and from controls (Fig. 3a). As AMR determinants were equally distributed among cases and controls, all isolates were pooled for further analysis of lineage and region.
Fig. 3. AMR gene content is explained by region of isolation, not disease status or clonal group.

a, Frequencies of AMR-associated genes in aEPEC by case or control status. Genes encoding AMR are shown to the right of the graph and are grouped by drug class. b,c, Discriminant analysis of principal components based on known genetic determinants of AMR. The graphs display the discriminant functions (DF) that best discriminate isolates into region of isolation (b, n = 185) or clonal group (c, n = 137). Data points are coloured coded according to their demographic group (see legend) and the genetic determinants most correlated with the DFs are labelled on the DF axes. Eigenvalues, corresponding to the ratio of the variance between groups over the variance within groups for each principal component in the discriminant function, are displayed in the insets. d, Frequency of AMR genetic determinants that differed between Asia, East Africa and West Africa. Genes that contain point mutations that result in AMR and that are not acquired through horizontal gene transfer are indicated with a cross.
Discriminant analysis of principal components33 on the binary matrix of AMR genetic determinants (Supplementary Table 4; Fig. 3b,c) revealed that the first 20 principal components accounted for >93% of variation in AMR profiles and were retained for discriminant analysis by phylogenetic lineage (clonal groups as defined previously6; Supplementary Fig. 1) or by geographic origin (East Africa, West Africa and Asia; Fig. 3 and Supplementary Fig. 3).
Variation in AMR was not associated with clonal group, apart from CG378 which was characterized by the absence of the most common AMR genes: blaTEM variants, sul2, tet(A) and tet(B), and the presence of the uncommon catA gene, detected in 7 of 9 CG378 isolates compared to 15 of 176 non-CG378 (P < 10-4, Fisher’s exact test, two-tailed). Variation in AMR determinants was associated with the region of origin (Fig. 3b and Supplementary Fig. 3b). Discriminant function 1 (DF1) separated Asian from African isolates and was associated with gyrA single nucleotide polymorphisms; DF2 separated East from West African isolates and was associated with dfrA8, dfrA5, tet(A) and sul2. Figure 3d shows the distribution of the dfrA alleles across the GEMS sites. For example, dfrA1 predominated at Asian sites and dfrA8 was most common at West African sites, whereas dfrA14 and dhfr7 were common in Mozambique and Kenya, respectively. Further, tet(A) was more common at West and East African sites than in Asia. These genetic differences were reflected in AMR phenotypes, as resistance to ciprofloxacin (n = 8, 12%) and third-generation cephalosporins (ceftazidime and ceftriaxone, both n = 6; 9%) was identified only in strains from Asia, while resistance to tetracycline was more common in African (East Africa, n = 44, 60%; West Africa, n = 33, 72%) than in Asian isolates, n = 27, 41 %; P < 0.05, Fisher’s exact test, two-tailed; Fig. 4a).
Fig. 4. AMR phenotypes by region and antimicrobial use at study sites.

a, AMR phenotypes of GEMS isolates, stratified by region of isolation. b,c, Percentage of antimicrobials prescribed to patients with watery diarrhoea (b) or dysentery (c) at each of the seven study sites.
Differences in local antimicrobial drug usage
The broad patterns of antimicrobial use in the treatment of diarrhoea across the GEMS study sites showed that trimethoprim/sulphamethoxazole and penicillins were used more frequently at African sites, whereas macrolides (azithromycin) and fluoroquinolones (in particular ciprofloxacin) were used more frequently in Asia (Fig. 4b,c). These patterns of antimicrobial use showed some association with AMR phenotypes, insofar as we observed higher levels of both usage and resistance for azithromycin and fluoroquinolones at Asian sites, and to trimethoprim at East African sites (Fig. 4). We could not formally test these associations, however, due to the small numbers of observations at some study sites and minor variations in usage of most drugs between the sites. We therefore investigated the associations between usage and resistance for the two drugs that showed substantial usage (>10%) at three or more study sites: ciprofloxacin and trimethoprim. Across the seven sites, ciprofloxacin usage was significantly associated with the prevalence of substitutions in the quinolone resistance-determining regions (QRDRs), gyrA and parC (Coefficient of determination (R2) = 0.87, P = 0.002; Fig. 5). By contrast, trimethoprim usage was not associated with the prevalence of horizontally acquired dfr genes that confer resistance to the drug (R2 = 0.04, P > 0.5).
Fig. 5. Relationship between the use of ciprofloxacin at GEMS study sites and ciprofloxacin resistance in aEPEC.
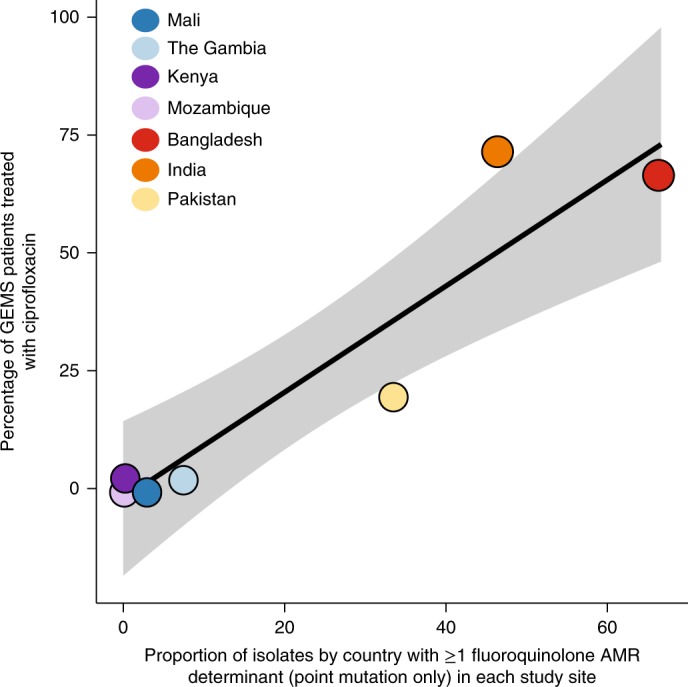
Linear regression (mean ± 95% confidence interval shaded in grey) of the proportion of aEPEC containing genetic determinants of ciprofloxacin resistance (from point mutations only) versus the proportion of watery diarrhoea and dysentery cases treated with ciprofloxacin at GEMS study sites.
Discussion
AMR and usage data reported here were collected at seven study sites in Asia and Africa, using the same protocols thus enabling comparisons between the sites26,34. No national AMR surveillance data are available from these countries; and AMR data on E. coli in these countries pertain mostly to isolates causing extra-intestinal infections, and to a limited number of drugs (mainly third-generation cephalosporins and fluoroquinolones)14,19,21,22,35. Furthermore, publicly available background data on antimicrobial usage at the seven study sites are limited. For example, the IMS Health MIDAS database includes usage data for India, Pakistan and Bangladesh reported in aggregate without detailed methods of data collection or interpretation; and no data for Mozambique, The Gambia, Kenya and Mali.
Nearly half of all isolates were resistant to penicillins, trimethoprim and tetracyclines. The rates of resistance to these drugs were generally lower amongst Asian isolates (21–62%) than in Africa isolates (59–84%); whereas resistance to newer drugs such as ceftriaxone, fluoroquinolones and azithromycin were detected at the Asian sites (Fig. 4a). These patterns were broadly consistent with the antimicrobial usage data at the corresponding study sites, which showed that ciprofloxacin and azithromycin were commonly used to treat diarrhoea in Asia, whereas trimethoprim-sulphamethoxazole was the mainstay of treatment in Africa (Fig. 4b,c). The high frequencies of ciprofloxacin resistance at the Asian sites are similar to rates previously reported among clinical cases of intestinal E. coli (including multiple pathotypes) in these countries17,19,21,22,36. We detected ESBL-producing isolates at the Asian, but not the African, study sites. Much higher levels have been reported from extra-intestinal E. coli infections (including bacteraemia) in hospitals in Asia and Africa16,19,35, we speculate that this may reflect selection due to use of third-generation cephalosporin at higher rates in hospitals than in the community, and/or the dissemination of the ESBL-producing extra-intestinal E. coli lineages, such as ST131.
In agreement with our data relating to AMR phenotypes, we found that the genetic determinants of resistance were similar in bacteria isolated from diarrhoeal cases and asymptomatic controls (Fig. 3a, Supplementary Table 6) and were not associated with the clonal lineage of the strain, but associated with the geographic region where the bacteria were isolated (Fig. 3b,c; Supplementary Fig. 3). These findings are consistent with the hypothesis that differences in the frequencies of AMR-encoding genes in different regions reflect selection due to differences in antimicrobial exposure (Fig. 4b,c). In strong support of this explanation, mutations in the QRDRs of gyrA and parC were significantly associated with the frequency of ciprofloxacin use across the seven study sites (Fig. 5). The same pattern of point mutations in the QRDRs of chromosomal genes has been observed in other Enterobacteriaceae associated with South Asia, including Salmonella enterica serovar Typhi37,38 and Shigella sonnei39.
The situation was more complex for horizontally transferred AMR genes associated with resistance to older drugs. Although individual dfr alleles were distributed differently across sites (Fig. 3d) and contributed to orthogonal components of the regional discriminant function (Fig. 3b), the overall prevalence of dfr genes was relatively high (50–90%) at each site and not significantly associated with use of trimethoprim for diarrhoea (Figs. 3d and 4b,c). Similarly, genes encoding resistance to ampicillin, streptomycin and tetracycline were common at sites where these drugs were seldom or never used for the treatment of diarrhoea. We also found evidence of several common elements mediating AMR to these older drugs, including small plasmids and class I integrons (Fig. 2). This could be due to: (1) a lack of fitness cost associated with these resistances, resulting in maintenance of the genes in the E. coli population after drug usage declines8,10,30,40,41; (2) co-selection for resistance to multiple drugs whose associated genes are present on the same mobile elements42,43; and/or (3) selection due to drug exposure unrelated to the treatment of diarrhoea. We could not distinguish between hypotheses using our data, although exposure to antimicrobials from other sources is quite likely. For example, although we found that trimethoprim, alone or combined with sulphamethoxazole, was used less frequently in India than alternative agents for the treatment of diarrhoea, other studies have reported frequent use of trimethoprim in hospitals, community settings36 and agriculture in India; there is also evidence of its presence in the environment, particularly surface waters44. Future studies would benefit from additional data on clinical and agricultural antimicrobial exposures, and assays for selected antimicrobials agents in urine and the environment.
We were also unable to determine the precise location of most AMR genes and associated mobile elements from our short-read sequence data. Further experiments such as conjugation or long-read sequencing38,45,46 could resolve this in future. It is notable, however, that the pCERC-like plasmids (which often carried resistance to streptomycin, sulphonamides and trimethoprim) are common in E. coli, possibly because their small size (~6 kb) imposes a low fitness cost29,47. It is also notable that these and many of the other AMR genes we detected are also associated with composite transposons that can integrate into the bacterial chromosome, where they are maintained at lower fitness cost than large resistance-encoding plasmids27,31,38. Many of the mobile elements we detected have been reported widely in E. coli and other Enterobacteriaceae from human intestinal and extra-intestinal samples and animal samples29,38,48–50. Atypical enteropathogenic E. coli has multiple reservoirs and our collection included extensive phylogenetic diversity within the E. coli population (90 unique lineages6); and we found no differences in AMR between isolates cultured from cases and asymptomatic colonization. Thus, although the isolates were not collected for the specific purpose of AMR surveillance, they may be broadly representative of intestinal E. coli in these settings.
Our data fill important information gaps concerning the prevalence of AMR among intestinal E. coli in children from developing countries in Africa and Asia. In particular, our study showed that resistance to multiple ‘older’ drugs (ampicillin, tetracycline, streptomycin and trimethoprim-sulphamethoxazole) was common at all sites, and that resistance to ‘newer’ antimicrobials, such as fluoroquinolones and azithromycin, has emerged only in Asian sites where these drugs are used in the management of diarrhoea. Resistance to older drugs was also common at these sites, such that only Asian isolates were resistant to seven or eight categories of antimicrobials, indicating that changing patterns of antimicrobial use leads to an accumulation of resistance determinants rather than their replacement.
Methods
aEPEC isolates and corresponding whole-genome sequences
A total of 185 confirmed aEPEC isolates from children aged 0–5 years at GEMS sites located in The Gambia, Mali, Kenya, Mozambique, Bangladesh, India and Pakistan34 were included in the analysis1,6,26,34. Their collection, selection for sequencing, whole-genome sequencing and phylogenomic analysis have been described previously6. Briefly, the isolates were mostly from fecal samples in which aEPEC alone (or with Giardia lamblia) was the only pathogen detected, where a pure culture could be obtained. All such isolates from diarrhoeal cases were sequenced (n = 94); controls matched for age, sex and study site were also included (n = 88). Three isolates were from children whose case/control status was uncertain. Faecal samples were collected at the study sites before antimicrobial treatment, although previous exposure to antimicrobials from other sources cannot be ruled out. Control children were also not receiving any antimicrobial treatment.
Whole-genome sequences were generated for all 185 aEPEC isolates at the Wellcome Trust Sanger Institute using the Illumina HiSeq platform (100 bp paired-end reads) and assembled using Velvet, as described previously6,51. Details of the individual isolates, accession numbers for the corresponding genome sequence reads and assemblies (deposited collectively under BioProject ERP001141), and associated metadata are provided in Supplementary Table 1.
Phenotypic characterization of AMR profiles
Antimicrobial susceptibility testing to 16 antimicrobials was performed using the VITEK2 (bioMérieux) system or an agar-dilution method. A summary of the drugs, testing methods, and the minimum inhibitory concentration (MIC) breakpoints used to determine susceptible, intermediate or resistant status for each drug is shown in Supplementary Table 2. The controls used were three reference S. enterica isolates with known resistance profiles that were kindly provided by the Microbiological Diagnostic Unit Public Health Laboratory. These strains had the following profiles: (1) susceptible to all drugs tested; (2) resistant to ampicillin, streptomycin, tetracycline, chloramphenicol, sulphathiazole, trimethoprim, kanamycin, spectinomycin and gentamicin; and (3) resistant to streptomycinmod, tetracycline, kanamycin, nalidixic acid and ciprofloxacin.
For VITEK2 assays, pure isolates were streaked on MacConkey agar plates and incubated at 37 °C overnight. Isolates were then subcultured onto horse blood agar (HBA) plates for fresh culture and incubated overnight at 37 °C. One to three colonies were selected from each HBA plate and suspended in saline to an absorbancy of ~0.5 MacFarlane Units before being subjected to VITEK2 analysis. The raw MIC data from the VITEK2 assays are shown in Supplementary Table 7.
Susceptibility to streptomycin, chloramphenicol, azithromycin and tetracycline were determined using an agar-dilution method. Bacterial suspensions were prepared as described above. To each of 32 stainless steel wells, 450 µl nutrient broth containing 0.05% agar was added, followed by 50 µl bacterial suspension. Each Mueller Hinton agar antimicrobial-containing plate for susceptibility testing and two control Mueller Hinton and MacConkey agar plates were inoculated using a 32-pin replicator. Each pin delivered 2 µl to the plate such that the final number of colony-forming units in each sample was ~104. Plates were incubated overnight at 37 °C and inspected the next day. Growth on an antimicrobial-containing Mueller Hinton plate was recorded as phenotypically resistant to the drug, whereas no growth was recorded as susceptible.
The European Committee on Antimicrobial Susceptibility Testing (EUCAST) MIC breakpoints (version 6) were used where available52. Differences exist between the EUCAST and Clinical and Laboratory Standards Institute (CLSI) guidelines in terms of MIC breakpoints and the drugs to be tested. As tetracycline does not have defined MIC breakpoints under the EUCAST scheme, we used the CLSI MIC breakpoint. Streptomycin and azithromycin do not have established MIC breakpoints under either scheme53,54. Previous research proposed a breakpoint of 16 µg ml-1 for streptomycin in E. coli54. Little information is available on the MIC distribution of azithromycin for E. coli. A breakpoint of 16 µg ml-1 has been proposed for S. enterica based on a study in which the majority of isolates displayed MICs of 4–8 µg ml-1(ref. 53). For the present study, the breakpoint MIC for each of these drugs was set at the conservative value of 16 µg ml-1. The antimicrobial susceptibility data for each isolate are shown in Supplementary Table 3.
Detection of AMR genes
An SRST2-formatted version of the ARG-ANNOT AMR gene database55 was downloaded from https://github.com/katholt/srst2. All sequence read sets were screened against the database using SRST2 to detect the presence of known acquired resistance genes in each genome56. The β-lactamase genes, ampC1, ampC2 and ampH, were excluded from analysis, as in E. coli they are core genes that normally do not confer antibiotic resistance. The results were transformed into a binary table in R to indicate presence/absence of acquired resistance gene alleles (Supplementary Table 4).
Detection of single nucleotide polymorphisms conferring resistance to fluoroquinolones and nitrofurantoin
Chromosomal mutations, known to be associated with resistance to fluoroquinolones in E. coli, were extracted from the genome-wide single nucleotide polymorphism calls obtained previously based on mapping the reads to the E. coli strain 12009 O103:H2 reference genome6. These included specific mutations in the quinolone resistance-determining regions of gyrA, gyrB, parC and parE57, and non-synonymous substitutions in nfsA (residues 11–15) that confer resistance to nitrofurantoin58,59.
Statistical analysis of AMR phenotype prediction from genotype
All statistical analyses were performed using the R Stats Package version 3.4.0. The ability of genotypes to predict drug susceptibility phenotypes was assessed by comparing antimicrobial susceptibility phenotypes (S, I and R) with the presence of known AMR-associated genes and mutations. Errors in predicting antimicrobial susceptibility were characterized as very major (calling a resistant isolate susceptible) or major (calling a susceptible isolate resistant)60,61. The currently accepted standards for very major error and major error rates are <1.5% and <3%, respectively60. Here, very major errors were said to have occurred when an isolate was phenotypically resistant, but no known resistance genes or mutations were detected, while major errors were made when an isolate carried known resistance determinant(s) but was phenotypically susceptible. Statistical analysis to determine sensitivity, specificity, PPV and NPV were calculated in the epiR package (v0.9-93)62 for R, using the epi.stats function with 0.95 confidence intervals for each antimicrobial tested.
Genomic reconstruction of demographic groups by discriminant analysis of principal components
The matrix of AMR genetic determinants (specifically the data in Supplementary Table 4 with only AMR genetic determinants used and isolates with two point mutations in gyrA treated as present to generate a binary matrix) was used as the input for discriminant analysis of principal components that was implemented in the adegenet package (v2.1.0) in R33. The first 20 principal components, which together explained >93% of the variance in AMR gene content, were retained for discriminant analysis to explore the ability of principal components to discriminate between groups of strains defined by geographic region of origin (West Africa: The Gambia and Mali; East Africa: Kenya and Mozambique; and Asia: Bangladesh, India and Pakistan) or clonal group membership. The two principal components contributing the most to discriminant analysis were plotted and labelled with the genetic determinants whose variation contributed the most to those components. The posterior group membership probabilities for each discriminant function were also plotted.
Construction of co-occurrence network
A pairwise co-occurrence matrix of acquired AMR genes was constructed by transforming the binary AMR gene content matrix in R. The co-occurrence relationships were visualised between all pairs of genes using the pheatmap package (v1.0.8) in R (https://CRAN.R-project.org/package=pheatmap) (Supplementary Fig. 2). Networks of co-occurring genes, in which nodes represent genes and edges represent a frequency of co-occurrence exceeding a given threshold (set to ≥20, ≥33, ≥46, ≥100 genomes), were visualized in R using the igraph package (v1.1.2)63 in R.
Plasmid replicon screening
An SRST2-formatted version of the PlasmidFinder database64 was downloaded from https://github.com/katholt/srst2 for the detection of 80 known plasmid replicon marker sequences. All sequence read sets were screened against the database using SRST2 to detect the presence of these replicons in each genome. The results were transformed into a binary table in R to indicate presence/absence.
Visualization of AMR and plasmid genotypes against a core gene tree
A subtree representing the relationships between the 185 GEMS aEPEC isolates was extracted from the full core phylogeny we published previously6 by pruning all other tips using R packages ape (v5.1)65 and geiger (v2.0.6)66. The presence of acquired AMR genes, mutations and plasmid replicons was plotted as a heatmap against the phylogeny using the plotTree function for R (https://github.com/katholt/plotTree).
Investigation of mechanisms of AMR gene mobilisation
Common AMR-associated genes that were shown to co-occur, specifically blaTEM-198, sul1, sul2, strA, strB, multiple dfrA alleles, tet(A) and tet(B), were further investigated in the aEPEC genome assemblies to determine whether they were carried on the same mobile elements. The aEPEC genome assemblies generated previously6 were interrogated with BLAST (v2.3.30), using as queries the AMR genes and the sequences of the plasmids pCERC1 (accession JN012467) and pCERC2 (accession KX291024), and the transposons Tn6029 (accession GQ150541)27, Tn1721 (accession X61367)67 and Tn10 (accession AF223162)68. For example, if the pCERC2 backbone and AMR genes were all detected on a single contig in the genome assembly, we inferred that these genes were moving together on a pCERC2-related plasmid. Two representative aEPEC isolates that were identified as harbouring a pCERC2-like plasmid backbone with different dfr gene insertions (strains 402635 and 400879) were selected as representatives for further analysis. These genomes were re-assembled with Unicycler (v0.2.0)69, annotated using Prokka (v1.12)70 and compared to the reference sequences for pCERC1 and pCERC2 using BLAST. The comparisons were then explored using Artemis Comparison Tool71 and plotted with genoplotR (v0.8.7) in R72.
Atypical enteropathogenic E. coli isolates were inferred to be likely carriers of Tn6029 or related transposons if the entire region of repC to strB was detected by BLAST in a single contig and a blaTEM variant (predominately blaTEM-198) was also detected in the genome. (Note that it is not possible for the complete transposon sequence to be assembled from short reads, as blaTEM is separated from the rest of the transposon by repeat copies of IS26 which cause a break in the assembly graph). A representative strain matching this pattern (401596) was re-assembled with Unicycler69 and the connectivity of Tn6029 genes in the resulting assembly graphs were visualised using Bandage (v0.8.1)73.
The distributions of class I integrons with different cassette regions were explored by extracting the DNA sequences spanning from int1 to sul1 genes using BLAST and MUMmer (v3.23)74. Different dfrA alleles were identified within the resulting sequences via BLAST searches of the ARG-ANNOT database55. Representatives of each distinct class I integron sequence (defined by the dfrA gene carried) were re-assembled with Unicycler69 and submitted to the Repository of Antibiotic-resistance Cassettes (RAC) website75 for detailed annotation.
Antimicrobial usage data and correlation with resistance
Data on the use of antimicrobials at each of the seven GEMS sites were collected as part of the original GEMS protocol1,26. These data included details of the antimicrobials prescribed to all cases presenting with watery diarrhoea or dysentery at the study clinics and were documented by a member of the GEMS clinical team26. Two of the recorded drugs were excluded from the current analysis: pivmecillinam, because it was not used at any discernible level, and metronidazole, which is active against obligate anaerobic protozoa and bacteria only76 and therefore does not pertain to E. coli, which is intrinsically resistant to this agent. The frequency of prescriptions for each drug at each site was visualized in R, using the ggplot2 package (v2.2.1)33.
The relationship between frequencies of ciprofloxacin and trimethoprim usage and associated genetic determinants was investigated via linear regression modelling in R using the lm function. For ciprofloxacin, the genetic determinants were either one or more quinolone resistance-associated point mutations in gyrA (point mutations in parC only occurred when gyrA mutations were also present), or the presence of the plasmid-borne genes qepA or qnrS. Genetic evidence of trimethoprim/sulphamethoxazole resistance required the combination of at least one dfrA gene together with sul1 or sul2. The data were visualized in R using the ggplot2 package33.
Data availability
Accession numbers for the short-read data and associated metadata are listed in Supplementary Table 1. The phenotypic resistance data are provided in Supplementary Tables 3 and 7 and the genotypic resistance profiles are shown in Supplementary Table 4.
Supplementary information
Supplementary Note, Supplementary Figures 1–3, Supplementary Tables 2, 5 and 6.
Details of 185 isolates of atypical enteropathogenic E. coli analysed in this study.
Antimicrobial susceptibility profiles of all isolates, inferred from VITEK and agar plate dilution methods.
Presence/absence of point mutations, acquired AMR genes and plasmid replicon genes in all isolates.
MIC data from VITEK2 for 185 isolates of atypical enteropathogenic E. coli (aEPEC).
Acknowledgements
This work was supported by the NHMRC of Australia (Project Grants 1043830 to K.E.H., and 1009296 and 1067428 to R.M.R-B.; Fellowship 1061409 to K.E.H.) and the Bill & Melinda Gates Foundation (Grant 38874 to M.M.L.). We thank G. Dougan and the sequencing teams at the Wellcome Trust Sanger Institute for sequencing the aEPEC isolate collection.
Author contributions
D.J.I., K.E.H. and R.M.R.-B. contributed to the design of the study and data interpretation. D.J.I. performed the experimental analyses. D.J.I. performed the majority of bioinformatics analyses with input from K.E.H. M.M.L. and K.L.K. oversaw the original GEMS study and provided bacterial isolates and associated metadata. All authors contributed to the writing of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally to this work: Kathryn E. Holt, Roy M. Robins-Browne.
Electronic supplementary material
Supplementary information is available for this paper at 10.1038/s41564-018-0217-4.
References
- 1.Kotloff KL, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Robins-Browne RM, et al. Are Escherichia coli pathotypes still relevant in the era of whole-genome sequencing? . Front. Cell. Infect. Microb. 2016;6:5222–5229. doi: 10.3389/fcimb.2016.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afset JE, Bevanger L, Romundstad P, Bergh K. Association of atypical enteropathogenic Escherichia coli (EPEC) with prolonged diarrhoea. J. Med. Microbiol. 2004;53:1137–1144. doi: 10.1099/jmm.0.45719-0. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen RN, Taylor LS, Tauschek M, Robins-Browne RM. Atypical enteropathogenic Escherichia coli infection and prolonged diarrhea in children. Emerg. Infect. Dis. 2006;12:597–603. doi: 10.3201/eid1204.051112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vieira MA, et al. Atypical enteropathogenic Escherichia coli as aetiologic agents of sporadic and outbreak-associated diarrhoea in Brazil. J. Med. Microbiol. 2016;65:998–1006. doi: 10.1099/jmm.0.000313. [DOI] [PubMed] [Google Scholar]
- 6.Ingle DJ, et al. Evolution of atypical enteropathogenic E. coli by repeated acquisition of LEE pathogenicity island variants. Nat. Microbiol. 2016;1:15010. doi: 10.1038/nmicrobiol.2015.10. [DOI] [PubMed] [Google Scholar]
- 7.Davis MA, et al. Genotypic-phenotypic discrepancies between antibiotic resistance characteristics of Escherichia coli isolates from calves in management settings with high and low antibiotic use. Appl. Environ. Microbiol. 2011;77:3293–3299. doi: 10.1128/AEM.02588-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glenn LM, et al. Analysis of antimicrobial resistance genes detected in multiple-drug-resistant Escherichia coli isolates from broiler chicken carcasses. Microb. Drug Resist. 2012;18:453–463. doi: 10.1089/mdr.2011.0224. [DOI] [PubMed] [Google Scholar]
- 9.Medina A, et al. Phenotypic and genotypic characterization of antimicrobial resistance in enterohemorrhagic Escherichia coli and atypical enteropathogenic E. coli strains from ruminants. J. Vet. Diagn. Invest. 2011;23:91–95. doi: 10.1177/104063871102300114. [DOI] [PubMed] [Google Scholar]
- 10.Ramírez Castillo FY, et al. Presence of multi-drug resistant pathogenic Escherichia coli in the San Pedro River located in the State of Aguascalientes, Mexico. Front. Microbiol. 2013;4:1–16. doi: 10.3389/fmicb.2013.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaletsky IC, Souza TB, Aranda KR, Okeke IN. Genetic elements associated with antimicrobial resistance in enteropathogenic Escherichia coli (EPEC) from Brazil. BMC Microbiol. 2010;10:25. doi: 10.1186/1471-2180-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stoesser N, et al. Evolutionary history of the global emergence of the Escherichia coli epidemic clone ST131. mBio. 2016;7:e02162-15 . doi: 10.1128/mBio.02162-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magiorakos AP, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2014;18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 14.Antibiotic Resistance: Global Report on Surveillance 2014 (WHO, 2014).
- 15.Lim C, et al. Epidemiology and burden of multidrug- resistant bacterial infection in a developing country. eLife. 2016;5:e18082. doi: 10.7554/eLife.18082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musicha P, et al. Trends in antimicrobial resistance in bloodstream infection isolates at a large urban hospital in Malawi (1998–2016): a surveillance study. Lancet Infect. Dis. 2017;17:1042–1052. doi: 10.1016/S1473-3099(17)30394-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathai E, et al. Antimicrobial resistance surveillance among commensal Escherichia coli in rural and urban areas in Southern India. Trop. Med. Int. Health. 2008;13:41–45. doi: 10.1111/j.1365-3156.2007.01969.x. [DOI] [PubMed] [Google Scholar]
- 18.Avasthi TS, et al. Genome of multidrug-resistant uropathogenic Escherichia coli strain NA114 from India. J. Bacteriol. 2011;193:4272–4273. doi: 10.1128/JB.05413-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta S, Kapur S, Padmavathi DV. Comparative prevalence of antimicrobial resistance in community-acquired urinary tract infection cases from representative states of Northern and Southern India. J. Clin. Diagn. Res. 2014;8:9–12. doi: 10.7860/JCDR/2014/9349.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shah M, et al. Prevalence, seasonal variation, and antibiotic resistance pattern of enteric bacterial pathogens among hospitalized diarrheic children in suburban regions of central Kenya. Trop. Med. Health. 2016;44:39. doi: 10.1186/s41182-016-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Begum YA, et al. Resistance pattern and molecular characterization of enterotoxigenic Escherichia coli (ETEC) strains isolated in Bangladesh. PLoS ONE. 2016;11:e0157415-11. doi: 10.1371/journal.pone.0157415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalil U, et al. Phenotypic and genotypic characterization of enteroaggregative Escherichia coli isolates from pediatric population in Pakistan. APMIS. 2016;124:872–880. doi: 10.1111/apm.12577. [DOI] [PubMed] [Google Scholar]
- 23.van Schaik W. The human gut resistome. Phil. Trans. R. Soc. B. 2015;370:20140087. doi: 10.1098/rstb.2014.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 25.Traa BS, Walker CLF, Munos M, Black RE. Antibiotics for the treatment of dysentery in children. Int. J. Epidemiol. 2010;39:i70–i74. doi: 10.1093/ije/dyq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotloff KL, et al. The Global Enteric Multicenter Study (GEMS) of diarrheal disease in infants and young children in developing countries: epidemiologic and clinical methods of the case/control study. Clin. Infect. Dis. 2012;55:S232–S245. doi: 10.1093/cid/cis753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reid CJ, Chowdhury PR, Djordjevic SPT. Tn6026 and Tn6029 are found in complex resistance regions mobilised by diverse plasmids and chromosomal islands in multiple antibiotic resistant Enterobacteriaceae. Plasmid. 2015;80:127–137. doi: 10.1016/j.plasmid.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Cain AK, Hall RM. Evolution of a multiple antibiotic resistance region in IncHI1 plasmids: reshaping resistance regions in situ. J. Antimicrob. Chemother. 2012;67:2848–2853. doi: 10.1093/jac/dks317. [DOI] [PubMed] [Google Scholar]
- 29.Anantham S, Hall RM. pCERC1, a small, globally disseminated plasmid carrying the dfrA14 cassette in the strA gene of the sul2-strA-strB gene cluster. Microb. Drug Resist. 2012;18:364–371. doi: 10.1089/mdr.2012.0008. [DOI] [PubMed] [Google Scholar]
- 30.Partridge SR, Hall RM. Evolution of transposons containing blaTEM genes. Antimicrob. Agents Chemother. 2005;49:1267–1268. doi: 10.1128/AAC.49.3.1267-1268.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cain AK, Liu X, Djordjevic SP, Hall RM. Transposons related to Tn1696 in IncHI2 plasmids in multiply antibiotic resistant Salmonella enterica serovar Typhimurium from Australian animals. Microb. Drug Resist. 2010;16:197–202. doi: 10.1089/mdr.2010.0042. [DOI] [PubMed] [Google Scholar]
- 32.Arredondo-Alonso S, Willems RJ, van Schaik W, Schürch AC. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microb. Genom. 2017;3:e000128. doi: 10.1099/mgen.0.000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically. BMC Genet. 2010;11:1–15. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Panchalingam S, et al. Diagnostic microbiologic methods in the GEMS-1 case/control study. Clin. Infect. Dis. 2012;55:S294–S302. doi: 10.1093/cid/cis754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sangare SA, et al. Very high prevalence of extended-spectrum beta-lactamase-producing Enterobacteriaceae in bacteriemic patients hospitalized in teaching hospitals in Bamako, Mali. PLoS ONE. 2017;12:e0172652-11. doi: 10.1371/journal.pone.0172652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laxminarayan R, Chaudhury RR. Antibiotic resistance in India: drivers and opportunities for action. PLoS Med. 2016;13:e1001974-7. doi: 10.1371/journal.pmed.1001974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pham Thanh D, et al. A novel ciprofloxacin-resistant subclade of H58 Salmonella Typhi is associated with fluoroquinolone treatment failure. . eLife. 2016;5:e14003. doi: 10.7554/eLife.14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong VK, et al. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat. Genet. 2015;47:632–639. doi: 10.1038/ng.3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.The HC, et al. South Asia as a reservoir for the global spread of ciprofloxacin-resistant Shigella sonnei: a cross-sectional study. . PLoS Med. 2016;13:e1002055-12. doi: 10.1371/journal.pmed.1002055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennett PM. Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol. 2008;153:S347–S357. doi: 10.1038/sj.bjp.0707607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pallecchi L, et al. Population structure and resistance genes in antibiotic-resistant bacteria from a remote community with minimal antibiotic exposure. Antimicrob. Agents Chemother. 2007;51:1179–1184. doi: 10.1128/AAC.01101-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 2010;8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 43.Carattoli A. Plasmids and the spread of resistance. Int. J. Med. Microbiol. 2013;303:298–304. doi: 10.1016/j.ijmm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Shimizu A, et al. Ubiquitous occurrence of sulfonamides in tropical Asian waters. Sci. Total Environ. 2013;452-453:108–115. doi: 10.1016/j.scitotenv.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 45.Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017;3:e000132. doi: 10.1099/mgen.0.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sheppard AE, et al. Nested Russian doll-like genetic mobility drives rapid dissemination of the carbapenem resistance gene blaKPC. Antimicrob. Agents Chemother. 2016;60:3767–3778. doi: 10.1128/AAC.00464-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Touchon M, et al. Antibiotic resistance plasmids spread among natural isolates of Escherichia coli in spite of CRISPR elements. Microbiology. 2012;158:2997–3004. doi: 10.1099/mic.0.060814-0. [DOI] [PubMed] [Google Scholar]
- 48.Holt KE, et al. Multidrug-resistant Salmonella enterica serovar Paratyphi A harbors IncHI1 plasmids similar to those found in serovar Typhi. J. Bacteriol. 2007;189:4257–4264. doi: 10.1128/JB.00232-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen CM, et al. High diversity of antimicrobial resistance genes, class 1 integrons, and genotypes of multidrug-resistant Escherichia coli in beef carcasses. Microb. Drug Resist. 2017;23:915–924. doi: 10.1089/mdr.2016.0223. [DOI] [PubMed] [Google Scholar]
- 50.Oliveira-Pinto C, et al. Occurrence and characterization of class 1 integrons in Escherichia coli from healthy individuals and those with urinary infection. J. Med. Microbiol. 2017;66:577–583. doi: 10.1099/jmm.0.000468. [DOI] [PubMed] [Google Scholar]
- 51.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kahlmeter G. European harmonization of MIC breakpoints for antimicrobial susceptibility testing of bacteria. J. Antimicrob. Chemother. 2003;52:145–148. doi: 10.1093/jac/dkg312. [DOI] [PubMed] [Google Scholar]
- 53.Sjolund-Karlsson M, et al. Antimicrobial susceptibility to azithromycin among Salmonella enterica isolates from the United States. Antimicrob. Agents Chemother. 2011;55:3985–3989. doi: 10.1128/AAC.00590-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sunde M, Norström M. The genetic background for streptomycin resistance in Escherichia coli influences the distribution of MICs. J. Antimicrob. Chemother. 2005;56:87–90. doi: 10.1093/jac/dki150. [DOI] [PubMed] [Google Scholar]
- 55.Gupta SK, et al. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2013;58:212–220. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Inouye M, et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6:90. doi: 10.1186/s13073-014-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eaves DJ, et al. Prevalence of mutations within the quinolone resistance-determining region of gyrA, gyrB, parC, and parE and association with antibiotic resistance in quinolone-resistant Salmonella enterica. Antimicrob. Agents Chemother. 2004;48:4012–4015. doi: 10.1128/AAC.48.10.4012-4015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whiteway J, et al. Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J. Bacteriol. 1998;180:5529–5539. doi: 10.1128/jb.180.21.5529-5539.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xavier BB, et al. Complete genome sequences of nitrofurantoin-sensitive and -resistant Escherichia coli ST540 and ST2747 strains. Genome Announc. 2014;2:e00239. doi: 10.1128/genomeA.00239-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jorgensen JH, Ferraro MJ. Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin. Infect. Dis. 2009;49:1749–1755. doi: 10.1086/647952. [DOI] [PubMed] [Google Scholar]
- 61.Köser CU, et al. Routine Use of Microbial Whole Genome Sequencing in Diagnostic and Public Health Microbiology. PLoS Pathog. 2012;8:e1002824-9. doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stevenson, M. et al. epiR: Tools for the Analysis of Epidemiological Data (R Foundation for Statistical Computing, 2017); https://CRAN.R-project.org/package=epiR
- 63.Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal, Complex Systems. 2006;1695:1–9. [Google Scholar]
- 64.Carattoli A, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014;58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 66.Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W. GEIGER: investigating evolutionary radiations. Bioinformatics. 2007;24:129–131. doi: 10.1093/bioinformatics/btm538. [DOI] [PubMed] [Google Scholar]
- 67.Allmeier H, Cresnar B, Greck M, Schmitt R. Complete nucleotide sequence of Tn1721: gene organization and a novel gene product with features of a chemotaxis protein. Gene. 1992;111:11–20. doi: 10.1016/0378-1119(92)90597-i. [DOI] [PubMed] [Google Scholar]
- 68.Lawley TD, Burland V, Taylor DE. Analysis of the complete nucleotide sequence of the tetracycline-resistance transposon Tn10. Plasmid. 2000;43:235–239. doi: 10.1006/plas.1999.1458. [DOI] [PubMed] [Google Scholar]
- 69.Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017;13:e1005595-22. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. [Google Scholar]
- 71.Carver T, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guy L, Kultima JR, Andersson SGE. genoPlotR: comparative gene and genome visualization in R. Bioinformatics. 2010;26:2334–2335. doi: 10.1093/bioinformatics/btq413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 2015;31:3550–3552. doi: 10.1093/bioinformatics/btv383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol.5, R2 (2004). [DOI] [PMC free article] [PubMed]
- 75.Partridge SR, Tsafnat G, Coiera E, Iredell JR. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol. Rev. 2009;33:757–784. doi: 10.1111/j.1574-6976.2009.00175.x. [DOI] [PubMed] [Google Scholar]
- 76.Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob. Agents Chemother. 1999;43:1533–1541. doi: 10.1128/aac.43.7.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Note, Supplementary Figures 1–3, Supplementary Tables 2, 5 and 6.
Details of 185 isolates of atypical enteropathogenic E. coli analysed in this study.
Antimicrobial susceptibility profiles of all isolates, inferred from VITEK and agar plate dilution methods.
Presence/absence of point mutations, acquired AMR genes and plasmid replicon genes in all isolates.
MIC data from VITEK2 for 185 isolates of atypical enteropathogenic E. coli (aEPEC).
Data Availability Statement
Accession numbers for the short-read data and associated metadata are listed in Supplementary Table 1. The phenotypic resistance data are provided in Supplementary Tables 3 and 7 and the genotypic resistance profiles are shown in Supplementary Table 4.