

Abstract
The aim of this study was to investigate whether telmisartan protects the heart from the ischaemia/reperfusion damage through a local microRNA‐1 modulation. Studies on the myocardial ischaemia/reperfusion injury in vivo and on the cardiomyocyte hypoxia/reoxygenation damage in vitro were done. In vivo, male Sprague‐Dawley rats administered for 3 weeks with telmisartan 12 mg/kg/d by gastric gavage underwent ischaemia/reperfusion of the left descending coronary artery. In these rats, infarct size measurement, ELISA, immunohistochemistry (IHC) and reverse transcriptase real‐time polymerase chain reaction showed that expressions of connexin 43, potassium voltage‐gated channel subfamily Q member 1 and the protein Bcl‐2 were significantly increased by telmisartan in the reperfused myocardium, paralleled by microRNA‐1 down‐regulation. In vitro, the transfection of cardiomyocytes with microRNA‐1 reduced the expressions of connexin 43, potassium voltage‐gated channel subfamily Q member 1 and Bcl‐2 in the cells. Telmisartan (50 µmol/L) 60 minutes before hypoxia/reoxygenation, while not affecting the levels of miR‐1 in transfected cells in normoxic condition, almost abolished the increment of miR‐1 induced by the hypoxia/reoxygenation to transfected cells. All together, telmisartan cardioprotected against the myocardial damage through the microRNA‐1 modulation, and consequent modifications of its downstream target connexin 43, potassium voltage‐gated channel subfamily Q member 1 and Bcl‐2.
Keywords: Bcl‐2, connexin 43, hypoxic H9c2 cells, KCNQ1, miR‐1, myocardial ischaemia/reperfusion, telmisartan
1. INTRODUCTION
Myocardial hypoxia or ischaemia‐associated hypoxia is a major determinant of complex pathological changes in the heart, since a constant supply of oxygen is indispensable for cardiac viability and function.1 The hypoxic state leads to a continuous cardiomyocyte structural and electrophysiological remodelling.2, 3 This is characterized by modifications of the ATP‐sensitive potassium (KATP) channels, important sensors for cardiac action potential repolarization3, 4, 5, 6 and modifications of connexin 43 (Cnx43), the main constitutional protein of the gap junctions into the ventricle.7, 8 These modifications are induced through changes in the local expression of microRNA‐1 (miR‐1)9, 10 which targets both Cnx43 and the potassium voltage‐gated channel subfamily Q member 1 (KCNQ1 or Kv7.1),11, 12, 13, 14 even if their combined role is not fully understood.
Telmisartan, an angiotensin II type 1 (AT1) receptor blocker and partial peroxisome proliferator‐activated receptor gamma (PPAR‐γ) agonist, has recently shown protective effects in the treatment of hypoxic/ischaemic cardiac damage15, 16, 17 related to increased levels of cardiac Cnx43 and reduced apoptosis.18, 19 However, it has not yet been studied whether the effects of telmisartan on Cnx43 have been exerted through modifications of the expression of the cardiac miR‐1. Therefore, the first objective of the present study was to study the effects of telmisartan treatment on the cardiac expression of miR‐1, Cnx43 and KCNQ1 in a rat model of myocardial ischaemia/reperfusion (I/R) injury. Then, to better define the molecular mechanisms determined by telmisartan, the cardiac expression of miR‐1, Cnx43 and KCNQ1 was monitored in hypoxic/reoxygenated embryonic rat ventricle H9c2 cells. This in vitro setting was further used to evaluate the putative effects of telmisartan on the transfected miR‐1 levels and thus on both Cnx43 and KCNQ1 expression. Furthermore, the anti‐apoptotic protein Bcl‐2, a further target of the silencing activity of the miR‐1, was monitored.20
2. MATERIALS AND METHODS
2.1. Animal treatment and surgical procedure
All the experimental procedures were approved by the Animal Ethics Committee of University of Campania ‘Luigi Vanvitelli’ of Naples (Protocol Number 2109, 27/7/12). Animal care was in compliance with Italian (DL 116/92) and European Commission (OJ of EC L358/1 18/12/86) guidelines on the use and protection of laboratory animals. All efforts were made to reduce the number of animals used and to minimize animal suffering. Male Sprague‐Dawley rats (220 ± 15 g, n = 40 total) were fed with tap water ad libitum and a standard chow diet for 3 weeks. The animals were randomly divided into four experimental groups (n = 10 per group): (a) untreated rats subjected to thoracotomy only and used as control (SHAM group); (b) untreated rats subjected to myocardial ischaemia/reperfusion (I/R) injury (I/R group); (c) rats administered for 3 weeks by gastric gavage with vehicle (1% methylcellulose, 4 mL/kg/d) and then subjected to I/R injury (I/R veh group); and (d) rats administered for 3 weeks by gastric gavage with telmisartan 12 mg/kg/d (Boehringer Ingelheim) and then subjected to I/reperfusion injury (I/R Tel group). According to previous evidence, this dose was effective in experimental myocardial ischaemia/reperfusion (I/R) injury.15, 16 At the end of the 3 weeks, rats were anaesthetized with intraperitoneal urethane (1.2 g/kg) and underwent thoracotomy or myocardial injury (I/R), as previously described with modifications.15 Briefly, in order to permit artificial ventilation when required, the rats were subjected to tracheotomy by using a polythene cannula. Then, the left thoracotomy was performed between the fourth and the fifth ribs. After the pericardium removal, the heart was exteriorized and then a fine silk ligature was placed around the left anterior descending coronary artery (LADCA), close to its origin. After 25 minutes of ischaemia, a 2‐hour reperfusion was performed. Rats were kept under artificial ventilation with room air at a rate of 56 strokes/min, a stroke volume of 1.0‐1.5 mL/100 g and a positive‐end expiratory pressure of 0.5‐1 cm H2O. Overall mortality was <5% throughout the entire study, in line with the usual procedures in our laboratory.21
2.2. Measurement of area at risk and infarct size
After the 2‐hour reperfusion period, the ratios between the weights of area at risk (AR) and left ventricle (LV) (AR/LV), infarct size (IS) and area at risk (IS/AR) and infarct size and left ventricle (IS/LV) were measured with Evans Blue dye as previously described.15 Selected experiments (n = 5 for each experimental group) were repeated monitoring AR without the staining procedures to measure IS. After the AR collection, half of each specimen was immediately frozen in liquid nitrogen for biochemical analyses and the other half was fixed by immersion in 10% buffered formalin and paraffin‐embedded for IHC.
2.3. Immunohistochemistry
A xylene substitute (HEMO‐De; Fisher Scientific) was used to remove paraffin from tissue sections and then rehydrated with ethanol gradient washes. A 3% hydrogen peroxide aqueous solution was used to sequentially quench the tissue sections, blocked for 1 hour at room temperature with phosphate‐buffered saline (PBS) 6% non‐fat dry milk (Bio‐Rad) and then incubated with specific antibodies anti‐Cnx43 (diluted 1:100; Santa Cruz; Cat. No. sc‐271837) and anti‐KCNQ1 (diluted 1:200; LifeSpan Biosciences; Cat. No. LS‐C405102), according to the recommended dilutions. After PBS washes, sections were incubated with secondary biotin‐conjugated goat anti‐rabbit IgG and avidin–biotin peroxidase complex (diluted 1:200; DBA; Cat. No. BA‐1000). An expert pathologist, blinded to the experimental protocol, analysed the specimens (intra‐observer variability 6%). The antigen expression was automatically calculated by using Image program LEICA IM500 and LEICA QWIN statistic program. Five distinct preparations for each group of animals were carried out, by analysing in each of them 20 microscope fields, for a total area of 2.4755 e+004 at 400× magnifications.
2.4. Hypoxic H9c2 cell culture
Embryonic rat ventricle H9c2 (2‐1) cardiomyocytes (Sigma; Cat. No. 88092904) were grown at 37°C under an atmosphere of 5% CO2 in 5.5 mmol/L glucose Dulbecco's modified Eagle's medium (DMEM; Aurogene; Cat. No. AU‐L0101‐500), supplemented with 10% heat‐inactivated foetal bovine serum (FBS) (Aurogene; Cat. No. AU‐S181H‐500), 5% l‐glutamine (Aurogene; Cat. No. AU‐X0550‐100) and 5% penicillin–streptomycin solution (Aurogene; Cat. No. AU‐L0022‐100).14
Hypoxic conditions were established according to a previous method by exposing H9c2 cells to cobalt chloride (CoCl2; Sigma; Cat. No. 15862) 400 µmol/L for 6 hours (H/R group) in DMEM 10%.22, 23, 24, 25 Hypoxia was confirmed by Western blot analysis of HIF‐1α protein expression. In order to investigate the effects of telmisartan treatment on hypoxic conditions alone, and not in combination with the serum and glucose deprivation (SGD), following the 6‐hour hypoxia the medium with CoCl2 was replaced by DMEM without CoCl2 for 2‐hour reoxygenation.26, 27 Sixty minutes before the hypoxia/reoxygenation,23 cells were exposed to a dose of telmisartan (50 µmol/L; H/R Tel group) already reported as effective on H9c2 cells, and to its vehicle dimethyl sulfoxide (DMSO 1% in cell medium; H/R DMSO group).18 At the end of the hypoxia/reoxygenation period, cell morphology was observed with optic microscopy (Leica DMi1, Germany). According to previous experience, cells were seeded at 5 × 103 cells/cm2 in 96‐well plates for the viability assay; at 1 × 106 cells/cm2 in 10 cm cell culture dishes for total RNA and protein detection; and at 1 × 104 cells/cm2 in 24‐well plates for immunofluorescence assay.28 Three independent experiments were performed. In a single experiment, each treatment was repeated three times.
2.5. Cell viability assay
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay was used to measure cell viability.14 Briefly, the addition of MTT solution (1:10 in culture medium) to each well was followed by a 3‐hour incubation at 37°C. Once MTT solution was removed, each well was washed for 20 minutes at room temperature with isopropanol‐HCl 0.2 N. A 96‐well plate reader (iMark; Bio‐Rad Laboratories) was used to detect the optical density (OD) values at 570 nm.
2.6. miR‐1 mimic transfection
Normoxic H9c2 cells were transfected with miR‐1 mimic (mim1) 5 nmol/L (Qiagen; Cat. No. MSY0003125) (mim1 group) or negative control (mim1‐ group), using Lipofectamine 2000 reagent (Life Technologies; Cat. No. 11668‐027) according to the manufacturer's protocol. After 24 hours of transfection, miR‐1‐transfected cells were exposed to hypoxia/reoxygenation alone (H/R mim1 group) and to DMSO 1% or telmisartan 50 µmol/L before hypoxia reoxygenation (respectively, H/R mim1 DMSO and H/R mim1 Tel groups).
2.7. qRT‐PCR
miRNA isolation was performed with the miRNeasy Mini Kit (Qiagen; Cat. No. 217004), according to the manufacturer's protocol ‘Purification of Total RNA, including Small RNAs, from Animal Cells'. In order to monitor the miRNA recovery efficiency and to normalize miRNA expression in the real‐time PCR analysis, 5 μL of Syn‐cel‐miRNA‐39 miScript miRNA Mimic 5 nmol/L (Qiagen; Cat. No. MSY0000010) was spiked into each sample, before nucleic acid preparation. RNA concentration and quality were determined with NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific). Mature miRNAs were converted to cDNA with miScript II RT kit (Qiagen; Cat. No. 218161). The CFX96 Touch™ Real‐Time PCR Detection System (Bio‐Rad Laboratories) was used to monitor miR‐1 levels (MIMAT0000416). Each reaction was carried out with SYBR Green PCR Kit (Qiagen; Cat. No. 218073) and specific miScript Primer Assays for miR‐1 (Qiagen; Cat. No. MS00012943) and Syn‐cel‐miR‐39 (Qiagen; Cat. No. MS00019789). ΔCt value was as CtmiR‐1 − CtmiR‐39 in order to obtain miR‐1 levels as 2−ΔCt. Fold change was then obtained as 2−ΔΔCt and calculated as 2−ΔCt of the treatment group/2−ΔCt of the control group. For fold change greater than 1, fold regulations were reported equal to the fold change values. For fold changes lower than 1, fold regulations were reported as the negative inverse of the fold change values.
2.8. Protein isolation and quantization
Heart tissues were homogenized in RIPA lysis buffer (Sigma; Cat. No. R0278) including a protease inhibitor cocktail (Roche; Cat. No. 11873580001). H9c2 cells were washed with cold PBS (Aurogene; AU‐L0615), scraped in 150 µL of cold RIPA lysis buffer (Sigma; Cat. No. R0278) including a protease inhibitor cocktail (Roche; Cat. No. 1187358000) and centrifuged at 13 800 g for 10 minutes at 4°C, to purify the protein supernatants from nucleic acids.14 Total protein concentration was determined following the Bio‐Rad protein assay protocol (Bio‐Rad Laboratories; Cat. No. 500‐0006) and used for Western blotting and ELISAs.
2.9. Western blotting assay
Thirty microgram of protein sample was separated on a 12% separation gel, electrotransferred onto a polyvinylidenedifluoride (PVDF) membrane (Merck Millipore; Cat. No. IPFL10100) and then blocked for 1 hour at room temperature with 5% non‐fat dry milk (Euroclone; Cat. No. EMR180500). The following specific primary antibodies were used for the overnight incubation of the blots using the recommended dilutions: anti‐HIF‐1α (diluted 1:200; Santa Cruz; Cat. No. sc‐8711), anti‐Cnx43 (diluted 1:500; Santa Cruz; Cat. No. sc‐271837), anti‐KCNQ1 (diluted 1:200; LifeSpan Biosciences; Cat. No. LS‐C405102) and anti‐actin (diluted 1:500; Santa Cruz; Cat. No.sc‐8432). Blots were then incubated for 1 hour at room temperature with horseradish peroxidase‐conjugated secondary anti‐rabbit (diluted 1:2000; Santa Cruz; Cat. No. sc‐2004), anti‐goat (diluted 1:2000; Santa Cruz; Cat. No. sc‐2020) or antimouse (diluted 1:2000; Santa Cruz; Cat. No. sc‐2005) antibodies. The signal was detected using the VisionWorks™ LS Analysis Software (Analytik Jena AG) and then expressed as densitometric units (DU).
2.10. ELISAs
Troponin I as marker of myocardial infarction and Bcl‐2 protein levels were quantified by using the Rat Cardiac Troponin I (cTnI) ELISA Kit (MyBioSource; Cat. No. MBS727624) and the Rat B‐cell CLL/lymphoma 2, Bcl‐2 ELISA Kit (MyBioSource; Cat. No. MBS704498), according to the manufacturer's protocols.
2.11. Immunofluorescence
H9c2 cells were fixed with 4% paraformaldehyde and washed with PBS (Aurogene; Cat. No. AU‐L0615). In order to inhibit non‐specific antibody binding, cells were incubated for 30 minutes in blocking solution (1% BSA in PBS). Cells were then incubated overnight at 4°C with the following primary antibodies, diluted in PBS blocking buffer: anti‐Cnx43 (diluted 1:100; Santa Cruz; Cat. No. sc‐271837) and anti‐KCNQ1 (diluted 1:200; Bioss Inc; Cat. No. bs‐6760R). Specific antigens were located using a fluorescein isothiocyanate (FITC)‐conjugated anti‐rabbit (diluted 1:1000; Immunoreagents; Cat. No. GTXRB‐003‐D488N). After the counterstaining H9c2 cells with pentahydrate bisbenzimide (Hoechst 33258; Sigma; Cat. No. 23491‐45‐4) and mounting with 90% glycerol in PBS, immunofluorescence images were obtained using a fluorescence microscope (Leica) and a fluorescence confocal microscope (LSM 710 Zeiss). Leica FW4000 (Leica) and Zen Zeiss (Zeiss) softwares were used to analyse the images. An observer blinded to the treatment performed the labelling quantization, by calculating the percentage of positive cells/total cells counted. This was calculated in each microscope field as the mean of labelled positive cells/400 cells counted. Four different fields were analysed for each treatment. Only bisbenzimide‐counterstained cells were considered as positive profiles in order to avoid overcounting cells.
2.12. Statistical analysis
The results are presented as mean ± standard error of the mean (SEM) of five observations per group in vivo, and as mean ± SEM of nine observations in vitro. Statistical significance was determined using ANOVA followed by Bonferroni's test. A P‐value <.05 was considered significant to reject the null hypothesis.
3. RESULTS
3.1. Myocardial tissue damage after telmisartan administration
Untreated Sprague‐Dawley rats subjected to I/R procedure (I/R group) exhibited an AR equal to the 63% of the LV. The IS was 65% of the AR and 40% of the LV (Figure 1A). The IS/AR values were not affected by the administration of vehicle (I/R veh) but were significantly reduced following treatment with telmisartan 12 mg/kg (I/R Tel; −38%, P < .01 vs I/R veh), as well as IS/LV ratios (−25%, P < .01 vs I/R veh; Figure 1A). Cardiac troponin I (cTnI) levels were significantly increased by I/R procedure (+67.9%, P < .05 vs SHAM) and significantly reduced in rats receiving telmisartan 12 mg/kg only (−31%, P < .05 vs I/R veh; Figure 1B).
Figure 1.
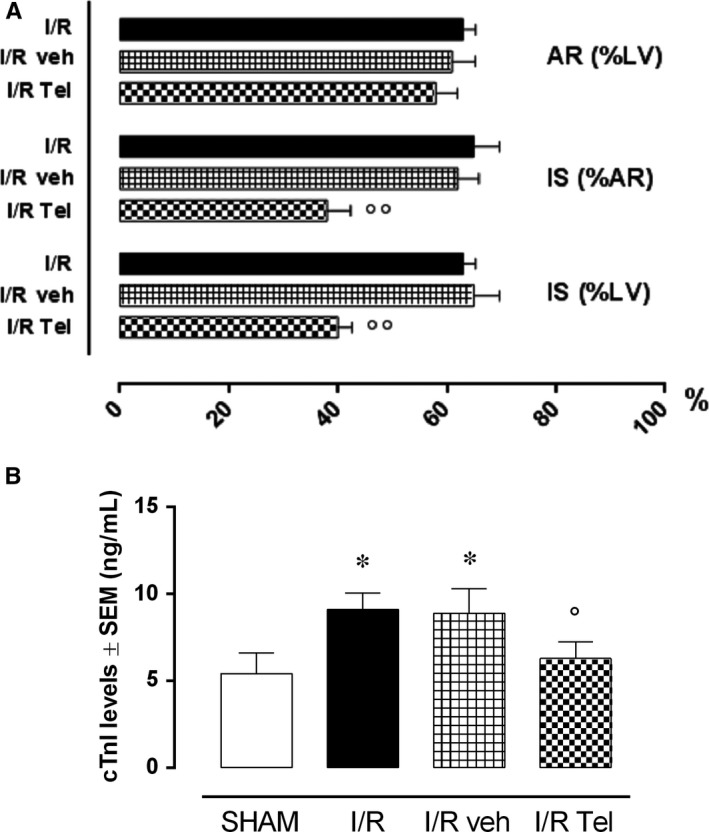
Effects of telmisartan on myocardial damage after I/R injury. A, AR/LV, IS/AR and IS/LV ratios and (B) cTnI levels in the different experimental groups. SHAM, thoracotomy only; I/R, ischaemia/reperfusion; veh, 1% methylcellulose 4 mL/kg/d; Tel, telmisartan 12 mg/kg; AR, area at risk; LV, left ventricle; IS, infarct size. Results are expressed as mean ± SEM of five observations. *P < .05 vs SHAM; °P < .05 and °°P < .01 vs I/R veh
3.2. Effects of telmisartan on Cnx 43, KCNQ1 and Bcl‐2 expression in I/R hearts
Following the I/R procedure, the IHC showed a significant reduction in Cnx43 and KCNQ1 expressions in I/R rats compared with SHAM group (respectively, −61.3% and −50%, P < .01 vs SHAM; Figure 2A,B). These were not significantly modified in I/R veh rats compared with I/R group (respectively, +6.9% and −5%), whereas I/R Tel group exhibited a consistent increase in both Cnx 43 and KCNQ1 levels compared with I/R group (respectively, +89.7% and +70%, P < .01 vs I/R veh; Figure 2A,B). The same trend was shown by Bcl‐2 levels, significantly decreased in I/R group (−70.6%, P < .01 vs SHAM; Figure 2C). These were markedly increased by telmisartan treatment +140% P < .05 vs I/R veh) and not by its vehicle (−4% vs I/R veh; Figure 2C).
Figure 2.
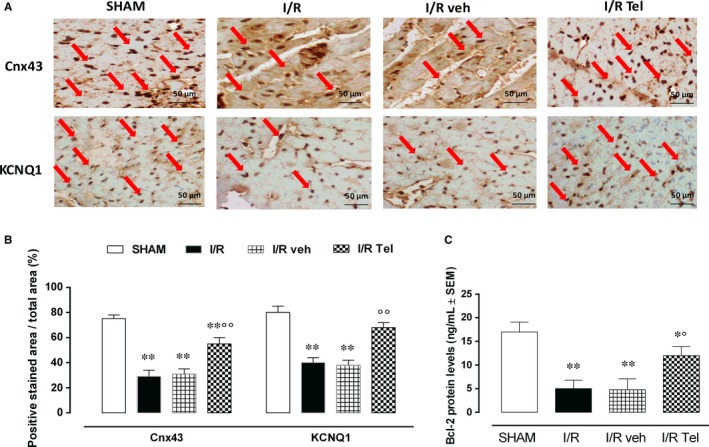
Effects of telmisartan on Cnx 43, KCNQ1 and Bcl‐2 expression in acute myocardial I/R injury. A, Representative images for immunochemical detection of Cnx43 and KCNQ1 levels. B, Bar graph showing Cnx43 and KCNQ1 expressed as percentage of positive stained area/total area. C, Bcl‐2 protein levels detected by ELISA. SHAM, thoracotomy only; I/R, ischaemia/reperfusion; veh, 1% methylcellulose 4 mL/kg/d; Tel, telmisartan 12 mg/kg. Results are expressed as mean ± SEM of five observations. **P < .01 vs SHAM; °P < .05 and °°P < .01 vs I/R veh. Scale bar = 50 µmol/L; 400× magnification
3.3. miR‐1 modulation by telmisartan in I/R hearts
qRT‐PCR analysis showed a significant increase in miR‐1 levels in rat hearts subjected to I/R procedure compared with SHAM rats (+100%, P < .05 vs SHAM; Figure 3A). Infarcted hearts exhibited an evident reduction in miR‐1 levels when administered with telmisartan 12 mg/kg (−36.4%, P < .05 vs I/R) and not with 1% methylcellulose vehicle (−2.3% vs I/R; Figure 3A). This miR‐1 down‐regulation was paralleled by an increased expression of Cnx43, KCNQ1 and Bcl‐2 in I/R Tel group (Figure 3B).
Figure 3.
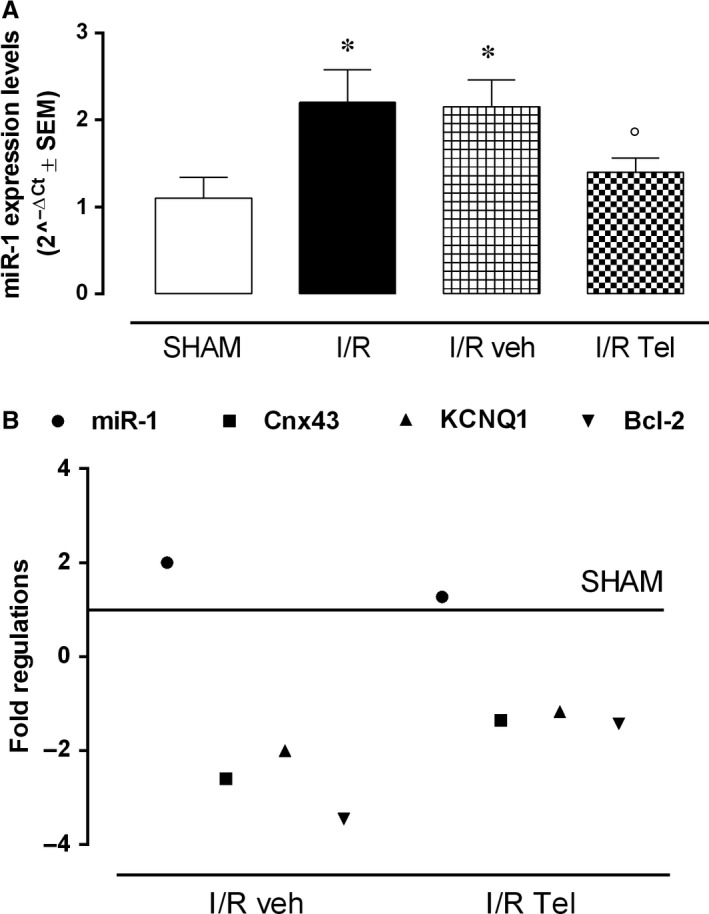
Effects of telmisartan on miR‐1 expression pattern in acute myocardial I/R injury. A, Bar graph showing qRT‐PCR miR‐1 levels expressed as 2−ΔCt ± SEM (B) Fold regulation of miR‐1, Cnx43, KCNQ1 and Bcl‐2 in the different experimental groups. SHAM, thoracotomy only; I/R, ischaemia/reperfusion; veh, 1% methylcellulose 4 mL/kg/d; Tel, telmisartan 12 mg/kg. Results are expressed as mean ± SEM of five observations. *P < .05 vs Sham; °P < .05 vs I/R veh
3.4. Cell morphology and viability in hypoxic/reoxygenated H9c2 cells exposed to telmisartan
H9c2 cells exposed to 400 µmol/L CoCl2 for 6 hours and to 2‐hour reoxygenation (H/R group) were severely damaged compared with normoxic cells (CTRL group), by exhibiting edge blurring, shedding, shrunken and floating in the cell culture medium (Figure 4A). They also had reduced cell viability (−61%, P < .01 vs CTRL) following the hypoxia/reoxygenation procedure (Figure 4B). Unmodified by DMSO exposure, the percentage of cell viability was significantly increased in H/R H9c2 cells pretreated with telmisartan 50 µmol/L (+96,4%, P < .01 vs H/R DMSO), although still reduced compared with CTRL (−24.4%, P < .01 vs CTRL) (Figure 4B). This was accompanied by a reduction in the damaged cell morphology (Figure 4A). Telmisartan and DMSO at the doses used did not modify the cell morphology and viability of normoxic H9c2 cells (data not shown).
Figure 4.

Effects of telmisartan on H/R H9c2 cell morphology and survival. A, Representative optical microscopy images at 10×, 20× and 40× magnifications. B, MTT assay for the determination of viable cell number. CTRL, normoxic cells; H/R, cells exposed to hypoxia/reoxygenation; H/R DMSO, H/R + DMSO 1%; H/R Tel, HR + telmisartan 50 µmol/L. Results are expressed as mean ± SEM of nine observations. **P < .01 vs CTRL; °°P < .01 vs H/R DMSO
3.5. Cnx 43, KCNQ1 and Bcl‐2 expression in H/R H9c2 cardiomyocytes
Hypoxic conditions because of CoCl2 exposure were confirmed in H9c2 cells by detecting HIF‐1α cell protein levels through Western blotting analysis: This was significantly increased in all the experimental conditions compared with normoxic H9c2 cells (H/R, +438.5%; HR DMSO, +400.0%; H/R Tel, +234.6%, P < .01 vs CTRL; Figure 5A). Interestingly, telmisartan treatment (50 µmol/L) 60 minutes before hypoxia/reoxygenation model significantly reduced HIF‐1α levels in untreated hypoxic group (−37.9%, P < .01 vs H/R DMSO; Figure 5A). In addition to this, cells KCNQ1 and Cnx43 were found to be significantly reduced in H/R group (KCNQ1, −89.2%; Cnx43, −71.7%, P < .01 vs CTRL) compared with normoxic cells (Figure 5A). Also, Bcl‐2 protein levels showed this trend (−53.3%, P < .05 vs CTRL; Figure 5B). All these factors were not affected by DMSO pretreatment (respectively, +7.1%; −7.7% and −3.6% vs H/R DMSO), while markedly increased by telmisartan exposure (KCNQ1, +471.4%, P < .01 vs H/R DMSO; Cnx43, +130.8% and Bcl‐2, +325%, P < .05 vs H/R DMSO; Figure 5A,B). In contrast, both the vehicle DMSO and telmisartan at the doses used did not affect the factors monitored in normoxic H9c2 cells (data not shown).
Figure 5.
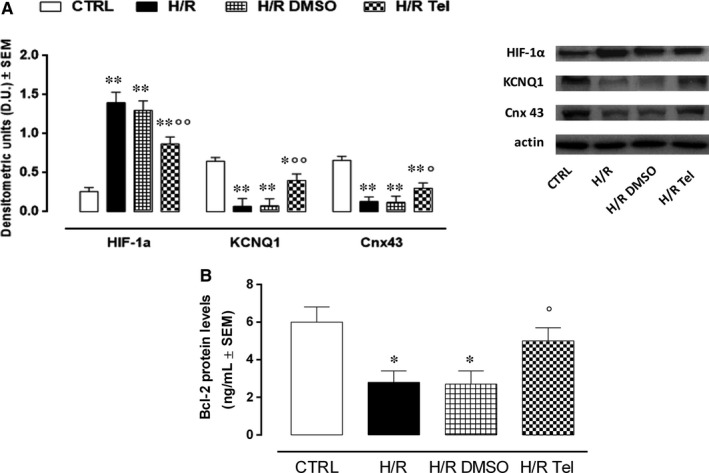
Modulation of Cnx43, KCNQ1 and Bcl‐2 by telmisartan in H7R H9c2 cells. A, Bar graph and representative Western blotting images, showing cell HIF‐1α, Cnx43 and KCNQ1 levels calculated as protein target/actin ratio. B, Bcl‐2 protein levels detected by ELISA. CTRL, normoxic cells; H/R, cells exposed to hypoxia/reoxygenation; H/R DMSO, H/R + DMSO 1%; H/R Tel, HR + telmisartan 50 µmol/L. Results are expressed as mean ± SEM of nine observations. *P < .05 and **P < .01 vs CTRL; °P < .05 and °°P < .01 vs H/R DMSO
3.6. miR‐1 modulation by telmisartan in hypoxic/reoxygenated H9c2
miR‐1 levels were strongly induced by hypoxia/reoxygenation in H9c2 cells (+237.7%, P < .01 vs CTRL), unmodified by DMSO (+6,1% vs H/R) and markedly reduced by telmisartan exposure (−63.2 P < .01 vs H/R DMSO) (Figure 6A).This drug did not produce any effect on miR‐1 in normoxic cells (data not shown).
Figure 6.

miR‐1 down‐regulation by telmisartan in myocardial hypoxia/reoxygenation in H9c2 cells. A, Bar graph showing qRT‐PCR miR‐1 levels expressed as 2−ΔCt ± SEM in non‐transfected H9c2 cells. CTRL, normoxic cells; H/R, cells exposed to hypoxia/reoxygenation; H/R DMSO, H/R + DMSO 1%; H/R Tel, HR + telmisartan 50 µmol/L; B, transfected miR‐1 levels before and after hypoxia/reoxygenation, expressed as 2−ΔCt ± SEM mim1‐, normoxic cells transfected with negative control mimic; mim1, normoxic cells transfected with miR‐1 mimic 5 nmol/L; H/R mim1, cells transfected with miR‐1 mimic 5 nmol/L and exposed to hypoxia/reoxygenation; H/R mim1 DMSO, H/R mim1 cells exposed to DMSO 1%; H/R mim1 Tel, H/R mim1 cells exposed to telmisartan 50 µmol/L. Results are expressed as mean ± SEM of nine observations. *P < .05 and **P < .01 vs CTRL; °°P < .01 vs H/R DMSO; ## P < .01 vs mim1‐; ^^P < .01 vs mim1; §§ P < .01 vs H/R mim1 DMSO
3.7. miR‐1 modulation in miR‐1‐transfected hypoxic/reoxygenated H9c2
miR‐1 levels were quantified in miR‐1‐transfected cardiomyocytes. As expected, normoxic H9c2 cells transfected with miR‐1 mimic (mim+) exhibited significantly higher miR‐1 levels compared with cells transfected with the negative control (+900%, P < .05 vs mim1‐; Figure 6B). Hypoxia/reoxygenation further increased miR‐1 levels (+200%, P < .01 vs H/R mim1), as well as DMSO exposure before hypoxia/reoxygenation (205%, P < .01 vs H/R mim1). Interestingly, telmisartan while not affecting the levels of miR‐1 in transfected cells in normoxic condition, it almost abolished the increment of miR‐1 induced by the H/R procedure to these cells (−62.3%, P < .01 vs H/R mim1 DMSO; Figure 6B).
3.8. Cnx43, KCNQ1 and Bcl‐2 modulation by telmisartan in miR‐1‐transfected cardiomyocytes exposed to hypoxia/reoxygenation
After hypoxia/reoxygenation, immunofluorescence analysis of miR‐1‐transfected H9c2 cardiomyocytes showed that telmisartan treatment caused a marked increase in Cnx43 (+400%, P < .01 vs H/R mim1 DMSO) and KCNQ1 compared with transfected cells unexposed to telmisartan (+253%, P < .01 vs H/R mim1 DMSO) (Figure 7A,B). In line with these, Bcl‐2 protein levels were significantly increased by telmisartan (+300%, P < .01 vs H/R mim1 DMSO) (Figure 7C).
Figure 7.

Telmisartan effects on hypoxic H9c2 cardiomyocytes transfected with miR‐1 mimic. A, Representative immunofluorescence images, showing Cnx3 and KCNQ1 levels in miR‐1‐transfected cells exposed to hypoxia/reperfusion. Cell nuclei are labelled in blue with Hoechst, whereas cells positive to Cnx43 or KCNQ1 antibodies are labelled in green. B, Bar graph showing the percentage of Cnx or KCNQ1‐positive cells/total counted cells. C, Bcl‐2 protein levels in miR‐1‐transfected H/R cardiomyocytes, detected by ELISA. mim1‐, normoxic cells transfected with negative control mimic; mim1, normoxic cells transfected with miR‐1 mimic 5 nmol/L; H/R mim1, cells transfected with miR‐1 mimic 5 nmol/L and exposed to hypoxia/reoxygenation; H/R mim1 DMSO, H/R mim1 cells exposed to DMSO 1%; H/R mim1 Tel, H/R mim1 cells exposed to telmisartan 50 µmol/L. Results are expressed as mean ± SEM of nine observations. # P < .05 vs mim1‐; ## P < 0.01 vs mim1‐;^P < .05 vs mim1; §§ P < .01 vs H/R mim1 DMSO. Scale bar = 10 µmol/L; 20× magnification
4. DISCUSSION
Here, we report that telmisartan protected the infarcted rat heart by locally increasing two important players in hypoxia‐induced cell survival,29 Cnx43 and KCNQ1 potassium channel. In contrast, telmisartan reduced miR‐1 expression within the infarcted heart along with increased expression of the known apoptotic marker20, 30 B‐cell lymphoma 2 (Bcl‐2). This action resulted in a lower degree of tissue damage. Telmisartan also copied this effect on embryonic rat ventricle H9c2 cells exposed to a hypoxia/reoxygenation procedure.
The cardioprotection afforded by telmisartan has been widely reported in myocardial I/R injury15, 16, 17, 18, 19, 31; however, the present study for the first time sheds light on the involvement of the potassium channel KCNQ1 and Cnx43 with respect to telmisartan action. As it modulates cardiac cell susceptibility to hypoxia32, 33 and regulates the impulse propagation and electrical synchronization between cardiomyocytes,34, 35 Cnx43 seems to have a key role in the setting of functional I/R damage. Similarly, recent evidence reported that Cnx43 together with KCNQ1 is part of the same signalling pathway that provides cell protection after myocardial infarction by acting in a functionally dependent manner.3, 29, 36 Indeed, mutations in both genes encoding for KCNQ1 and Cnx43 have been associated with clinical sudden infant death syndrome (SIDS), for which hypoxia is a major risk factor.37, 38 The data presented, therefore, identified telmisartan as a regulatory candidate of these two important factors in the infarcted myocardium, Cnx43 and KCNQ1. Moreover, they highlighted that telmisartan cardioprotection is exerted by reducing the apoptosis within the myocardium relating to tissue protection. Indeed, apoptosis of cardiomyocytes, associated with quantitative disorders of proteins, aggravates myocardial I/R injury.39 It should also be noted that telmisartan decreased the resident miR‐1, which controls both the cardiac structure and the functionality. miR‐1 predominantly regulates the electrical and contractile activity of the heart, by modulating atrioventricular and ventricular conduction at multiple levels.40, 41, 42 Alterations of its expression levels result in atrioventricular block, impaired contractile function and increase in ROS levels following cardiac I/R injury.9, 43, 44, 45 This microRNA targets both Cnx43 and KCNQ1,11, 13, 14, 40, 41 and, when down‐regulated, improves KCNQ1 expression in H9c2 cells,14 in line with the results achieved here. Again, this is the first time that a study reports miR‐1 involvement in a telmisartan‐induced cardioprotection. One hypothesis that could be formulated for this phenomenon is that the miR‐1 down‐regulation could be linked to the blocking activity of telmisartan on the AT1 receptors, which notoriously stimulate the expression of cellular miRNAs under hypoxia conditions.46, 47 Induced by hypoxia, the AT1 receptors promote miRNA expression through mechanisms depending on Gaq/11 and Erk1/2 activation and by blocking AT1 receptors, telmisartan could impair the activation of Gaq/11 and Erk1/2 G‐protein‐dependent signalling, leading to a reduced miR‐1 expression.46, 47 On another note, telmisartan‐induced miR‐1 down‐regulation also correlated with increased viability of hypoxic/reoxygenated H9c2 cells and increased expression of the anti‐apoptotic protein Bcl‐2 into the cells.
Summarizing these points, telmisartan cardioprotected from myocardial ischaemia/reperfusion injury by modifying the cardiac levels of the three factors Cnx43, KCNQ1 and miR‐1. What still required deeper evaluation were the cell types which telmisartan‐activated and the sequence of events. It is opinion that, although non‐cardiomyocyte cells of the heart express Cnx43 gap junctions and are able to influence cardiac electrophysiology, KCNQ1 channels are a predominant feature of cardiac myocytes.48, 49, 50 Similarly, it is opinion that there should be a coordinated and sequential action by telmisartan on the factors monitored, probably through miR‐1 first and then on the other two. Therefore, in order to investigate a possible Cnx43/KCNQ1 modulation afforded by telmisartan through miR‐1 regulation, we translated the research on cultured hypoxic/reoxygenated embryonic rat H9c2 cardiomyocytes exposed to the hypoxia‐mimetic chemical cobalt chloride (CoCl2), and treated with telmisartan. Ventricular H9c2 cells are a well‐known relevant cell model in the mimicking of IR injury: Being the closest cells to primary cardiomyocytes concerning their energy metabolism features, such as number and arrangements of mitochondria and presence of beta‐tubulin II,51, 52, 53 H9c2 cells are a simple validated and useful in vitro model for exploring the mechanisms driven by hypoxia/reoxygenation.23, 24, 25, 54, 55 These lead to the hydroxylase enzyme inactivation, hypoxia‐inducible factor HIF‐1α stabilization, ROS generation, apoptosis and increased Bax/Bcl‐2 ratio followed by activation of caspase‐3 cleavage.55, 56
The data obtained in the present study on H9c2 cells confirmed the reduction in Cnx43, KCNQ1 and Bcl‐2 expression following exposure to the hypoxia‐mimetic agent, while treatment with telmisartan restored them. This in line with other experiences showing that angiotensin II (ATII) exerts an inhibitory effect on Cnx43, KCNQ1 and Bcl‐2 expression, and PPAR‐γ agonism is effective in preventing the reduction in Cnx43 induced by ATII.57, 58, 59 Interestingly, the telmisartan‐activated response on Cnx43, KCNQ1 and Bcl‐2 exerted through a down‐regulation of miR‐1 was confirmed by mimic transfection of miR‐1. Thus suggesting in vivo crosstalk between Cnx43, KCNQ1, Bcl‐2 and miR‐1,3 one would argue that the properties exerted by telmisartan in hypoxic/ischaemic cardiac damage are exerted through miR‐1 and consequent modifications of Cnx43, potassium channel KCNQ1 and Bcl‐2 expression in the cardiomyocytes.
CONFLICT OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTION
MCT performed the research, FB performed immunofluorescence, AM analysed the data, IP performed the immunohistochemical analysis, EG and GP performed the surgical procedure, and GFN and MD designed the research and wrote the study.
Trotta MC, Ferraro B, Messina A, et al. Telmisartan cardioprotects from the ischaemic/hypoxic damage through a miR‐1‐dependent pathway. J Cell Mol Med. 2019;23:6635–6645. 10.1111/jcmm.14534
DATA ACCESSIBILITY
The authors confirm that the data supporting the findings of this study are available within the article.
REFERENCES
- 1. Giordano FJ. Oxygen, oxidative stress, hypoxia and heart failure. J Clin Invest. 2005;115:500‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mehta JL, Li DY. Inflammation in ischemic heart disease: response to tissue injury or a pathogenetic villain? Cardiovasc Res. 1999;43:291‐299. [DOI] [PubMed] [Google Scholar]
- 3. Waza AA, Bhat SA, Hussain MU, Ganai BA. Connexin 43 and ATP‐sensitive potassium channels crosstalk: a missing link in hypoxia/ischemia stress. Cell Tissue Res. 2018;371:213‐222. [DOI] [PubMed] [Google Scholar]
- 4. Hool LC. Differential regulation of the slow and rapid components of guinea‐pig cardiac delayed rectifier K+ channels by hypoxia. J Physiol. 2004;554:743‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimoda LA, Polak J. Hypoxia. 4. Hypoxia and ion channel function. Am J Physiol Cell Physiol. 2011;300:C951‐C967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lamothe SM, Song W, Guo J, et al. Hypoxia reduces mature hERG channels through calpain up‐regulation. FASEB J. 2017;31:5068‐5077. [DOI] [PubMed] [Google Scholar]
- 7. Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762‐H1770. [DOI] [PubMed] [Google Scholar]
- 8. Wu X, Huang W, Luo G, Alain LA. Hypoxia induces connexin 43 dysregulation by modulating matrix metalloproteinases via MAPK signaling. Mol Cell Biochem. 2013;384:155‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pan Z, Sun X, Ren J, et al. miR‐1 exacerbates cardiac ischemia‐reperfusion injury in mouse models. PLoS ONE. 2012;7:e50515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng Y, Tan N, Yang J, et al. A translational study of circulating cell‐free microRNA‐1 in acute myocardial infarction. Clin Sci (Lond). 2010;119:87‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Curcio A, Torella D, Iaconetti C, et al. MicroRNA‐1 downregulation increases connexin 43 displacement and induces ventricular tachyarrhythmias in rodent hypertrophic hearts. PLoS ONE. 2013;8:e70158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lei Y, Peng X, Li T, et al. ERK and miRNA‐1 target Cx43 expression and phosphorylation to modulate the vascular protective effect of angiotensin II. Life Sci. 2019;216:59‐66. [DOI] [PubMed] [Google Scholar]
- 13. Li Y, Yang CM, Xi Y, et al. MicroRNA‐1/133 targeted dysfunction of potassium channels KCNE1 and KCNQ1 in human cardiac progenitor cells with simulated hyperglycemia. Int J Cardiol. 2013;167:1076‐1078. [DOI] [PubMed] [Google Scholar]
- 14. Trotta MC, Salerno M, Brigida AL, et al. Inhibition of aldose‐reductase‐2 by a benzofuroxane derivative bf‐5m increases the expression of kcne1, kcnq1 in high glucose cultured H9c2 cardiac cells and sudden cardiac death. Oncotarget. 2018;9:17257‐17269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rinaldi B, Di Filippo C, Capuano A, et al. Adiponectin elevation by telmisartan ameliorates ischaemic myocardium in zucker diabetic fatty rats with metabolic syndrome. Diabetes Obes Metab. 2012;14:320‐328. [DOI] [PubMed] [Google Scholar]
- 16. Di Filippo C, Rossi C, Ferraro B, et al. Involvement of proteasome and macrophages M2 in the protection afforded by telmisartan against the acute myocardial infarction in Zucker diabetic fatty rats with metabolic syndrome. Mediators Inflamm. 2014;2014:972761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yuan X, Zhu D, Guo XL, et al. Telmisartan attenuates myocardial apoptosis induced by chronic intermittent hypoxia in rats: modulation of nitric oxide metabolism and inflammatory mediators. Sleep Breath. 2015;19:703‐709. [DOI] [PubMed] [Google Scholar]
- 18. Li X, Lan Y, Nie M, et al. Telmisartan suppresses cardiac hypertrophy by inhibiting cardiomyocyte apoptosis via the NFAT/ANP/BNP signaling pathway. Mol Med Rep. 2017;15:2574‐2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang HY, Li X, Tian Y. Telmisartan reduces arrhythmias through increasing cardiac connexin43 by inhibiting IL‐17 after myocardial infarction in rats. Eur Rev Med Pharmacol Sci. 2017;21:5283‐5289. [DOI] [PubMed] [Google Scholar]
- 20. Tang Y, Zheng J, Sun Y, et al. MicroRNA‐1 regulates cardiomyocyte apoptosis by targeting Bcl‐2. Int Heart J. 2009;50:377‐387. [DOI] [PubMed] [Google Scholar]
- 21. Di Filippo C, Perretti M, Rossi F, et al. Acute myocardial infarction in streptozotocin‐induced hyperglycaemic rats: protection by a carbon monoxide‐releasing molecule (CORM‐3). Naunyn Schmiedebergs Arch Pharmacol. 2012;385:137‐144. [DOI] [PubMed] [Google Scholar]
- 22. Tong XX, Wu D, Wang X, et al. Ghrelin protects against cobalt chloride‐induced hypoxic injury in cardiac H9c2 cells by inhibiting oxidative stress and inducing autophagy. Peptides. 2012;38:217‐227. [DOI] [PubMed] [Google Scholar]
- 23. Wu K, Xu W, You Q, et al. Increased expression of heat shock protein 90 under chemical hypoxic conditions protects cardiomyocytes against injury induced by serum and glucose deprivation. Int J Mol Med. 2012;30:1138‐1144. [DOI] [PubMed] [Google Scholar]
- 24. Gallo S, Gatti S, Sala V, et al. Agonist antibodies activating the Met receptor protect cardiomyoblasts from cobalt chloride‐induced apoptosis and autophagy. Cell Death Dis. 2014;5:e1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng CI, Lee YH, Chen PH, et al. Cobalt chloride induces RhoA/ROCK activation and remodeling effect in H9c2 cardiomyoblasts: involvement of PI3K/Akt and MAPK pathways. Cell Signal. 2017;36:25‐33. [DOI] [PubMed] [Google Scholar]
- 26. Jian L, Lu Y, Lu S, Lu C. Chemical chaperone 4‐phenylbutyric acid protects H9c2 cardiomyocytes from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress‐induced apoptosis. Mol Med Rep. 2016;13:4386‐4392. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y, Su W, Zhang Q, et al. Glycine protects H9c2 cardiomyocytes from high glucose‐ and hypoxia/reoxygenation‐induced injury via inhibiting PKCβ2 activation and improving mitochondrial quality. J Diabetes Res. 2018;2018:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trotta MC, Maisto R, Alessio N, et al. The melanocortin MC5R as a new target for treatment of high glucose‐induced hypertrophy of the cardiac H9c2 cells. Front Physiol. 2018;9:1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Waza AA, Andrabi K, Hussain MU. Protein kinase C (PKC) mediated interaction between conexin43 (Cx43) and K(+)(ATP) channel subunit (Kir6.1) in cardiomyocyte mitochondria: Implications in cytoprotection against hypoxia induced cell apoptosis. Cell Signal. 2014;26:1909‐1917. [DOI] [PubMed] [Google Scholar]
- 30. Grunefelder J, Miniati DN, Murata S, et al. Upregulation of Bcl‐2 through caspase‐3 inhibition ameliorates ischemia/reperfusion injury in rat. Circulation. 2001;104:I202‐I206. [DOI] [PubMed] [Google Scholar]
- 31. Saavedra JM, Sanchez‐Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Plotkin LI, Bellido T. Bisphosphonate‐induced, hemi‐channel‐mediated anti‐apoptosis through the Src/ERK pathway: a gap junction‐independent action of connexin 43. Cell Comm Adhes. 2001;8:377‐382. [DOI] [PubMed] [Google Scholar]
- 33. Lin JH, Yang J, Liu S, et al. Connexin mediates gap junction‐independent resistance to cellular injury. J Neurosci. 2003;23:430‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poelzing S, Akar FG, Baron E, Rosenbaum DS. Heterogeneous connexin43 expression produces electrophysiological heterogeneities across ventricular wall. Am J Physiol Heart Circ Physiol. 2004;286:H2001‐H2009. [DOI] [PubMed] [Google Scholar]
- 35. Ripplinger CM, Li W, Hadley J, et al. Enhanced transmural fiber rotation and Connexin 43 heterogeneity are associated with an increased upper limit of vulnerability in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res. 2007;101:1049‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Phys Heart Circ Phys. 2003;285:H921‐H930. [DOI] [PubMed] [Google Scholar]
- 37. Neary MT, Mohun TJ, Breckenridge RA. A mouse model to study the link between hypoxia, long QT interval and sudden infant death syndrome. Dis Model Mech. 2013;6:503‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Norstrand DW, Asimaki A, Rubinos C, et al. Connexin43 mutation causes heterogeneous gap junction loss and sudden infant death. Circulation. 2012;125:474‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krijnen PA, Nijmeijer R, Meijer CJ, et al. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002;55:801‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang B, Lin H, Xiao J, et al. The muscle‐specific microRNA miR‐1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13:486‐491. [DOI] [PubMed] [Google Scholar]
- 41. Zhao Y, Ransom JF, Li A, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA‐1‐2. Cell. 2007;129:303‐317. [DOI] [PubMed] [Google Scholar]
- 42. Rau F, Freyermuth F, Fugier C, et al. Misregulation of miR‐1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. 2011;18:840‐845. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y, Sun L, Zhang Y, et al. Overexpression of microRNA‐1 causes atrioventricular block in rodents. Int J Biol Sci. 2013;9:455‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ai J, Zhang R, Gao X, et al. Overexpression of microRNA‐1 impairs cardiac contractile function by damaging sarcomere assembly. Cardiovasc Res. 2012;95:385‐393. [DOI] [PubMed] [Google Scholar]
- 45. Wang L, Yuan Y, Li J, et al. MicroRNA‐1 aggravates cardiac oxidative stress by post‐transcriptional modification of the antioxidant network. Cell Stress Chaperones. 2015;20:411‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sodhi CP, Kanwar YS, Sahai A. Hypoxia and high glucose upregulate AT1 receptor expression and potentiate ANG II‐induced proliferation in VSM cells. Am J Physiol Heart Circ Physiol. 2003;284:H846‐H852. [DOI] [PubMed] [Google Scholar]
- 47. Jeppesen PL, Christensen GL, Schneider M, et al. Angiotensin II type 1 receptor signalling regulates microRNA differentially in cardiac fibroblasts and myocytes. Br J Pharmacol. 2011;164:394‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rasmussen HB, Møller M, Knaus HG, et al. Subcellular localization of the delayed rectifier K(+) channels KCNQ1 and ERG1 in the rat heart. Am J Physiol Heart Circ Physiol. 2004;286:H1300‐H1309. [DOI] [PubMed] [Google Scholar]
- 49. Kim C, Maijdi M, Xia P, et al. Non‐cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell‐derived cardiomyocytes during differentiation. Stem Cells Dev. 2010;19:783‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Johnson RD, Camelliti P. Role of non‐myocyte gap junctions and connexin hemichannels in cardiovascular health and disease: novel therapeutic targets? Int J Mol Sci. 2018;19:866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuznetsov AV, Javadov S, Sickinger S, et al. H9c2 and HL‐1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia‐reoxygenation. Biochim Biophys Acta. 2015;1853:276‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Monge C, Beraud K, Tepp S, et al. Comparative analysis of the bioenergetics of adult cardiomyocytes and nonbeating HL‐1 cells: respiratory chain activities, glycolytic enzyme profiles, and metabolic fluxes. Can J Physiol Pharmacol. 2009;87:318‐326. [DOI] [PubMed] [Google Scholar]
- 53. Guzun R, Karu‐Varikmaa G‐G, et al. Mitochondria‐cytoskeleton interaction: distribution of beta‐tubulins in cardiomyocytes and HL‐1 cells. Biochim Biophys Acta. 2011;1807:458‐469. [DOI] [PubMed] [Google Scholar]
- 54. Hescheler J, Meyer R, Plant S, et al. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res. 1991;69:1476‐1486. [DOI] [PubMed] [Google Scholar]
- 55. Wang G, Cui J, Guo Y, Wang Y, Kang L, Liu L. Cyclosporin A protects H9c2 cells against chemical hypoxia‐induced injury via inhibition of MAPK signaling pathway. Int Heart J. 2016;57:483‐489. [DOI] [PubMed] [Google Scholar]
- 56. Mao SY, Meng XY, Xu ZW, et al. The role of ZFP580, a novel zinc finger protein, in TGF mediated cytoprotection against chemical hypoxia‐induced apoptosis in H9c2 cardiac myocytes. Mol Med Rep. 2017;15:2154‐2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sovari AA, Iravanian S, Dolmatova E, et al. Inhibition of c‐Src tyrosine kinase prevents angiotensin II‐mediated connexin‐43 remodeling and sudden cardiac death. Am Coll Cardiol. 2011;58:2332‐2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang B, Cui X, Jin HH, et al. Ginsenoside Re prevents angiotensin II‐induced gap‐junction remodeling by activation of PPARγ in isolated beating rat atria. Life Sci. 2017;190:36‐45. [DOI] [PubMed] [Google Scholar]
- 59. Yasuno S, Kuwahara K, Kinoshita H, et al. Angiotensin II type 1a receptor signalling directly contributes to the increased arrhythmogenicity in cardiac hypertrophy. Br J Pharmacol. 2013;170:1384‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.