

Abstract
Background
Patients with Brugada syndrome (BrS) are known to have arrhythmic events after alcohol drinking and are recommended to avoid its excessive intake. Mechanisms underlying the alcohol‐induced cardiac events are however unknown. This study aimed to test the hypothesis whether activity of alcohol‐metabolizing enzymes determines fatal arrhythmic events after drinking alcohol.
Methods
A total of 198 Japanese patients with BrS were enrolled in this study. These patients were classified into symptomatic (n = 90) and asymptomatic (n = 108) groups. The former was divided into an alcohol‐related group (syncope after alcohol drinking, n = 16) and an alcohol‐unrelated group (n = 74). Polymerase chain reaction was performed to determine genetic variants of genes encoding alcohol dehydrogenase 1B (ADH1B) and aldehyde dehydrogenase 2 (ALDH2).
Results
The genotype distribution for ALDH2 was not significantly different between symptomatic and asymptomatic groups and between alcohol‐related and alcohol‐unrelated groups. The genotype distribution for ADH1B was not significantly different between symptomatic and asymptomatic groups, but the genotype ADH1B His/His was significantly more prevalent in the alcohol‐related group than in the alcohol‐unrelated group (81.3% vs 50%, P = .023). In multivariate logistic regression analysis, the genotype of ADH1B His/His was independently associated with syncope after alcohol drinking (odds ratio, 5.746; 95% confidence interval, 1.580‐28.421; P = .007).
Conclusions
Arrhythmic events after alcohol drinking was associated with enhanced activity of alcohol‐metabolizing enzyme ADH1B in our cohort of BrS. Therefore, the lifestyle change to avoid the excessive alcohol intake deserves attention.
Keywords: alcohol, alcohol‐metabolizing enzymes, Brugada syndrome, polymorphism, syncope
1. INTRODUCTION
Brugada syndrome (BrS) is diagnosed as a coved‐type ST‐segment elevation in the right precordial leads.1 Patients with BrS develop symptoms including ventricular fibrillation (VF), aborted sudden death, syncope, or nocturnal agonal respiration. Previous reports2, 3 and consensus guideline1 have shown that alcohol drinking is associated with occurrence of syncope in BrS. Alcohol taken into a body is metabolized by two enzymes, namely alcohol dehydrogenase 1B (ADH1B) and aldehyde dehydrogenase 2 (ALDH2). Alcohol is metabolized to acetaldehyde by ADH1B and sequentially converted to acetate by ALDH2 (Figure 1). The activity of the two enzymes individually varies depending on genetic variants of the enzymes.
Figure 1.
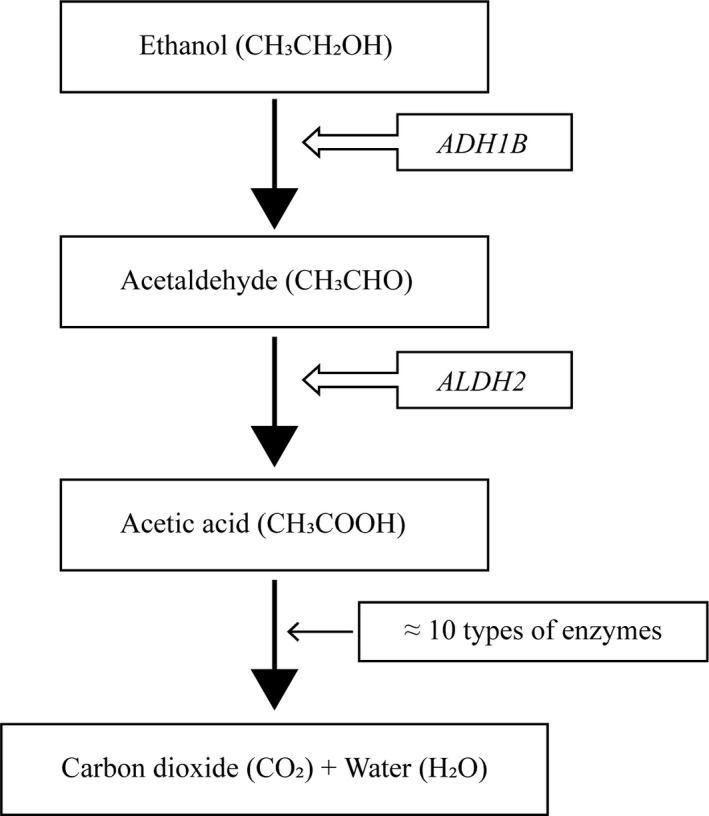
A scheme showing the process of alcohol taken into a body metabolized by two enzymes
The prevalence of BrS is relatively higher in the Southeast Asian countries including Japan compared to the Western countries.1, 4, 5 This geographical prevalence of BrS is similar to that of loss‐of‐function type variants in alcohol‐metabolizing enzymes: ADH1B and ALDH2, which are more common in Asians than in Caucasians.6, 7, 8 More recently, Watanabe et al9 reported that habitual excessive alcohol drinkers among Japanese BrS patients with implantable cardioverter‐defibrillator (ICD) implantation have more fatal arrhythmic events. Therefore, we hypothesized that genetic polymorphisms of the alcohol‐metabolizing enzymes would be associated with arrhythmic events in BrS, thus contributing to the pathophysiology and arrhythmogenesis of the disease. The purpose of this study was to test this hypothesis.
2. METHODS
2.1. Study population (Figure 2)
The study subjects consisted of 198 BrS Japanese patients (176 men with a mean age of 44.9 ± 15.8 years and 22 women with a mean age of 47.4 ± 17.6 years). The diagnosis of BrS was made on the basis of electrocardiogram (ECG) findings according to the expert consensus statement.1 In 198 patients, 90 patients (45.5%: 85 men, mean 43.1 ± 15.2 years; 5 women, mean 39.8 ± 19.1 years) had history of syncope and/or aborted sudden cardiac death caused by ventricular tachycardia or fibrillation (symptomatic group). According to medical records, after other causes of syncope like neutrally mediated syncope had been excluded, all the symptomatic patients were thought to have “arrhythmic syncope.” When fatal arrhythmias were recorded by ECG, they were either transient VT/VF or cardiopulmonary arrest due to continued VF. In contrast, remaining 108 patients (54.5%: 91 men, mean 46.5 ± 16.3 years; 17 women, mean 49.7 ± 17.2 years) did not have any episodes of syncope (asymptomatic group) (Figure 2).
Figure 2.
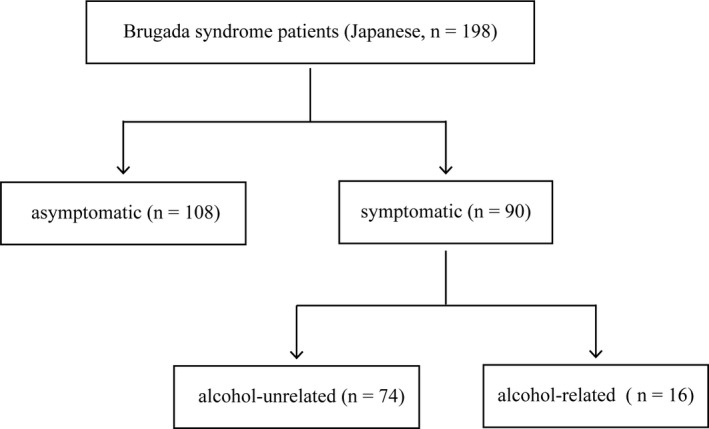
Subgroup of study population
The patients of the symptomatic group were further classified into two groups: 16 patients (all men, mean 46.8 ± 13.3 years) who suffered from syncope after drinking alcohol (alcohol‐related group) and 74 patients (69 men, mean 42.3 ± 15.5 years; 5 women, mean 39.8 ± 19.1 years) who had no history of syncope after drinking alcohol (alcohol‐unrelated group) (Figure 2). Syncope after alcohol drinking is defined as follows: syncope occurred within 6 hours after alcohol drinking, and the amount of drinking alcohol was not specified. Whether or not family history of sudden death was present was evaluated in all patients enrolled in this study. Family history was defined as at least one first‐degree family member had died suddenly with a type 1 Brugada ECG pattern during their ante mortem or before the age of 45 years in the absence of known heart disease. The study was approved by the ethics committee of Shiga University of Medical Science (approval number: 27‐161), and written informed consent was obtained from each patient.
2.2. Determination of ADH1B and ALDH2 genotypes
Genomic DNA was extracted from blood lymphocyte as previously reported.10 Subsequently, genotyping was performed using TaqMan single‐nucleotide polymorphism (SNP) genotyping assay (Applied Biosystems) in a real‐time PCR system (StepOnePlus™; Applied Biosystems) for ADH1B (His48Arg; rs1229984) and ALDH2 (Glu504Lys; rs671). Amplification conditions were 95°C for 20 seconds, 40 cycles of 95°C for 3 seconds, and 60°C for 20 seconds. The genotyping was performed with a blind method to the identification of the study subjects.
2.3. Statistical analysis
The baseline clinical data were expressed as the mean ± standard deviation for continuous variables. A comparison between two groups was performed with Student's t test or the nonparametric Mann‐Whitney's U test, as appropriate. For discrete variables, the data were expressed as counts and percentages. Categorical variables were compared with chi‐squared test. Allele frequencies determined by direct gene counting and genotype distributions were checked for departure from Hardy‐Weinberg equilibrium using the Pearson's chi‐squared test. A two‐tailed value of P < .05 was considered to be statistically significant. A multiple logistic regression analysis was performed to determine the risk factors of syncope after drinking alcohol. Independent variables were included on the basis of theoretical and clinical grounds. Data analyses were conducted using JMP® 10 (SAS Institute Inc.).
3. RESULTS
3.1. Characteristics of the study patients
Table 1A shows the comparison of the clinical characteristics between symptomatic and asymptomatic groups. There was no significant difference in age, family history of sudden death, the frequency of spontaneous type1 Brugada ECG pattern between the two groups. The prevalence of male gender was significantly higher in the symptomatic than in the asymptomatic group.
Table 1.
Comparison of clinical characteristics
| A. Symptomatic group vs asymptomatic group | |||
|---|---|---|---|
| Variables | Symptomatic group (n = 90) | Asymptomatic group (n = 108) | P‐value |
| Age (years) | 42.6 ± 15.3 | 47.0 ± 16.4 | .077 |
| Male gender, n (%) | 85 (94.4) | 91 (84.3) | .023 |
| FH of SD, n (%) | 9 (10) | 10 (9.3) | .860 |
| Spontaneous of type 1 ECG, n (%) | 50 (55.6) | 46 (42.6) | .069 |
| B. Alcohol‐related group vs alcohol‐unrelated group | |||
|---|---|---|---|
| Variables | Alcohol‐related group (n = 16) | Alcohol‐unrelated group (n = 74) | P‐value |
| Age (years) | 46.8 ± 13.3 | 42.1 ± 15.7 | .275 |
| Male gender, n (%) | 16 (100) | 69 (93.2) | .285 |
| FH of SD, n (%) | 0 (0) | 9 (12.2) | .141 |
| Spontaneous of type 1 ECG, n (%) | 10 (62.5) | 40 (54.1) | .538 |
| Arrhythmia Syncope | 16 (100) | 74 (100) | — |
| Aborted sudden death | 0 (0) | 2 (2.7) | .506 |
| Documented VF | 7 (43.75) | 34 (45.95) | .973 |
| VF induced by EPS | 2 (12.5) | 15 (20.27) | .472 |
| ICD | 8 (50) | 46 (62.2) | .368 |
| Appropriate shock deliveries | 2/8 (25) | 3/46 (6.5) | .096 |
Data are expressed as mean ± standard deviation or n (%).
Abbreviations: FH, family history; EPS, electrophysiologic study; ICD, implantable cardioverter defibrillator; SD, sudden death; VF, ventricular fibrillation.
Table 1B shows the comparison of the clinical characteristics between alcohol‐related and unrelated groups. There was no significant difference in age, gender, family history of sudden death, the frequency of spontaneous type1 Brugada ECG pattern, aborted sudden death, documented VT/VF, VF induced by electrophysiology study, and ICD implantation between the two groups. Based on ICD follow‐up, although statistically not significant, VF recurrence rate was higher in the alcohol‐related group (25% vs 6.5%, P = .096). Indeed, one of our cases experienced appropriate ICD therapy for four episodes of recurrent VF, which were all associated with alcohol drinking. In this group, average time interval between the start of drinking and cardiac events was 3.5 hours.
3.2. Genotype distribution of ADH1B and ALDH2
Table 2A shows distribution of genotypes for ADH1B and ALDH2 in the symptomatic and the asymptomatic groups. A vast majority of the study patients had the variant ADH1B His genotype (ADH1B Arg/His or ADH1B His/His) and the variant ALDH2 Glu genotype (ALDH2 Glu/Glu or ALDH2 Glu/Lys) in both groups (ADH1B His: 92.3% vs 95.4%, P = .355; ALDH2 Glu: 91.1% vs 93.5%, P = .524). The genotype distributions of the two genes did not deviate from the Hardy‐Weinberg equilibrium: ADH1B gene, χ 2 = 0.223, P = .64 for the symptomatic group and χ 2 = 1.029, P = .31 for the asymptomatic group; ALDH2 genes, χ 2 = 3.324, P = .07 for the symptomatic group and χ 2 = 0.017, P = .90 for the asymptomatic group. There was no significant overall difference in genotype distribution for ADH1B and ALDH2 in the two groups (ADH1B: χ 2 = 1.013, P = .564, Figure 3A; ALDH2: χ 2 = 2.065, P = .379, Figure 3B).
Table 2.
Frequency distribution of genotypes for ADH1B and ALDH2
| A. Symptomatic group vs asymptomatic group | |||
|---|---|---|---|
| Variables | Symptomatic group (n = 90) | Asymptomatic group (n = 108) | P‐value |
| ADH1B gene | |||
| ADH1B Arg/Arg, n (%) | 7 (7.8) | 5 (4.6) | .355 |
| ADH1B Arg/His, n (%) | 33 (36.7) | 45 (41.7) | .473 |
| ADH1B His/His, n (%) | 50 (55.6) | 58 (53.7) | .794 |
| ALDH2 gene | |||
| ALDH2 Glu/Glu, n (%) | 56 (62.2) | 59 (54.6) | .281 |
| ALDH2 Glu/Lys, n (%) | 26 (28.9) | 42 (38.9) | .140 |
| ALDH2 Lys/Lys, n (%) | 8 (8.9) | 7 (6.5) | .524 |
| B. Alcohol‐related group vs alcohol‐unrelated group | |||
|---|---|---|---|
| Variables | Alcohol‐related group (n = 16) | Alcohol‐unrelated group (n = 74) | P‐value |
| ADH1B gene | |||
| ADH1B Arg/Arg or ADH1B Arg/His, n (%) | 3 (18.8) | 37 (50.0) | .023 |
| ADH1B His/His, n (%) | 13 (81.2) | 37 (50.0) | |
| ALDH2 gene | |||
| ALDH2 Glu/Glu, n (%) | 10 (62.5) | 46 (62.2) | .980 |
| ALDH2 Glu/Lys, n (%) | 5 (31.3) | 21 (28.4) | .818 |
| ALDH2 Lys/Lys, n (%) | 1 (6.3) | 7 (9.5) | .683 |
Data are expressed as number (%).
Figure 3.
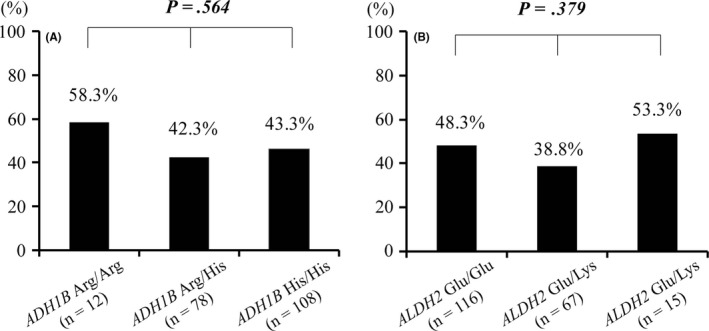
Comparison of frequency of symptomatic patients to all patients enrolled in this study according to genotypes of ADH1B and ALDH2. A, There was no difference in the frequency of the symptomatic patients by ADH1B genotype. B, There was no difference in the frequency of the symptomatic patients by ALDH2 genotype. Abbreviations: ADH1B, alcohol dehydrogenase subunit; ALDH2, aldehyde dehydrogenase 2
Table 2B shows distribution of genotypes for ADH1B and ALDH2 in the alcohol‐related syncope group and the alcohol‐unrelated group. A vast majority of the study patients had the variant ADH1B His genotype and the ALDH2 Glu genotype in both0groups (ADH1B His: 93.8% vs 91.9%, P = .801; ALDH2 Glu: 93.8% vs 90.6%, P = .683). The genotype distribution of the two genes did not deviate from the Hardy‐Weinberg equilibrium: ADH1B gene, χ 2 = 2.939, P = .09 for the alcohol‐related group and χ 2 = 0.019, P = .89 for the alcohol‐unrelated group; ALDH2 gene, χ 2 = 0.118, P = .73 for the alcohol‐related group and χ 2 = 3.394, P = .07 for the alcohol‐unrelated group. There was no significant overall difference in genotype distribution for ADH1B and ALDH2 between the two groups (ADH1B: χ 2 = 5.470, P = .065, Figure 4A; ALDH2: χ 2 = 0.190, P = .301, Figure 4B).
Figure 4.
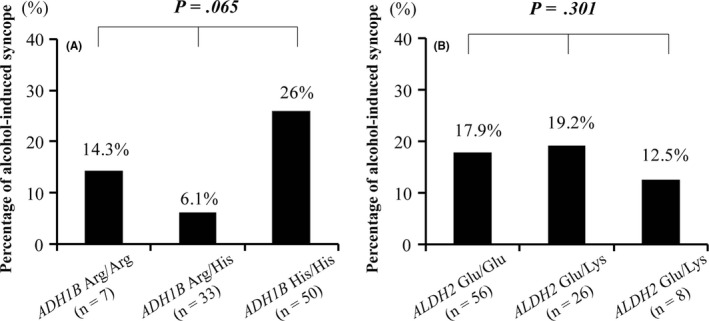
Comparison of frequency of patients with alcohol‐related syncope to symptomatic patients in this study according to genotypes of ADH1B and ALDH2. A, There was no difference in the frequency of the patients with alcohol‐related syncope by ADH1B genotype. B, There was no difference in the frequency of the patients with alcohol‐related syncope by ALDH2 genotype. Abbreviations: ADH1B, alcohol dehydrogenase subunit; ALDH2, aldehyde dehydrogenase 2
The prevalence of ADH1B His/His was significantly higher in the alcohol‐related group than in the alcohol‐unrelated group (81.3% vs 50%, P = .023). When compared to those carrying ADH1B Arg/His or ADH1B Arg/Arg (Figure 5), frequency of alcoholic‐related syncope was significantly higher in ADH1B His/His (26% vs 7.5%; odds ratio, 4.330; 95% confidence interval, 1.140–16.475; P = .023).
Figure 5.
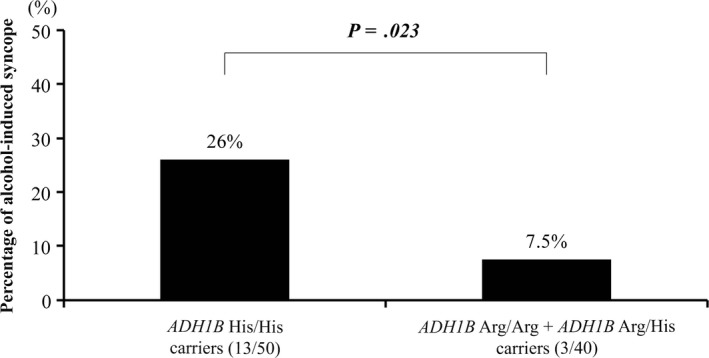
Comparison of frequency of patients with alcohol‐related syncope to patients with symptomatic patients in this study according to genotypes of ADH1B His/His The frequency of ADH1B His/His was significantly higher than genotypes of ADH1B Arg/His and ADH1B Arg/Arg. Abbreviation: ADH1B, alcohol dehydrogenase subunit
Multivariate regression logistic analysis revealed that the genotype of ADH1B His/His was significantly and independently associated with syncope after drinking alcohol, after adjustment for age, ADH1B His/His genotypes, spontaneous type1 Brugada ECG pattern (ADH1B His/His gene, odds ratio, 5.746; 95% confidence interval, 1.580‐28.421; P = .007; Table 3). Because patients in the alcohol‐related group were all male and did not have family history of sudden death, both gender and family history were not included as the confounding factors.
Table 3.
Multivariate logistic regression analysis for syncope after drinking alcohol
| Variable | OR | P > |z| | 95%CI |
|---|---|---|---|
| Age | 0.109 | .141 | 0.004‐2.046 |
| ADH1B His/His | 5.746 | .007 | 1.580‐28.421 |
| Spontaneous of type1 ECG | 0.579 | .357 | 0.169‐1.840 |
Abbreviations: CI, confidence interval; OR, odds ratio.
4. DISCUSSION
The present study found: (a) ~20% of fatal arrhythmic events were related to alcohol drinking in our cohort of symptomatic BrS, (b) the distribution for ALDH2 genotypes was not significantly different between symptomatic and asymptomatic groups or between alcohol‐related and alcohol‐unrelated syncope groups, (c) the distribution for ADH1B genotypes was not significantly different between symptomatic and asymptomatic groups, but the genotype ADH1B His/His was significantly more frequent in the alcohol‐related group compared to the alcohol‐unrelated group, and (d) the genotype ADH1B His/His was independently associated with syncope after drinking alcohol (Table 3).
Alcohol (ethanol) exerts various physiological responses (Figure 1).11 It is metabolized by two enzymes, namely ADH1B and ALDH2. The former metabolizes ethanol to acetaldehyde in the cytosol; the latter acetaldehyde to acetic acid in the mitochondria. The ADH1B gene is located on chromosome 4, and its single‐nucleotide polymorphism (SNP: rs1229984) replaces arginine to histidine at residue 47 (Arg47His). The ALDH2 on chromosome 12 has also a SNP (rs671) which substitutes glutamate with lysine at position 504 (Glu504Lys). These two SNPs exert loss‐of‐function effects and are more common in East Asians than in Caucasians and play essential roles in alcohol‐induced physical reactions.11, 12, 13
The variant ADH1B His/His is a super‐active subunit of ADH1B and possesses ~40 times more efficient ethanol metabolizing activity compared to ADH1B Arg/Arg,11, 14 and is seen in 60% of Japanese.15, 16 In contrast, 57% of Japanese are homozygous for the ALDH2 Glu/Glu which has normal enzymatic activity. Homozygotes ALDH2 Lys/Lys occur in 5%–10% and possess only ~4% of normal enzymatic activity.17
Among Japanese BrS patients implanted with ICD, Watanabe et al9 reported that habitual excessive alcohol drinkers showed significantly more frequent recurrence of fatal arrhythmic events. Because carriers with ADH1B His/His have stronger ability of alcohol metabolism and higher level of alcohol consumption,11, 18 their report may suggest the same results as our findings in that the ADH1B His/His genotype was significantly more frequent among the patients suffering alcohol‐related fatal arrhythmic events. Our results also support the recommendation of 2014 Expert Consensus Statement1 to avoid the excessive alcohol intake to prevent the arrhythmic risk as a lifestyle change.
The super‐active ADH1B variant results in an earlier and higher peak of blood acetaldehyde level. Acetaldehyde then stimulates the sympathetic nervous tone through the release of catecholamine from sympathetic nervous terminals.19, 20 The patients carrying this SNP might have an earlier and larger increase in sympathetic tone than those with other SNP carriers. On cessation of ethanol drinking, the larger sympathetic stimulation declines with disappearance of acetaldehyde, and the following stronger parasympathetic activation could be possible to occur. The strongest alcohol metabolizers may have a large meal concurrently with a large amount of alcohol consumption, producing a full stomach condition. It is well known that, a full stomach test can unmask type1 ECG by through the increased vagal activity.21
It is well known that autonomic function influences the severity of BrS.22, 23, 24, 25 Both experimental and clinical studies have shown that ST‐segment elevation, a marker of increased risk of arrhythmic events, is mitigated by the administration of beta‐adrenergic agonists and enhanced by parasympathetic agonists.26, 27 In this connection, Makimoto et al28 reported that 34 out of 93 patients with BrS showed significant ST segment elevation in V1 to V3 leads during the recovery phase after exercise tolerance test, indicating the involvement of parasympathetic reactivation. In these 34 patients, clinical prognosis was significantly worse and SCN5A mutations were more frequently identified.
There are several reports on excessive alcohol consumption and increased risk of sudden cardiac death.29, 30 In an experimental study, excessive alcohol directly exerts proarrhythmic effects by inhibiting sodium and calcium channels.31, 32
4.1. Limitations
Although it is known that alcohol‐induced flushing33, 34 and frequency of habitual drinking are useful information to learn if a subject tolerates alcohol intake, the medical record was not available. The ethnicity‐dependent difference of frequency of alcohol‐related syncope in BrS remains unknown because our study population was limited to the Japanese.
5. CONCLUSION
Arrhythmic events after alcohol drinking in BrS patients were associated with enhanced activity of alcohol‐metabolizing enzyme caused by ADH1B His/His, supporting the recommendation of 2014 Expert Consensus Statement in that the excessive alcohol intake should be avoided.1
CONFLICTS OF INTEREST
Authors declare no conflict of interests for this article.
ACKNOWLEDGEMENTS
We thank Ms Kazu Toyooka and Ms Arisa Ikeda for excellent technical assistance. This work was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (M.H., 15H04818, S.O., 15K09689d, H.H., 17K09495) and the Japan Ministry of Health, Labor and Welfare (M.H., H24‐033, H26‐040, H27‐032).
Wu Q, Hayashi H, Hira D, et al. Genetic variants of alcohol‐metabolizing enzymes in Brugada syndrome: Insights into syncope after drinking alcohol. J Arrhythmia. 2019;35:752–759. 10.1002/joa3.12227
REFERENCES
- 1. Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Executive Summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. J Arrhythm. 2014;30:29–47. [DOI] [PubMed] [Google Scholar]
- 2. Ohkubo K, Nakai T, Watanabe I. Alcohol‐induced ventricular fibrillation in a case of Brugada syndrome. Europace. 2013;15(7):1058. [DOI] [PubMed] [Google Scholar]
- 3. Achaiah A, Andrews N. Intoxication with alcohol: an underestimated trigger of Brugada syndrome? JRSM Open. 2016;7:2054270416640153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2:429–40. [DOI] [PubMed] [Google Scholar]
- 5. Holst AG, Jensen HK, Eschen O, Henriksen FL, Kanters J, Bundgaard H, et al. Low disease prevalence and inappropriate implantable cardioverter defibrillator shock rate in Brugada syndrome: a nationwide study. Europace. 2012;14:1025–9. [DOI] [PubMed] [Google Scholar]
- 6. Li H, Mukherjee N, Soundararajan U, Tárnok Z, Barta C, Khaliq S, et al. Geographically separate increases in the frequency of the derived ADH1B*47His allele in eastern and western Asia. Am J Hum Genet. 2007;81:842–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H, Borinskaya S, Yoshimura K, et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann Hum Genet. 2009;73:335–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Osier MV, Pakstis AJ, Soodyall H, Comas D, Goldman D, Odunsi A, et al. A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am J Hum Genet. 2002;71:84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watanabe T, Kabutoyai T, Watanabe H, et al. Alcohol drinking was associated with arrhythmic events in Brugada syndrome. Shinzo. 2017;49(2):103–9. [Google Scholar]
- 10. Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, et al. A novel SCN5A gain‐of‐function mutation M1875T associated with familial atrial fibrillation. J Am Coll Cardiol. 2008;52:1326–34. [DOI] [PubMed] [Google Scholar]
- 11. Goodman LS, Gilman A, Brunton LL. Goodman & gilman's manual of pharmacology and therapeutics. New York: McGraw‐Hill Medical; 2008. [Google Scholar]
- 12. Macgregor S, Lind PA, Bucholz KK, Hansell NK, Madden P, Richter MM, et al. Associations of ADH and ALDH2 gene variation with self report alcohol reactions, consumption and dependence: an integrated analysis. Hum Mol Genet. 2009;18(3):580–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Husemoen L, Fenger M, Friedrich N, Tolstrup JS, Beenfeldt Fredriksen S, Linneberg A. The association of ADH and ALDH gene variants with alcohol drinking habits and cardiovascular disease risk factors. Alcohol Clin Exp Res. 2008;32(11):1984–91. [DOI] [PubMed] [Google Scholar]
- 14. Yoshida A, Hsu LC, Yasunami M. Genetics of human alcohol‐metabolizing enzymes. Prog Nucleic Acid Res Mol Biol. 1991;40:255–87. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki Y, Fujisawa M, Ando F, Niino N, Ohsawa I, Shimokata H, et al. Alcohol dehydrogenase 2 variant is associated with cerebral infarction and lacunae. Neurology. 2004;63(9):1711–3. [DOI] [PubMed] [Google Scholar]
- 16. Higuchi S, Matsushita S, Murayama M, Takagi S, Hayashida M. Alcohol and aldehyde dehydrogenase polymorphisms and the risk for alcoholism. Am J Psychiatry. 1995;152(8):1219–21. [DOI] [PubMed] [Google Scholar]
- 17. Yokoyama A, Muramatsu T, Ohmori T, et al. Alcohol‐related cancers and aldehyde dehydrogenase‐2 in Japanese alcoholics. Carcinogenesis. 1998;19:1383–7. [DOI] [PubMed] [Google Scholar]
- 18. Crous‐Bou M, Rennert G, Cuadras D, Salazar R, Cordero D, Saltz Rennert H, et al. Polymorphisms in alcohol metabolism genes ADH1B and ALDH2, alcohol consumption and colorectal cancer. PLoS ONE. 2013;8(11):e80158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanhoutte PM, Verbeuren TJ, Webb RC. Local modulation of adrenergic neuroeffector interaction in the blood vessel well. Physiol Rev. 1981;61(1):151–247. [DOI] [PubMed] [Google Scholar]
- 20. Chiba S, Tsukada M. Possible mechanism of acetaldehyde‐induced noradrenaline release from sympathetic nerve terminals in isolated blood vessels. Br J Pharmacol. 1988;95(1):177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ikeda T, Abe A, Yusu S, Nakamura K, Ishiguro H, Mera H, et al. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17:602–7. [DOI] [PubMed] [Google Scholar]
- 22. Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–70. [DOI] [PubMed] [Google Scholar]
- 23. Kasanuki H, Ohnishi S, Ohtuka M, Matsuda N, Nirei T, Isogai R, et al. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation. 1997;95:2277–85. [DOI] [PubMed] [Google Scholar]
- 24. Nakazawa K, Sakurai T, Takagi A, Kishi R, Osada K, Nanke T, et al. Autonomic imbalance as a property of symptomatic Brugada syndrome. Circ J. 2003;67:511–4. [DOI] [PubMed] [Google Scholar]
- 25. Wichter T, Matheja P, Eckardt L, Kies P, Schäfers K, Schulze‐Bahr E, et al. Cardiac autonomic dysfunction in Brugada syndrome. Circulation. 2002;105:702–6. [DOI] [PubMed] [Google Scholar]
- 26. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST‐segment elevation. Circulation. 1999;100:1660–6. [DOI] [PubMed] [Google Scholar]
- 27. Mizumaki K, Fujiki A, Tsuneda T, Sakabe M, Nishida K, Sugao M, et al. Vagal activity modulates spontaneous augmentation of ST elevation in the daily life of patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:667–73. [DOI] [PubMed] [Google Scholar]
- 28. Makimoto H, Nakagawa E, Takaki H, Yamada Y, Okamura H, Noda T, et al. Augmented ST‐segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome. J Am Coll Cardiol. 2010;56(19):1576–84. [DOI] [PubMed] [Google Scholar]
- 29. Selb Semerl J, Selb K. Coffee and alcohol consumption as triggering factors for sudden cardiac death: case‐crossover study. Croat Med J. 2004;45:775–80. [PubMed] [Google Scholar]
- 30. Wannamethee G, Shaper AG. Alcohol and sudden cardiac death. Br Heart J. 1992;68:443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klein G, Gardiwal A, Schaefer A, Panning B, Breitmeier D. Effect of ethanol on cardiac single sodium channel gating. Forensic Sci Int. 2007;171:131–5. [DOI] [PubMed] [Google Scholar]
- 32. Habuchi Y, Furukawa T, Tanaka H, Lu LL, Morikawa J, Yoshimura M. Ethanol inhibition of Ca2+ and Na+ currents in the guinea‐pig heart. Eur. J. Pharmacol. 1995;292:143–9. [DOI] [PubMed] [Google Scholar]
- 33. Peng GS, Chen YC, Wang MF, Lai CL, Yin SJ. ALDH2*2 but not ADH1B*2 is a causative variant gene allele for Asian alcohol flushing after a low‐dose challenge: correlation of the pharmacokinetic and pharmacodynamic findings. Pharmacogenet Genomics. 2014;24:607–17. [DOI] [PubMed] [Google Scholar]
- 34. Yokoyama A, Mizukami T, Yokoyama T. Genetic polymorphisms of alcohol dehydrogense‐1B and aldehyde dehydrogenase‐2, alcohol flushing, mean corpuscular volume, and aerodigestive tract neoplasia in Japanese drinkers. Adv Exp Med Biol. 2015;815:265–79. [DOI] [PubMed] [Google Scholar]