

Abstract
The photophysical studies of gum arabic (GA) in the presence of urea, 1,3-dimethylurea (DMU), tetramethylurea (TMU), guanidine hydrochloride (GuHCl), formamide (FA), acetamide (AA), and dimethyl formamide (DMF) were carried out by monitoring the emission, three-dimensional emission contour, and time-correlated fluorescence lifetime techniques. On addition of only 1 × 10–3 M urea, 75.0% of the fluorescence of GA is quenched, while the same occurs in GuHCl at 3.0 M. FA quenched 50% of the fluorescence of GA at 5.0 M. However, DMU, TMU, AA, and DMF resulted in a fluorescence enhancement. The unusual fluorescence trends reveal the existence of CH...π interactions in the proteins of GA. The experimental results and the structural aspects of proteins in GA led us to propose that the aggregation of polyproline helices in GA, through several CH...π interactions, would have a major role to play in the emulsification mechanism of GA.
Introduction
Gum arabic (GA) is recognized as an efficient emulsifying agent of the food industry. GA stabilizes the oil-in-water emulsions over a wide range of pH, temperature, and ionic strength.1,2 It is considered to be the “gold standard” for beverage emulsion manufacture as it can stabilize the concentrated and extensively diluted flavor oil emulsions.3,4 The major advantage of GA as an emulsifying agent is that it is a reliable emulsifier as the emulsions stay stable for a very long time,5 while the main disadvantage is its cost. A high gum/oil ratio (∼1:1) is required to generate the emulsions.6,7 This is because only the protein portion of the gum is directly involved during emulsification.8,9
GA is a highly branched, neutral or slightly acidic, polysaccharidic complex, containing about 2% of polypeptide. The composition, structure, conformation, and characterization of GA have been extensively explored and reported in the literature.10−20 In general, three main fractions have been isolated by hydrophobic interaction chromatography.12 The three fractions (F) are arabinogalactan-peptide or AG, arabinogalactan-protein or AGP, and glycoprotein or GP. F1 (AG) makes 88% of the gum but has the least protein content of 1.1%. It is a thin oblate ellipsoid, branched disclike structure15 with a molecular weight of 2.86 × 105 g mol–1. The branches are mainly composed of 1,3-linked β-d-galactopyranosyl units with 1,6-linked β-d-galactopyranosyl side chains to which there are linked many arabinosyl, uronic acid, and rhamnose residues,15,16 where an approximately 43 amino acid residue peptide sequence was supposed to be totally buried. Circular dichroism (CD) studies reveal that the peptide in the AG fraction had no secondary structures.21 F2 (AGP) makes 10.3% of the gum with a protein content of 9%. It is a linear chain with branched building blocks, which has been described in terms of a wattle blossom macromolecular assembly.22 The AGP consists of polysaccharide domains of Mw ∼1.8 × 106 g mol–1 held together by a short polypeptide backbone chain of approximately 250–400 amino acids. The presence of secondary structures, PPII helices, and β-strands was identified by circular dichroism studies.21 F3 (GP) makes 1.3% of the gum with a protein content of 24.6%. It consists of spheroidal ringlike monomers of hydroxyproline (Hyp)-arabinogalactan (AG) subunits and more anisotropic oligomers resulting from monomer self-association (MW, 2.95 × 105 g mol–1). At the molecular level, the GP fraction has secondary structures mainly made of β-sheets and turns but to a lesser extent of PPII and α-helices.21,23
It is reported that the AGP fraction determines the emulsifying and interfacial properties of the gum.8,9 In this study, the protein microenvironment of GA is explored to find an explanation for the emulsifying ability of GA. The urea derivatives, amide derivatives, and GuHCl are common protein denaturants. The denaturation of the proteins in GA by urea, 1,3-dimethylurea (DMU), tetramethylurea (TMU), guanidine hydrochloride (GuHCl), formamide (FA), acetamide (AA), and dimethyl formamide (DMF) was monitored using fluorescence spectral techniques.
Denaturation of many proteins by urea and guanidine hydrochloride (GuHCl) has been reported in the literature.24−26 The goal of such studies is to obtain an insight into the stabilization mechanism and the folding process of the proteins. Initial studies report two models for urea-induced denaturation. In the first one, direct interactions between urea and the protein backbone are considered.27−31 The second model is based on the indirect interactions, where urea-induced changes in the water structure drive the unfolding process.32−34 Down the years, the direct interaction model gained increased acceptance31,34−39 in the scientific community. However, recent studies performed using the molecular simulation signify that urea denatures the proteins mainly by destabilizing the hydrophobic interactions than by forming hydrogen bonds with the protein backbone.40,41 On the other hand, the unfolding mechanism of GuHCl is associated with the Coulombic interaction between GuHCl and the protein. The charged Gu+ and Cl– ions denature the protein by masking the positively and negatively charged amino acid side chains.42,43
A wide range of research activities are being carried out for many decades to understand the emulsification mechanism of GA. Many reports have suggested that the presence of aggregations and the hydrophobic interactions in the protein microenvironment of the gum could be the reason for its remarkable emulsifying properties. Herein, by exploring the protein microenvironments of GA using photophysical studies, we propose that these aggregations are primarily caused by the hydrophobic CH...π interactions. The study further elaborates on the role of CH...π interactions in the emulsifying ability of GA.
Results and Discussion
Emission Studies
Fluorescence measurements of GA in the presence of various denaturing agents were carried out (Figure 1A–F). The emission spectrum of GA exhibits a maximum at 315 nm, which is attributed to tyrosine amino acid.44,45 On addition of 1.0 × 10–3 M urea (Figure 1A), 75% of the emission intensity, centered at 315 nm, is quenched, which is accompanied with a shoulder at 350 nm and a new emission at 450 nm. Interestingly, GuHCl (Figure 1B) quenches the fluorescence gradually reaching a 75% quenching at 3.0 M. A similar observation was made for FA (Figure 1C), but only 50% of the fluorescence of GA was quenched by 5.0 M FA. The fluorescence of GA in the presence of DMU (Figure 1D), TMU (Figure S1), AA (Figure 1E), and DMF does not exhibit any quenching phenomenon, rather resulted in an enhancement of fluorescence accompanied with a significant red shift and an increasing tail region at around 450 nm, but no distinct peaks at 450 nm were observed.
Figure 1.

Emission spectra of GA (2.86 × 10–6 M) in the absence and presence of various concentrations of (A) urea, (B) GuHCl, (C) FA, (D) DMU, (E) AA, and (F) TMU.
The emission spectrum of GA in the presence of urea and GuHCl shows that [urea]3/4 = 1.0 × 10–3 M, [GuHCl]3/4 = 3.0 M, and [FA]1/2 = 5.0 M. As 88.0% of the gum is made of the AG fraction, which does not fluoresce,21 the fluorescence observed for GA is mainly attributed to the tyrosine amino acid present in AGP and GP fractions of GA. In particular, the fluorescence is reported to arise predominantly from Tyr amino acid.44,45 Had urea brought about the quenching of fluorescence by disturbing the polysaccharides in GA, a similar emission trend should be expected for FA as well because FA has a structure quite similar to that of urea. Since FA quenches the fluorescence gradually and urea quenches very rapidly, the disruptions in the polysaccharide sequence by FA and urea could not be the major reason for quenching. We rather presume that the solutes disrupt the protein sequence of GA.
The ability of the urea derivatives to disrupt the water molecules to trigger the indirect denaturing mechanism follows the order TMU > DMU > urea.46 Quenching is observed for urea, while a red shift accompanied with fluorescence enhancement is observed for DMU and TMU (Figure 1D,F). Had the quenching in urea was due to the solute indirect or direct interaction with the protein backbone, a similar observation would have been expected for DMU and TMU as well. This clearly reveals that urea is not interacting with the protein backbone but rather attacks the tyrosine amino acid present in the protein sequence directly.
A report suggests that urea denatures protein by binding favorably to hydrophobic amino acid side chains.40,41 The occurrence of 75% fluorescence quenching in GA on addition of a very low concentration of (1.0 × 10–3 M) urea clearly reveals that urea directly attacks the tyrosine amino acid in the protein sequence of the AGP and GP fractions of GA. The prominent red-shifted peak at such a low concentration of urea signifies its strong interaction with the tyrosine amino acid. The trend in the fluorescence spectra of GA in the presence of urea, FA, and GuHCl clearly reveals that urea is making a unique interaction with the aromatic amino acids of GA. To obtain a plausible explanation and a concrete mechanism for the unusual behavior of urea, the protein structure of GA was explored in detail.
The amino acid composition of GA is reported in the literature.21 Hydroxyproline (Hyp) is the most abundant followed by serine, proline (Pro), and threonine and many other amino acids in minor quantities. Because of the conformational properties of Pro and Hyp, it has been suggested that polyproline II helix (PPII) is a dominant conformation in the structure of the proline-rich regions in proteins.47 CD studies of GA reveal the presence of PPII helices.21,48 A PPII helix is a type of protein secondary structure, which occurs in proteins comprising repeating Pro and/or Hyp residues49 (Figure 2A). A left-handed PPII helix is formed when sequential residues all adopt (φ, ψ) backbone dihedral angles of roughly (−75, 150°) and have trans isomers (Figure 2A) of their peptide amide bonds.50 The PPII helix because of its extended character is relatively open and has no internal hydrogen bonding, as opposed to the more common helical secondary structures, the α-helix and β-helix.51,52 There are several reports on the importance of the PPII helix for the functional and structural machinery of peptides and proteins.47,53−56 PPII helices play a major role in protein–protein53,57 as well as protein–nucleic acid51,58 interactions.
Figure 2.
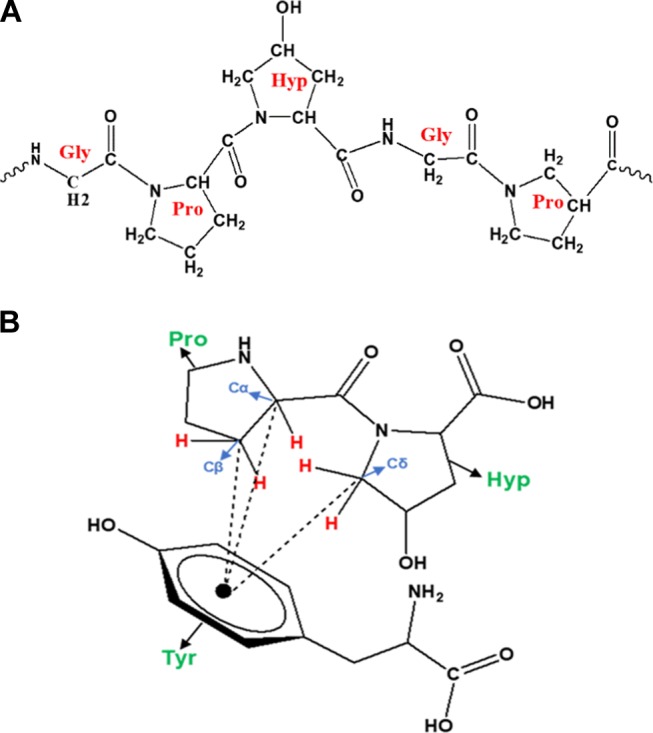
(A) PPII helix with trans amide bonds. (B) CH...π interactions between a two-amino acid (Pro and Hyp) peptide and tyrosine.
Aromatic amino acid residues (tryptophan (Trp), phenylalanine (Phe), and tyrosine (Tyr)) are relatively rare within the PPII helix, but they are known to play a unique role in the structure and function of the protein.59,60,63 Proline and aromatic amino acid residues can interact favorably with each other.64−66 When aromatic amino acid residues occur in a proline-rich sequence, there is a very high population of cis amide bonds due to multiple aromatic proline interactions.64,65,67−69 Nature selects against aromatic residues in proline-rich domains, which are usually PPII helices that accommodate only a trans amide bond.70 Hence, the possibility of the presence of aromatic residues in PPII helices is more in the terminals of a PPII helix or in adjacent protein molecules. The frequency of interactions in globular proteins increases when the Pro and aromatic residues occur in near vicinities. There are also well-documented examples in complexes where Pro is in one polypeptide chain and the aromatic amino acid in another. The high content of both Pro and Hyp (imino acids) on the surface of the collagen triple helix has been found to interact favorably with available Phe residues in adjacent molecules.61−64,71,72
The unusual affinity between proline and aromatic amino acids is termed as CH...π interactions.73,74 The proline and hydroxyproline amino acids take a ring form (Figure 2B) due to their two-point connection of the side chain to the electron-withdrawing protein backbone. The Cα and Cδ (Figure 2B) are the most acidic as they are present adjacent to the backbone amide, and they act as potent hydrogen bond donors. Thus, the electron-rich aromatic amino acids interact favorably with the partially positive Hα and Hδ. These CH...π interactions are sometimes referred to as CH...π hydrogen bonds.71,74 Since both the aromatic ring and the proline ring possess CH groups, CH...π interactions are also considered as hydrophobic interactions.73
The predominant fluorophore in GA is reported to be the tyrosine amino acid.44,45 L-tyrosine amino acid, in general, exhibits a fluorescence maximum at 305 nm and is prone to red shift due to hydrogen bonding interactions.75−79 The occurrence of fluorescence maximum at 315 nm for GA signifies that the fluorophores (Tyr, Phe) are probably involved in CH...π and π-π interactions. There are also several lines of evidence reported in the literature for the presence of molecular associations and aggregations in GA primarily in the AGP fraction. Initially, Sanchez et al. reported some rheological evidence for self-association of the molecules in aqueous solution of GA.80 Reports suggested that molecular assemblies are internal structural characteristics of AGP.81,82 The statement was further seconded by Renard et al.22 In addition, spray-drying was found to increase the molecular weight of the AGP fraction due to AGP aggregations.83 Recently, Evans et al. reported a high adsorbed amount of GA on the limolene oil droplets and it was interpreted to be due to some multilayer adsorption.84 This observation was envisaged by Dickinson85 to be due to significant intermolecular association within the adsorbed layer. The aggregations reported to be present in the AGP fraction are presumed to be the aggregations of several PPII helices occurring due to multiple CH...π interactions. The proline and hydroxyproline (imino acids) amino acids in GA together make approximately 300 residues, while the aromatic amino acids phenylalanine and tyrosine together make approximately 50 residues. Owing to the high concentration of imino acids, it can be presumed that all of the aromatic amino acids are involved in the favorable CH... π interactions. The fluorescence emission trends of GA in the presence of various solutes further support the assumption.
Recent studies suggest that urea denatures the protein by binding favorably to hydrophobic side chains.40,41 According to the mechanism, urea molecules replace water in the first shell around peptides. Urea has a planar distribution of charges, which enables it to occupy positions that are inaccessible to water.87,88 Hence, it is predicted that urea unfolds the protein by masking the hydrophobic interactions between the aliphatic and aromatic amino acid side chains.
The interactions between hydrophobic CH groups present in the hydrocarbon molecules are referred to as hydrophobic interactions. The interactions between aliphatic amino acids and aromatic amino acids fall into this category. The hydrophobic molecules, as the name suggests, detest water molecules and hence group together by inducing interactions among themselves. The hydrophobic interaction can be amplified by the π...π interaction, electrostatic interaction, hydrogen bonding interaction, and donor–acceptor interaction. The aromatic amino acids have a delocalized π electron cloud. Like charges repel, but these π...π interactions are favorable because a dipole with a partial positive charge and negative charges may be induced for a favorable interaction to take place. In GA, the aromatic amino acids are involved in π...π interactions among themselves and also in CH...π interactions with the Pro and Hyp iminoacids. The CH...π interactions are also hydrophobic interactions because they basically take place between CH groups. However, here there is no requirement to induce a dipole because there already exists a dipole between the π electron cloud and the partial positive charges of the CH groups in the proline amino acid. Therefore, these interactions could be more favorable than the π...π interactions. It should be for the same reason that urea, which has a dipole existing in its structure, is able to make a unique interaction with the aromatic amino acid side chains in GA.
In the structure of urea, the two NH2 moieties and the carbonyl carbon become partially positive due to the electron-withdrawing carbonyl group. The carbonyl oxygen bears a partial negative charge. The distribution of partial positive charges over the two NH2 moieties and the carbonyl carbon forms a positive cloud (Figure 3A), which is able to effectively interact with the delocalized electron cloud present in the aromatic amino acids. The CH groups present in the proline amino acid are reported to involve in favorable CH...π interactions with the aromatic amino acids and also in CH...O interactions with the carbonyl oxygen.89 The CH...π interactions are stronger than CH...O interactions. Two aromatic amino acids involved in π...π interactions rip apart on addition of urea. Each aromatic amino acid individually interacts with the positive cloud of the urea molecules. Hence, two interacting aromatic amino acids rip with ease by two urea molecules. In the case of CH...π interactions, the aromatic amino acid involved in CH... π interactions with the proline amino acid rips apart with ease by a single urea molecule. The urea molecules while interacting with its positive cloud with the π electron cloud of the aromatic amino acid simultaneously form a CH...O interaction with its carbonyl oxygen and the CH groups of the proline amino acid. Hence, we can conclude that urea is more effective in destabilizing the hydrophobic CH...π interactions than the hydrophobic π...π interactions. The crowding of many aromatic amino acids around a single proline amino acid, due to the favorable CH... π interactions, is also reported in the literature.89 In such a situation, urea, which is capable of effectively destabilizing the CH... π interactions, will have opportunity to sit amid several aromatic amino acids. In GA, the occurrence of rapid quenching at a low concentration of 1 × 10–3 M can be explained to be due to the presence of several aromatic amino acids within the quenching radius of urea molecules. Thus, the simultaneous effective binding of urea with aromatic amino acids and proline amino acids and the possibility of the existence of several aromatic amino acids (signified by the aggregations present in GA) within the quenching radius of a fewer urea molecules should be the reason for the observed remarkably efficient quenching.
Figure 3.
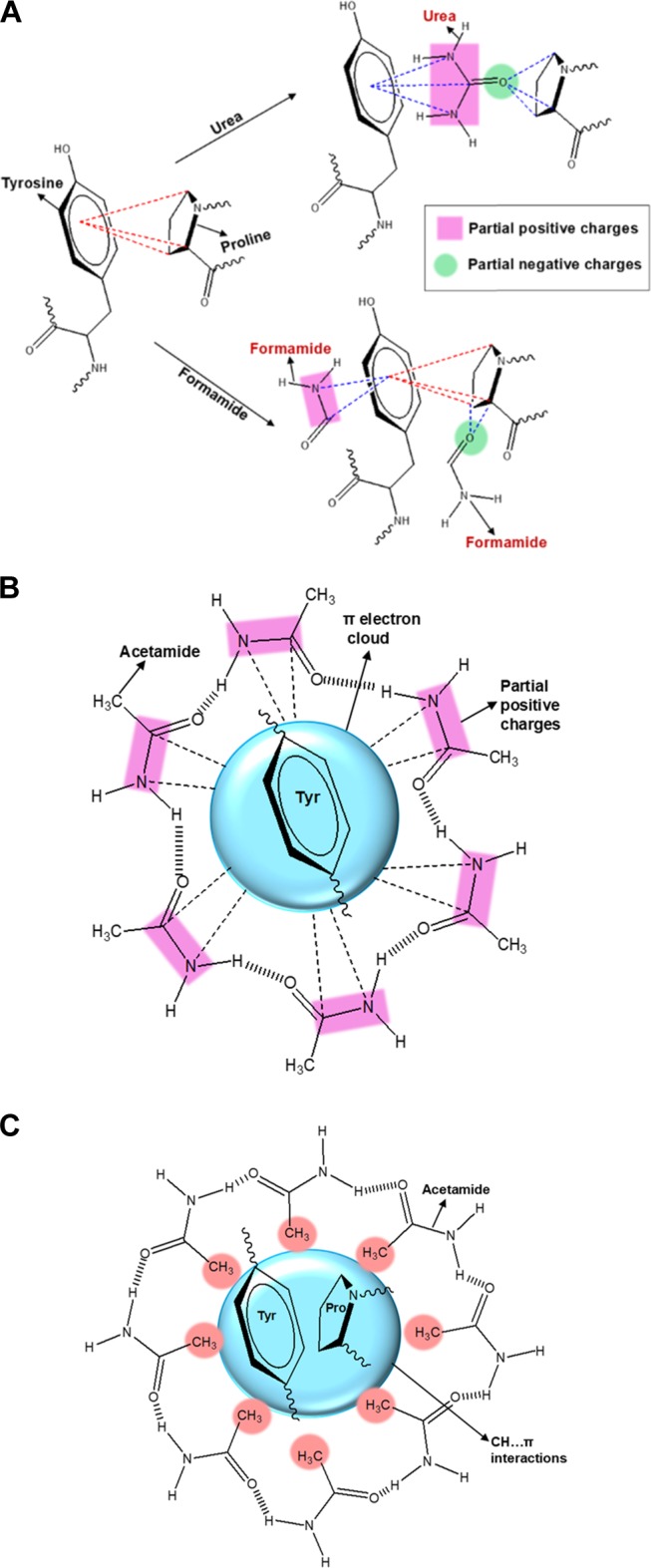
(A) Interaction of urea and FA with amino acids involved in CH...π interactions: red dotted lines, CH...π interactions; blue dotted lines, solute interaction with amino acids. (B) Acetamide interaction with Tyr present in the proteins where CH...π interactions are not possible. (C) Acetamide interaction with Tyr present in the proteins of GA involved in CH...π interactions.
A similar coexistence of amino and carbonyl groups is observed in FA as well, which results only in gradual quenching of fluorescence of GA (Figure 1C). Only a single carbonyl group is present in both urea and FA. Two amino groups are present in urea, while FA possesses only one amino group. Hence, the observation of rapid quenching for urea can be attributed to the presence of two amino groups in its structure. A big positive plane can be generated in urea due to the distribution of partial positive charges over three atoms, two amino nitrogen atoms and one carbonyl carbon atom (Figure 3A). FA can generate only a small positive plane as the partial positive charges are distributed over two atoms only, one amino nitrogen atom and one carbonyl carbon atom (Figure 3A). Hence, the presence of a big positive cloud in urea enables it to bind like a glue with the π electron cloud of the aromatic amino acid. However, the interactions between the small positive cloud of FA and the π cloud of the aromatic amino acid will not be effective enough. For the same reason, FA will not be able to rupture the CH...π interaction as easily as urea (Figure 3A) because the three CH groups Cα, Cβ, and Cδ of the proline amino acid bear a partial positive charge.36 Hence, the strong interactions between urea and the aromatic acids is the primary reason for the efficient rupturing of the CH...π interactions and also for the rapidly quenching fluorescence.
In solution, GuHCl exists as a stable guanidinium cation and chloride anion. The gradual quenching of fluorescence by GuHCl (Figure 1B) reveals that the guanidinium cation does not effectively pair up with the π electron cloud of the aromatic ring. On comparing the structural aspects of urea, FA, and GuHCl, it becomes evident that the aromatic π electron cloud requires a positive cloud (and not a cation) created by the distribution of partial positive charges over at least three atoms to make an effective interaction, and only urea was found to possess such an assembly.
Addition of AA, which has a structure similar to that of FA, except for the presence of a hydrophobic methyl group in place of hydrogen, results in enhancement of fluorescence even at very low concentrations (Figure 1E). A similar trend was observed for DMF as well. Even the urea derivatives DMU and TMU (Figure 1D,F) enhance the fluorescence of GA. Earlier studies on the BSA protein fluorescence in the presence of DMU90 and AA90 have reported a quenching phenomenon. Fluorescence studies of N-acetyl-l-tyrosinamide (a model for Tyr present in proteins) report a gradual quenching by urea and methyl acetamide.91 In the N-acetyl-l-tyrosinamide system, only Tyr amino acids are present and only π...π aromatic interactions are possible. Hence, the π electron cloud in the Tyr amino acid favorably interacts with the partial positive charges on the NH2 groups of urea and methyl acetamide (Figure 3B), resulting in quenching of the fluorescence. In GA, the aromatic amino acids are involved in π...π interactions but to a greater extent are involved in favorable CH...π interactions. From the emission results, it is obvious that only urea is able to perturb the CH...π interactions efficiently and results in rapid quenching. GuHCl and FA, as they do not possess any hydrophobic groups, are able to gradually quench the fluorescence. On addition of AA and DMU, hydrophobic methyl-group-containing systems are introduced into the microenvironment of GA.
As conveyed by the emission trends in the presence of various solutes, the favorable CH...π interactions would not desire to part that easily (unless they have the big positive plane containing urea in the medium); hence, they attract the methyl groups present in the solutes toward them to safeguard their favorable interaction forces. The hydrophobic microenvironment created by the accumulation of AA not only safeguards the CH... π interactions but also slightly shields the fluorophores from neighboring groups that quench the fluorescence, and as a result, AA and DMU enhance the fluorescence of GA (Figure 3C). On this basis, we can emphasize that on adding GA to the oil/water system, the amino acids involved in the hydrophobic CH...π interactions are naturally attracted toward the hydrophobic oil phase, while the hydrophilic polysaccharides protrude into the water phase, thereby forming stable emulsions.
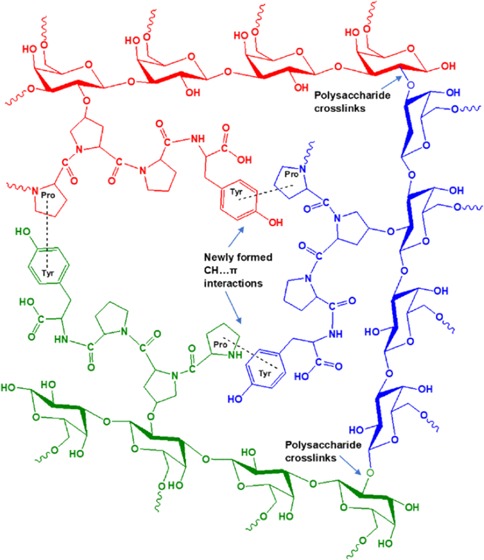
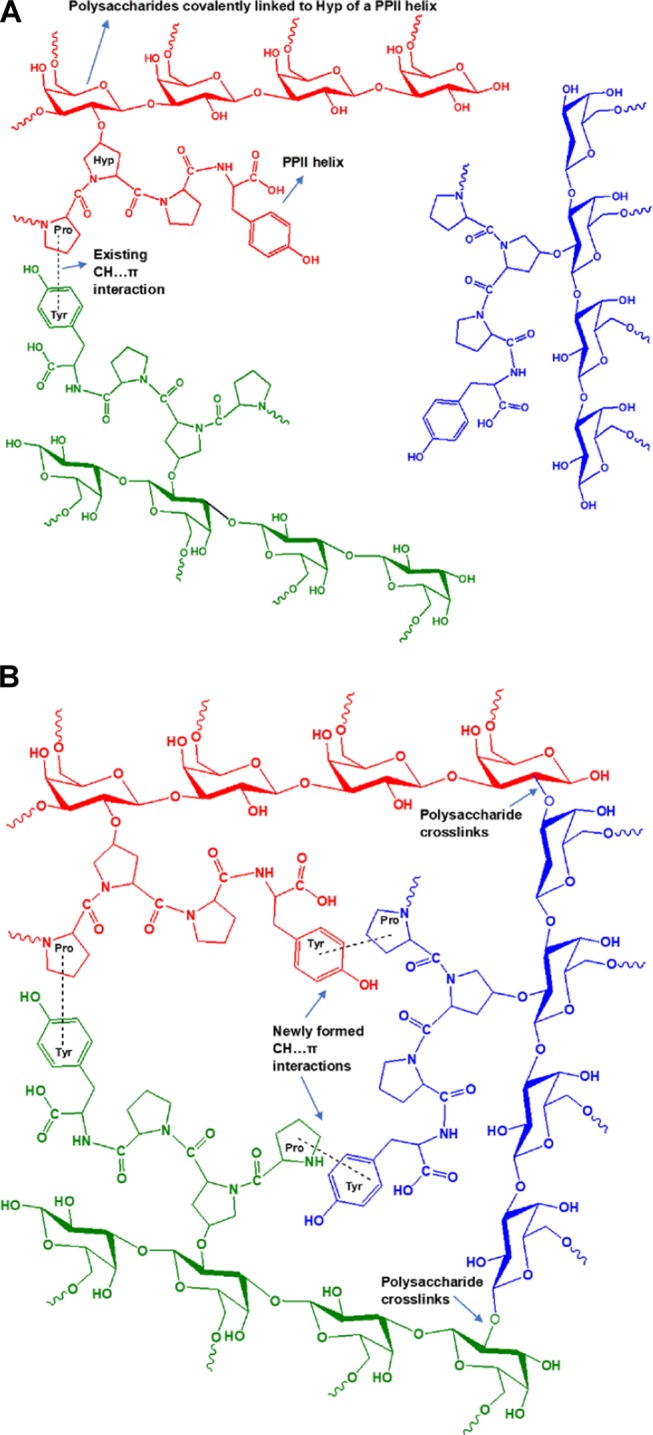
Phillips and his collaborators developed a series of “Supergum” products by subjecting the conventional GA to maturation technology.92,93 It is assumed that on maturation the proteinaceous material in GA is aggregated and that through the formation of several cross-links between the polysaccharide units, the low-molecular-weight AG fraction and the GP fraction are linked to the high-molecular-weight AGP fraction, thereby increasing the overall concentration and the molecular weight of AGP. We speculate that initially there exist aggregations, but all of the PPII helices and other protein secondary structures are not in an aggregated state due to the intervening highly branched polysaccharide units. During maturation under controlled conditions of temperature and pressure, the agitation can bring about an increase in the aggregation of polypeptides, which in turn may trigger the formation of cross-links between the polysaccharide units. The cross-linked polysaccharide units may relieve some stress, making room for more polypeptides to aggregate, and the process should go on, ultimately resulting in an increased concentration and molecular weight of the AGP fraction. This aggregated matured gum is reported to have substantial enhancement in the emulsification properties.93 We have proposed that these aggregations are occurring predominantly due to CH...π interactions. The amino acids involved in CH...π interactions have a unique affinity for the hydrophobic groups as confirmed by the emission trends in DMU and AA. The greater the number of CH...π interactions, the greater the affinity for hydrophobic groups and hence an increased emulsification ability. Therefore, we conclude that the aggregation of proteins in GA has a crucial role to play in the emulsification efficiency of the gum.
We have proposed two schemes to depict the microenvironment of GA before maturation (Scheme 1A) and after the maturation process (Scheme 1B). The scheme brings to light how the CH...π interactions formed during the maturation process can be the cause of aggregation and cross-linking in GA. Studies on various properties of the matured gums are reported in the literature, but the fluorescence properties are yet to be explored. A comparative fluorescence spectral study of naturally matured and industrially matured gums will reveal important insights regarding the emulsifying mechanism of GA.
Scheme 1. (A) GA Microenvironment before the Maturation Process; (B) GA Microenvironment after the Maturation Process.
In the emission spectrum of GA with increasing concentrations of urea (Figure 1A), the peak at 315 nm along with quenching shows a red shift to around 350 nm and a new emission is also observed at 450 nm. A similar trend is observed in the emission spectra for GA with increasing concentrations of GuHCl and FA (Figure 1B,C). The emission spectra of GA in DMU (Figure 1D), TMU (Figure 1F), AA (Figure 1E), and DMF also show a prominent red shift from 315 to around 350 nm, but there is no new emission at 450 nm. It has been reported that an emission above 400 nm accompanied with high lifetimes for protein molecules on denaturation confirms the existence of fluorescing amino acids in a heterogeneous environment.94−98 GA is a polysaccharide–protein complex. The microdomains of GA are heterogeneous due to the presence of various sugars and amino acids. Hence, the emission at 450 nm, observed for GA in the presence of urea, FA, and GuHCl, is attributed to the amino acid fluorophores exposed to the heterogeneous environments in GA due to denaturation, which was further authenticated by the three-dimensional (3D) emission contour studies.
The predominant fluorophore in GA is the Tyr amino acid.45 The red shift in the emission maximum (315 to around 350 nm) on addition of solute is attributed to the formation of hydrogen bonds between the −OH group in Tyr and the C=O or NH2 groups in the solutes.75−79 The lifetime decay analysis supports the formation of tyrosine hydrogen bonding with the solute molecules.
The emission spectrum of GA in the presence of TMU (Figure 1F) exhibited a remarkable enhancement of fluorescence accompanied with a significant red shift, which is attributed to the hydrophobic interaction between the methyl groups in TMU and the aromatic amino acids, creating a water-less hydrophobic cavity around the fluorophore. The inaccessibility of the fluorophore and hence the impossibility of fluorescence quenching by the water molecules and the nearby groups should explain the remarkably high fluorescence enhancement. A similar explanation should hold good for DMU, AA, and DMF as well. These solutes possess fewer methyl groups; hence, they create a hydrophobic environment but not a cavity and therefore result in moderate enhancement of fluorescence. The fluorescence trends for GA in the presence of DMU and TMU are further ascertained by the 3D emission contour studies.
Lifetime Studies
The fluorescence decay of aqueous solution of GA in the presence of urea (Figure 4A), GuHCl (Figure S7), DMU (Figure 4B), TMU (Figure 4C), and AA (Figure S8) was measured, and the corresponding decay analyses are provided in the supporting information (Tables S1–S5). The excitation wavelength was fixed at 280 nm and the decay wavelength at 315 nm. The fluorescence decay of GA is triexponential in the absence and presence of various denaturing agents. For GA, taken as such, the lifetimes of the three components are 0.11 (τ1), 0.97 (τ2), and 2.88 (τ3) ns (Table S1) and the relative amplitude percentages are 19.0 (A1), 70.0 (A2), and 11.0 (A3) %, respectively. On addition of urea (1.0 × 10–2 M), the relative amplitude percentages of the three components are 30.0 (A1), 35.0 (A2), and 31.0 (A3) % (Table S1). In the presence of urea, A2 has decreased drastically. With increasing concentrations of urea, the amplitude % does not show much variation. On addition of GuHCl [1.0 × 10–1 M], the relative amplitude percentages of the three components are 14.0 (A1), 74.0 (A2), and 13.0 (A3) %, respectively (Table S2). The amplitude % was almost similar to that of GA alone. With increasing concentrations of GuHCl, A2 decreases gradually to 34.0% at 5.0 M. The individual component lifetimes and the average lifetimes show only slight changes with increasing concentrations of both the denaturants. The trend observed in the amplitude distribution in the presence of these two denaturants reflects the trend in the emission spectrum.
Figure 4.
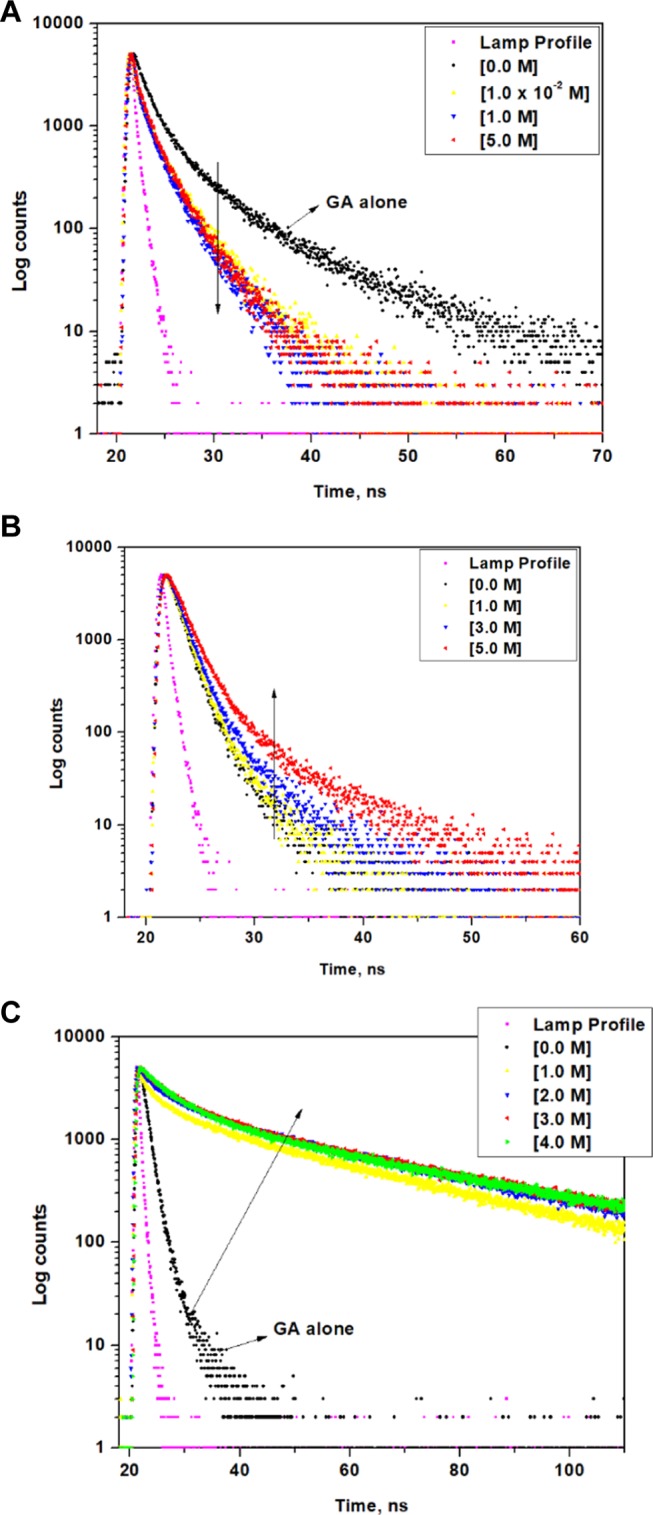
(A) Fluorescence decay of GA in the absence and presence of various concentrations of (A) urea, (B) DMU, and (C) TMU.
On addition of TMU (1.0 M) to GA, the lifetimes of the three components are 1.11(τ1), 4.51(τ2), and 26.43(τ3) ns and the relative amplitude percentages are 10.0 (A1), 14.0 (A2), and 76.0 (A3) %, respectively (Table S4). With increasing concentrations of TMU, the amplitude % shows only appreciable changes. The lifetime data for GA in TMU (Table S4) clearly reveals the formation of a high lifetime component (τ1), which supports the remarkable enhancement in the emission spectrum and the observation of single emitting region in the contour spectrum very well.
In all of the lifetime analysis tables, there is one common trend observed. The relative amplitude of component two (A2) decreases. In the case of urea and GuHCl, there is a simultaneous increase in component one (A1) and component three (A3). At 5 M urea and GuHCl, there is almost an equal distribution of A1, A2, and A3. In the case of DMU, TMU, and AA, the decrease in A2 is associated with a greater increase in A1 than in A3. In the emission spectrum of GA in the presence of urea and GuHCl, at 5 M, three distinct peaks (first at 315 nm, second at around 350 nm, and third at around 450 nm) are observed. However, the emission trends in the presence of DMU, TMU, and AA show a peak at 315 nm, which is red-shifted at higher concentrations to around 350 nm, and only an increasing tail region at around 450 nm, but no distinct peaks are observed at around 450 nm. The common red-shifted peak at around 350 nm observed in the emission spectrum of GA in the presence of all of the solutes is attributed to the tyrosine amino acid involved in hydrogen bonding interactions with the solute molecules. Similarly, in the decay analysis, the increase in A1 is a common observation for all of the solutes and so it is correlated to the tyrosine amino acid involved in hydrogen bonding. A big increase in A3 has occurred only for urea and GuHCl, whose corresponding emission spectra also show an evolution of a distinct peak at around 450 nm. As stated earlier, the tyrosine amino acid when exposed to a heterogeneous environment due to denaturation exhibits an unusual red-shifted emission associated with a high lifetime. The corresponding lifetime of A3 (τ3) for GA even in the absence of any solute is relatively high in comparison with that of τ2 and τ1. The τ3 values of GA in the presence of various solutes register even higher lifetime values. Hence, component A3 is correlated to the tyrosine amino acid exposed to the heterogeneous aqueous phase. Component A2 that is present at high percentage in GA alone (A2 = 70%) is correlated to the tyrosine amino acid in its native state in GA.
Three-dimensional Emission Contour Studies
To gain further insight into the microenvironment, 3D emission contour studies were performed for GA in the presence of various solutes. In the contour spectra of GA alone (Figure 5A), two major regions are observed. The first region (R1) corresponds to the excitation centered at 280 nm and emission centered at 315 nm. The second region (R2) corresponds to the excitation centered at around 400 nm and emission at 450 nm. R1 shows a strong fluorescence intensity, and R2 shows a weak fluorescence intensity. On addition of (1.0 × 10–3 M) urea (Figure 5B), the fluorescence intensity decreases at R1 and increases at R2. Thus, the fluorescence at R2 arises from fluorophores in the denatured protein exposed to the heterogeneous environment. A similar observation was made for GA in the presence of GuHCl (Figure S1).
Figure 5.
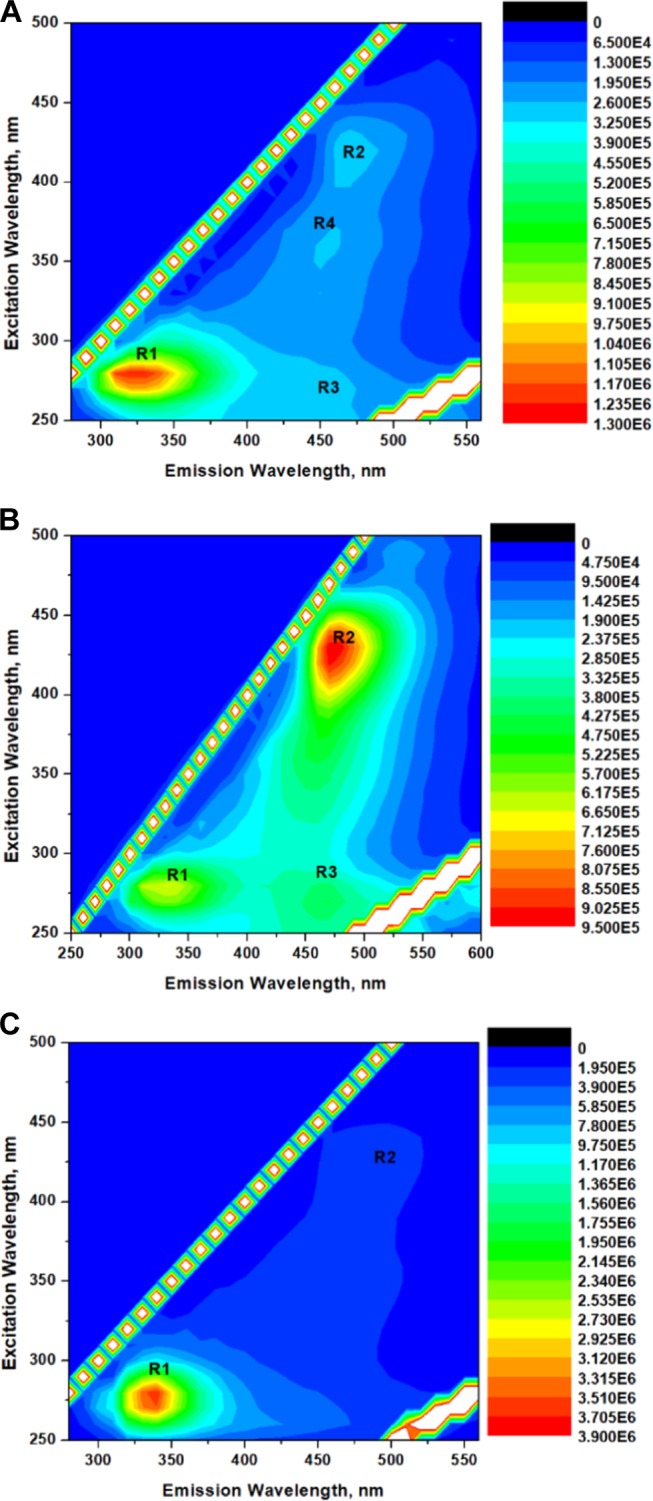
(A) Three-dimensional (3D) emission contour spectrum of GA (2.86 × 10–6 M). Three-dimensional (3D) emission contour spectra of GA (2.86 × 10–6 M) in the presence (B) of urea (1.0 × 10–3 M) and (C) of TMU (1 × 10–1 M).
In the contour spectra of GA in the presence of DMU (Figures S2–S4), the intensity of R1 increases and that of R2 decreases with increasing concentrations of DMU. On addition of TMU to GA (Figures 5C and S5,S6), even at low concentrations (Figure 5C), the region R2 completely disappears and only one region R1 is present. The observation of only one emitting region on addition of TMU signifies the aggregation of TMU (through the hydrophobic -CH3 groups) around the hydrophobic aromatic amino acids, creating a homogeneous microenvironment around the fluorophore. This phenomenon ascertains the fact that when the microenvironment around the aromatic amino acid is same, it emits in one region only, but when it is exposed to a heterogeneous microenvironment, the tyrosine amino acid shows several unusual red-shifted emissions. In the contour spectra of GA in the presence of DMU (Figure S2), the intensity in R2 is higher than that in GA alone (Figure 5A). This reveals that DMU denatures the proteins and the fluorophores are exposed to the heterogeneous phase, but as the concentration of DMU increases, the immediate environment of the fluorophore becomes homogeneous and detached from the external environment. As a result, we observe enhancement in the emission spectra and a single emitting region in the contour spectra. Hence, it becomes obvious that DMU and TMU safeguard the aromatic amino acids in the proteins, while they still denature the proteins by interacting with the protein backbone probably through an indirect mechanism.
Conclusions
The present investigation examines the fluorescence spectral properties of GA in the presence of urea, DMU, TMU, FA, AA, and DMF. The quenching phenomenon observed for GA in the presence of urea, FA, and GuHCl is attributed to the interaction between the aromatic π system, present in the tyrosine amino acid in the proteins of GA, and the partially positive NH2 groups in urea, FA, and GuHCl. The enhancement of fluorescence observed for GA in the presence of DMU, TMU, AA, and DMF is attributed to the hydrophobic interactions between the aromatic system of the tyrosine amino acid and the CH3 groups in DMU, TMU, AA, and DMF. The two unusual trends, that is, (i) the rapid quenching of fluorescence of GA by urea and (ii) the enhancement of fluorescence of GA by AA and DMU, enabled us to reveal the presence of CH...π interactions in GA. The affinity of the amino acids involved in CH...π interactions for the hydrophobic groups, as confirmed by the enhancement of fluorescence of GA by AA, is speculated to be the major driving force for the emulsification ability of GA. The increase in efficiency of the emulsification properties of the matured GA was owed to the increased aggregations of PPII helices through the favorable CH...π interactions during the maturation process.
Materials and Experimental Methods
Sample Preparation
The acacia gum (gum arabic) laboratory-grade sample (CAS No 9000-01-05) was purchased from Merck. A 2.0% (w/v) solution of GA was prepared. Urea (molecular biology grade) was obtained from Merck Chemicals, India. The urea derivatives 1,3-dimethylurea and tetramethylurea were purchased from Alfa Aesar Chemicals, U.K. GuHCl was purchased from Merck. FA and DMF were obtained from Qualigens, India Ltd. AA was purchased from Sigma-Aldrich. Solutions of GA and the solutes were prepared in triple distilled water.
Steady-State Fluorescence Measurements
The fluorescence emission and 3D spectral measurements were performed in a Fluoromax 4 P spectrofluorimeter (Horiba Jobin Yvon) using fluorescence software provided by the manufacturer. 3D contour plots were obtained by simultaneously scanning the excitation and emission monochromators.
Time-Correlated Single-Photon Counting Technique
The fluorescence decay measurements of GA solutions were recorded using an IBH time-correlated single-photon counting spectrometer with a microchannel plate photomultiplier tube (MCP-PMT) (Hamamatsu R3809U) as a detector and a 280 nm LED (Spectra Physics) as an excitation source.
Acknowledgments
The authors thank Prof. Dr P. Ramamurthy and Dr C. Selva-raju of the National Centre for Ultrafast Processes (NCUFP), University of Madras, Taramani campus, Chennai, for permitting and assisting us to avail the instrument facilities. The authors also thank Principal, Dr R. Ganesan, D.G. Vaishnav College (Autonomous), Chennai, and Shri. Ashok Kumar Mundra, Secretary, D.G. Vaishnav College (Autonomous), Chennai, for permitting us to avail the laboratory and instrumentation facilities.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01980.
Detailed information of the materials and experimental methods, 3D emission contour figures, fluorescence decay charts, and decay analysis tables (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chanamai R.; McClements D. J. Comparison of gum arabic, modified starch, and whey protein isolate as emulsifiers: Influence of pH, CaCl2 and temperature. J. Food Sci. 2002, 67, 120–125. 10.1111/j.1365-2621.2002.tb11370.x. [DOI] [Google Scholar]
- Charoen R.; Jangchud A.; Jangchud K.; Harnsilawat T.; Naivikul O.; McClements D. J. Influence of biopolymer emulsifier type on formation and stability of rice bran oil-in-water emulsions: Whey protein, gum arabic, andmodified starch. J. Food Sci. 2011, 76, 165–172. 10.1111/j.1750-3841.2010.01959.x. [DOI] [PubMed] [Google Scholar]
- Piorkowski D. T.; McClements D. J. Beverage emulsions: Recent developments in formulation, production and applications. Food Hydrocolloids 2014, 42, 5–41. 10.1016/j.foodhyd.2013.07.009. [DOI] [Google Scholar]
- Schultz M.Industry Requirements for Hydrocolloids in Beverage Emulsions. In Gums and Stabilisers for the Food Industry-15; Williams P. A., Phillips G. O., Eds.; Royal Society of Chemistry: Cambridge, U.K., 2010; pp 257–266. [Google Scholar]
- Tan C.-T.Beverage Emulsions. In Food Emulsions; 4th ed.; Friberg S. E., Larsson K., Sjöblom J., Eds.; Marcel Dekker: NY, 2004; Vol. 524, p 485. [Google Scholar]
- McNamee B. F.; O’Riordan E. A.; O’Sullivan M. Emulsification and encapsulation properties of gum arabic. J. Agric Food Chem. 1998, 46, 4551–4555. 10.1021/jf9803740. [DOI] [Google Scholar]
- Nakauma M.; Funami T.; Noda S.; Ishihara S.; Al-Assaf S.; Nishinari K.; et al. Comparison of sugar beet pectin, soybean soluble polysaccharide, and gum arabic as emulsifiers. 1. Effect of concentration, pH and salts on the emulsifying properties. Food Hydrocolloids 2008, 22, 1254–1267. 10.1016/j.foodhyd.2007.09.004. [DOI] [Google Scholar]
- Randall R. C.; Phillips G. O.; Williams P. A. The role of the proteinaceous component on the emulsifying properties of gum arabic. Food Hydrocolloids 1988, 2, 131–140. 10.1016/S0268-005X(88)80011-0. [DOI] [Google Scholar]
- Ray A. K.; Bird P. B.; Iacobucci G. A.; Clark B. C. Jr. Functionality of gum arabic: Fractionation, characterization and evaluation of gum fractions in citrus oil emulsions and model beverages. Food Hydrocolloids 1995, 9, 123–131. 10.1016/S0268-005X(09)80274-9. [DOI] [Google Scholar]
- a Jurasek P.; Kosik M.; Phillips G. O. The classification of natural gums. Part II. Characterisation of the gum arabic of commerce based on a chemometric study of amino-acid compositions. Food Hydrocolloids 1993, 7, 157–174. 10.1016/S0268-005X(09)80167-7. [DOI] [Google Scholar]; b Jurasek P.; Kosik M.; Phillips G. O. The classification of natural gums. III. Acacia senegal and related species (gum arabic). Food Hydrocolloids 1993, 7, 255–280. 10.1016/S0268-005X(09)80176-8. [DOI] [Google Scholar]
- Snowden M. J.; Phillips G. O.; Williams P. A. Functional characteristics of gum arabic. Food Hydrocolloids 1987, 1, 291–300. 10.1016/S0268-005X(87)80017-6. [DOI] [Google Scholar]
- a Randall R. C.; Phillips G. O.; Williams P. A. The role of the proteinaceous component on the emulsifying properties of gum Arabic. Food Hydrocolloids 1988, 2, 131–140. 10.1016/S0268-005X(88)80011-0. [DOI] [Google Scholar]; b Randall R. C.; Phillips G. O.; Williams P. A. Fractionation and characterization of gum from Acacia Senegal. Food Hydrocolloids 1989, 3, 65–75. 10.1016/S0268-005X(89)80034-7. [DOI] [Google Scholar]
- a Li X.; Fang Y.; Al-Assaf S.; Phillips G. O.; Nishinari K.; Zhang H. Rheological Study of Gum Arabic Solutions: Interpretation Based on Molecular Self-Association. Food Hydrocolloids 2009, 23, 2394–2402. 10.1016/j.foodhyd.2009.06.018. [DOI] [Google Scholar]; b Li X.; Fang Y.; Zhang H.; Nishinari K.; Al-Assaf S.; Phillips G. O. Rheological properties of gum arabic solution: From Newtonianism to thixotropy. Food Hydrocolloids 2011, 25, 293–298. 10.1016/j.foodhyd.2010.06.006. [DOI] [Google Scholar]
- Yadav M. P.; Igartuburu J. M.; Yan Y.; Nothnagel E. U. Chemical investigation of the structural basis of the emulsifying activity of gum Arabic. Food Hydrocolloids 2007, 21, 297–308. 10.1016/j.foodhyd.2006.05.001. [DOI] [Google Scholar]
- Sanchez C.; Schmitt C.; Kolodziejczyk E.; Lapp A.; Gaillard C.; Renard D. The acacia gum arabinogalactan fraction is a thin oblate ellipsoid: a new model based on small-angle neutron scattering and ab initio calculation. Biophys. J. 2008, 94, 629–639. 10.1529/biophysj.107.109124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D. M. W.; Stoddart J. F. Studies on uronic acid materials. Carbohydr. Res. 1966, 2, 104–114114. 10.1016/S0008-6215(00)81474-3. [DOI] [Google Scholar]
- Daoub R. M. A.; Elmubarak A. H.; Misran M.; Hassan E. A.; Osmana M. E. Characterization and functional properties of some natural Acacia gums. J. Saudi Soc. Agric. Sci. 2018, 17, 241–249. 10.1016/j.jssas.2016.05.002. [DOI] [Google Scholar]
- Grein A.; da Silva B. C.; Wendel C. F.; Tischer C. A.; Sierakowski M. R.; Dewes Moura A. B.; Iacomini M.; Gorin P. A. J.; Simas-Tosin F. F.; Riegel-Vidotti I. C. Structural characterization and emulsifying properties of polysaccharides of Acacia mearnsii de Wild gum. Carbohydr. Polym. 2013, 92, 312–320. 10.1016/j.carbpol.2012.09.041. [DOI] [PubMed] [Google Scholar]
- Martínez M.; Sanabria L.; de Pinto G. L.; Igartuburu J. M. 1D- and 2D-NMR spectroscopy studies of the polysaccharide gum from Spondias purpurea var. lutea. Food Hydrocolloids 2005, 19, 37–43. 10.1016/j.foodhyd.2003.09.007. [DOI] [Google Scholar]
- Anderson D. M. W.; Weiping W. Gum Arabic (Acacia Senegal) from Uganda: characteristic N.M.R. spectra, amino acid composition, and gum/oil cationic relationship. Int. Tree Crops J. 1992, 7, 167–179. 10.1080/01435698.1992.9752915. [DOI] [Google Scholar]
- Renard D.; Lavenant-Gourgeon L.; Ralet M. C.; Sanchez C. Acacia senegal gum: continuum of molecular species differing by their protein to sugar ratio, molecular weight, and charges. Biomacromolecules. 2006, 7, 2637–2649. 10.1021/bm060145j. [DOI] [PubMed] [Google Scholar]
- Renard D.; Garnierb C.; Lappc A.; Schmittd C.; Sancheze C. Corrigendum to “Structure of arabinogalactan-protein from Acacia gum: From porous ellipsoids to supramolecular architectures” [Carbohydr. Polym. 2012, 90, 322–332]. Carbohydr. Polym. 2013, 97, 864–867. 10.1016/j.carbpol.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Renard D.; Lepvrierb E.; Garnierb C.; Roblinc P.; Nigend M.; Sanchezd C. Structure of glycoproteins from Acacia gum: An assembly of ring like glycoproteins modules. Carbohydr. Polym. 2014, 99, 736–747. 10.1016/j.carbpol.2013.08.090. [DOI] [PubMed] [Google Scholar]
- Tanford C.; Kawahara K.; Lapanje S. Proteins in 6 M guanidine hydrochloride. Demonstration of random coil behaviour. J. Biol. Chem. 1966, 241, 1921–1923. [PubMed] [Google Scholar]
- Greene R. F.; Pace C. N. Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, a-chymotrypsin, and b-lactoglobulin. J. Biol. Chem. 1974, 249, 5388–5394. [PubMed] [Google Scholar]
- Fersht A.Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W. H. Freeman: NY, 1999. [Google Scholar]
- Aastrand P.-O.; Wallqvist A.; Karlstrom G. Molecular-dynamics simulations of 2-M aqueous urea solutions. J. Phys. Chem. A 1994, 98, 8224–8233. 10.1021/j100084a046. [DOI] [Google Scholar]
- Tirado-Rives J.; Orozco M.; Jorgensen W. L. Molecular Dynamics Simulations of the Unfolding of Barnase in Water and 8 M Aqueous Urea. Biochemistry. 1997, 36, 7313–7329. 10.1021/bi970096i. [DOI] [PubMed] [Google Scholar]
- Grdadolnik J.; Mare′chal Y. Urea and Urea–Water Solutions—an Infrared Study. J. Mol. Struct. 2002, 615, 177–189. 10.1016/S0022-2860(02)00214-4. [DOI] [Google Scholar]
- Mountain R. D.; Thirumalai D. Molecular Dynamics Simulations of End-to-End Contact Formation in Hydrocarbon Chains in Water and Aqueous Urea Solution. J. Am. Chem. Soc. 2003, 125, 1950–1957. 10.1021/ja020496f. [DOI] [PubMed] [Google Scholar]
- Klimov D. K.; Straub J. E.; Thirumalai D. Aqueous urea solution destabilizes Aβ16–22 oligomers. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 14760–14765. 10.1073/pnas.0404570101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank H. S.; Franks F. Structural Approach to the Solvent Power of Water for Hydrocarbons; Urea as a Structure Breaker. J. Chem. Phys. 1968, 48, 4746–4757. 10.1063/1.1668057. [DOI] [Google Scholar]
- Finer E. G.; Franks F.; Tait M. J. Nuclear magnetic resonance studies of aqueous urea solutions. J. Am. Chem. Soc. 1972, 94, 4424–4429. 10.1021/ja00768a004. [DOI] [Google Scholar]
- Hoccart X.; Turrell G. Raman spectroscopic investigation of the dynamics of urea–water complexes. J. Chem. Phys. 1993, 99, 8498–8503. 10.1063/1.465626. [DOI] [Google Scholar]
- Chitra R.; Smith P. E. Molecular Dynamics Simulations of the Properties of Cosolvent Solutions. J. Phys. Chem. B 2002, 106, 1491–1500. 10.1021/jp011462h. [DOI] [Google Scholar]
- Caballero-Herrera A.; Nordstrand K.; Berndt K. D.; Nilsson L. Effect of urea on peptide conformation in water: molecular dynamics and experimental characterization. Biophys. J. 2005, 89, 842–857. 10.1529/biophysj.105.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenbrink C.; van Gunsteren W. F. Methane clustering in explicit water: effect of urea on hydrophobic interactions. Phys. Chem. Chem. Phys. 2005, 7, 53–58. 10.1039/b413167c. [DOI] [PubMed] [Google Scholar]
- Lee M.-E.; van der Vegt N. F. A. Does Urea Denature Hydrophobic Interactions?. J. Am. Chem. Soc. 2006, 128, 4948–4949. 10.1021/ja058600r. [DOI] [PubMed] [Google Scholar]
- O’Brien E. P.; Dima R. I.; Brooks B.; Thirumalai D. Interactions between hydrophobic and ionic solutes in aqueous guanidinium chloride and urea solutions: lessons for protein denaturation mechanism. J. Am. Chem. Soc. 2007, 129, 7346–7353. 10.1021/ja069232+. [DOI] [PubMed] [Google Scholar]
- Stumpe M. C.; Grubmüller H. Interaction of Urea with Amino Acids: Implications for Urea-Induced Protein Denaturation. J. Am. Chem. Soc. 2007, 129, 16126–16131. 10.1021/ja076216j. [DOI] [PubMed] [Google Scholar]
- Su Z.; Dias C. Molecular interactions accounting for protein denaturation by urea. J. Mol. Liq. 2016, 228, 168–175. 10.1016/j.molliq.2016.10.022. [DOI] [Google Scholar]
- Monera O. D.; Kay C. M.; Hodges R. S. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 1994, 3, 1984–1991. 10.1002/pro.5560031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilloni C.; Rocco A. G.; Eberini I.; Gianazza E.; Broglia R. A.; Tiana G. Urea and guanidinium chloride denature protein L in different ways in molecular dynamics simulations. Biophys. J. 2008, 94, 4654–4661. 10.1529/biophysj.107.125799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhenadhayalan N.; Rajan M.; Rajendran K. Fluorescence spectral studies of Gum Arabic: Multi-emission of Gum Arabic in aqueous solution. J. Lumin. 2014, 155, 322–329. 10.1016/j.jlumin.2014.06.022. [DOI] [Google Scholar]
- Sethuraman S.; Rajendran K. Multicharacteristic Behavior of Tyrosine Present in the Microdomains of the Macromolecule Gum Arabic at Various pH Conditions. ACS Omega 2018, 3, 17602–17609. 10.1021/acsomega.8b02928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Fan Y.; Gao Y. Q. Effects of Urea, Tetramethyl Urea, and Trimethylamine N-Oxide on Aqueous Solution Structure and Solvation of Protein Backbones: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2010, 114, 557–568. 10.1021/jp9084926. [DOI] [PubMed] [Google Scholar]
- Williamson M. P. The structure and functions of proline-rich regions in proteins. Biochem. J. 1994, 297, 249–260. 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Torrez L.; Nigen M.; Williams P.; Doco T.; Sanchez C. Acacia senegal vs. Acacia seyal gums - Part 1: Composition and structure of hyperbranched plant exudates. Food Hydrocolloids 2015, 51, 41–53. 10.1016/j.foodhyd.2015.04.019. [DOI] [Google Scholar]
- Adzhubei A. A.; Sternberg M. J. E.; Makarov A. A. Polyproline-II Helix in Proteins: Structure and Function. J. Mol. Biol. 2013, 425, 2100–2132. 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Adzhubei A. A.; Sternberg M. J. E. Left-handed Polyproline II Helices Commonly Occur in Globular Proteins. J. Mol. Biol. 1993, 229, 472–493. 10.1006/jmbi.1993.1047. [DOI] [PubMed] [Google Scholar]
- Stapley B. J.; Creamer T. P. A survey of left-handed polyproline II helices. Protein Sci. 1999, 8, 587–595. 10.1110/ps.8.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubellis M. V.; Caillez F.; Blundell T. L.; Lovell S. C. Properties of polyproline II, a secondary structure element implicated in protein–protein interactions. Proteins 2005, 58, 880–892. 10.1002/prot.20327. [DOI] [PubMed] [Google Scholar]
- Siligardi G.; Drake A. F. The importance of extended conformations and, in particular, the PII conformation for the molecular recognition of peptides. Biopolymers 1995, 37, 281–292. 10.1002/bip.360370406. [DOI] [PubMed] [Google Scholar]
- Rath A.; Davidson A. R.; Deber C. M. The structure of “unstructured” regions in peptides and proteins: role of the polyproline II helix in protein folding and recognition. Biopolymers 2005, 80, 179–185. 10.1002/bip.20227. [DOI] [PubMed] [Google Scholar]
- Jenkins C. L.; Raines R. T. Insights on the conformational stability of collagen. Nat. Prod. Rep. 2002, 19, 49–59. 10.1039/a903001h. [DOI] [PubMed] [Google Scholar]
- Bella J.; Eaton M.; Brodsky B.; Berman H. M. Crystal and molecular structure of a collagen-like peptide at 1.9A° resolution. Science 1994, 266, 75–81. 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- Kay B. K.; Williamson M. P.; Sudol M. The importance of being proline: the interaction of proline rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. 10.1096/fasebj.14.2.231. [DOI] [PubMed] [Google Scholar]
- Hicks J. M.; Hsu V. L. The extended lefthanded helix: a simple nucleic acid-binding motif. Proteins. 2004, 55, 330–338. 10.1002/prot.10630. [DOI] [PubMed] [Google Scholar]
- Fraser R. D.; MacRae T. P.; Miller A. Molecular packing in type I collagen fibrils. J. Mol. Biol. 1987, 193, 115–125. 10.1016/0022-2836(87)90631-0. [DOI] [PubMed] [Google Scholar]
- Farndale R. W.; Lisman T.; Bihan D.; Hamaia S.; Smerling C. S.; Pugh N.; Konitsiotis A.; Leitinger B.; de Groot P. G.; Jarvis G. E.; Raynal N. Cell-collagen interactions: The use of peptide Toolkits to investigate collagen-receptor interactions. Biochem. Soc. Trans. 2008, 36, 241–250. 10.1042/BST0360241. [DOI] [PubMed] [Google Scholar]
- Helseth D. L. Jr.; Veis A. Collagen self-assembly in vitro. J. Biol. Chem. 1981, 256, 7118–7128. [PubMed] [Google Scholar]
- Kuznetsova N.; Leikin S. Does the triple helical domain of type I collagen encode molecular recognition and fiber assembly while telopeptides serve as catalytic domains? Effect of proteolytic cleavage on fibrillogenesis and on collagen-collagen interaction in fibers. J. Biol. Chem. 1999, 274, 36083–36088. 10.1074/jbc.274.51.36083. [DOI] [PubMed] [Google Scholar]
- Prockop D. J.; Fertala A. Inhibition of the self-assembly of collagen I into fibrils with synthetic peptides. Demonstration that assembly is driven by specific binding sites on the monomers. J. Biol. Chem. 1998, 273, 15598–15604. 10.1074/jbc.273.25.15598. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R.; Chakrabarti P. Stereospecific Interactions of Proline Residues in Protein Structures and Complexes. J. Mol. Biol. 2003, 331, 925–940. 10.1016/S0022-2836(03)00759-9. [DOI] [PubMed] [Google Scholar]
- Pal D.; Chakrabarti P. Cis Peptide Bonds in Proteins: Residues Involved, their Conformations, Interactions and Locations. J. Mol. Biol. 1999, 294, 271–288. 10.1006/jmbi.1999.3217. [DOI] [PubMed] [Google Scholar]
- Neidigh J. W.; Fesinmeyer R. M.; Andersen N. H. Designing a 20-residue protein. Nat. Struct. Biol. 2002, 9, 425–430. 10.1038/nsb798. [DOI] [PubMed] [Google Scholar]
- Stewart D. E.; Sarkar A.; Wampler J. E. Occurrence and role of cis peptide-bonds in protein structures. J. Mol. Biol. 1990, 214, 253–260. 10.1016/0022-2836(90)90159-J. [DOI] [PubMed] [Google Scholar]
- Yao J.; Dyson H. J.; Wright P. E. 3-Dimensional Structure of a Type-VI turn in a Linear Peptide in Water Solution - Evidence for Stacking of Aromatic Rings as a Major Stabilizing Factor. J. Mol. Biol. 1994, 243, 754–766. 10.1016/0022-2836(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Yao J.; Feher V. A.; Espejo B. F.; Reymond M. T.; Wright P. E.; Dyson H. J. Stabilization of a Type-VI Turn in a Family of Linear Peptides in Water Solution. J. Mol. Biol. 1994, 243, 736–753. 10.1016/0022-2836(94)90044-2. [DOI] [PubMed] [Google Scholar]
- Persikov A. V.; Ramshaw J. A. M.; Kirkpatrick A.; Brodsky B. Amino acid propensities for the collagen triple-helix. Biochemistry. 2000, 39, 14960–14967. 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- Brandl M.; Weiss M. S.; Jabs A.; Suhnel J.; Hilgenfeld R. C-H...π-interactions in proteins. J. Mol. Biol. 2001, 307, 357–377. 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- Steiner T.; Koellner G. Hydrogen bonds with π-acceptors in proteins: Frequencies and role in stabilizing local 3D structures. J. Mol. Biol. 2001, 305, 535–557. 10.1006/jmbi.2000.4301. [DOI] [PubMed] [Google Scholar]
- Kar K.; Ibrar S.; Nanda V.; Getz T. M.; Kunapuli S. P.; Brodsky B. Aromatic Interactions Promote Self-Association of Collagen Triple-Helical Peptides to Higher Order Structures. Biochemistry 2009, 48, 7959–7968. 10.1021/bi900496m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zondlo N. J. Aromatic–Proline Interactions: Electronically Tunable CH/π Interactions. Acc. Chem. Res. 2013, 46, 1039–1049. 10.1021/ar300087y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. B. A.; Laws W. R.; Rousslang K. W.; Wyssbrod H. R.. Topics in Fluorescence Spectroscopy: Biochemical Applications; Lakowicz J. R., Ed.; Plenum Press: NY, 2002; Vol. 3, pp 1–63. [Google Scholar]
- Gersten J. I.; Nitzan A. Spectroscopic properties of molecules interacting with small dielectric particles. J. Chem. Phys. 1981, 75, 1139–1152. 10.1063/1.442161. [DOI] [Google Scholar]
- Alev-Behmoaras T.; Toulme′ J.; He′lene C. Quenching of tyrosine fluorescence by phosphate ions: a model study for protein nucleic acid complexes. Photochem. Photobiol. 1979, 30, 533–539. 10.1111/j.1751-1097.1979.tb07177.x. [DOI] [Google Scholar]
- Shimizu O.; Watanabe J.; Imakubo K. Effect of phosphate ion on fluorescent characteristics of tyrosine and its conjugate base. Photochem. Photobiol. 1979, 29, 915–919. 10.1111/j.1751-1097.1979.tb07791.x. [DOI] [Google Scholar]
- Willis K. J.; Szabo A. G. Fluorescence decay kinetics of tyrosinate and tyrosine hydrogen-bonded complexes. J. Phys. Chem. A 1991, 95, 1585–1589. 10.1021/j100157a015. [DOI] [Google Scholar]
- Sanchez C.; Renard D.; Robert P.; Schmitt C.; Lefebvre J. Structural and rheological properties of acacia gum dispersions. Food Hydrocolloids 2002, 16, 257e267 10.1016/S0268-005X(01)00096-0. [DOI] [Google Scholar]
- Aoki H.; Al-Assaf S.; Katayama T.; Phillips G. O. Characterization and properties of Acacia senegal (L.) Willd. var. senegal with enhanced properties (Acacia (sen) SUPER GUM TM): Part 2—Mechanism of the maturation process. Food Hydrocolloids 2007, 21, 329–337. 10.1016/j.foodhyd.2006.04.002. [DOI] [Google Scholar]
- Al-Assaf S.; Sakata M.; McKenna C.; Aoki H.; Phillips G. O. Molecular associations in acacia gums. Struct. Chem. 2009, 20, 325–336. 10.1007/s11224-009-9430-3. [DOI] [Google Scholar]
- Al-Assaf S.; Andres-Brull M.; Cirre J.; Phillips G. O.. Structural Changes Following Industrial Processing of Acacia Gums. In Gum Arabic; Kennedy J. F., Phillips G. O., Williams P. A., Eds.; The Royal Society of Chemistry: London, 2012; pp 153–168. [Google Scholar]
- Evans M.; Ratcliffe I.; Williams P. A. Emulsion stabilization using protein polysaccharide complexes. Curr. Opin. Colloid Interface Sci. 2013, 18, 272–282. 10.1016/j.cocis.2013.04.004. [DOI] [Google Scholar]
- Dickinson E. Hydrocolloids acting as emulsifying agents – How do they do it?. Food Hydrocolloids 2017, 78, 2–14. 10.1016/j.foodhyd.2017.01.025. [DOI] [Google Scholar]
- Cui D.; Ou S.-C.; Patel S. Protein denaturants at aqueous-hydrophobic interfaces: self-consistent correlation between induced interfacial fluctuations and denaturant stability at the interface. J. Phys. Chem. B 2014, 119, 164–178. 10.1021/jp507203g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koishi T.; Yasuoka K.; Willow S. Y.; Fujikawa S.; Zeng X. C. Molecular insight into different denaturing efficiency of urea, guanidinium, and methanol: a comparative simulation study. J. Chem. Theory Comput. 2013, 9, 2540–2551. 10.1021/ct3010968. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya R.; Chakrabarti P. Stereospecific Interactions of Proline Residues in Protein Structures and Complexes. J. Mol. Biol. 2003, 331, 925–940. 10.1016/S0022-2836(03)00759-9. [DOI] [PubMed] [Google Scholar]
- a Kumaran R.; Perumal R. Denaturation mechanism of BSA by urea derivatives: evidence for hydrogen-bonding mode from fluorescence tools. J. Fluoresc. 2011, 21, 1499–1508. 10.1007/s10895-011-0836-0. [DOI] [PubMed] [Google Scholar]; b Kumaran R.; Perumal R. Photophysical studies on the interaction of amides with Bovine Serum Albumin (BSA) in aqueous solution: Fluorescence quenching and protein unfolding. J. Lumin. 2014, 148, 277–284. 10.1016/j.jlumin.2013.12.016. [DOI] [Google Scholar]
- Lee J.; Ross R. T. Absorption and Fluorescence of Tyrosine Hydrogen-Bonded to Amide-like Ligands. J. Phys. Chem. B 1998, 102, 4612–4618. 10.1021/jp9719927. [DOI] [Google Scholar]
- Al-Assaf S.; Katayama T.; Phillips G. O.; Sasaki Y.; Williams P. A. Quality control of gum arabic. Foods Food Ingredients J. Jpn. 2003, 208, 771–780. [Google Scholar]
- Phillips G. O.Giving Nature a Helping Hand. In Gums and Stabilisers for the Food Industry-14; Williams P. A., Phillips G. O., Eds.; Royal Society of Chemistry: Cambridge, U.K., 2008; pp 3–26. [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy; 3rd ed.; Kluwer Academic Plenum: NY, 1999; pp 530–605. [Google Scholar]
- Kumaran R.; Ramamurthy P. Denaturation mechanism of BSA by urea derivatives: evidence for hydrogen-bonding mode from fluorescence tools. J. Fluoresc. 2011, 21, 1499–1508. 10.1007/s10895-011-0836-0. [DOI] [PubMed] [Google Scholar]
- Royer C. A. Probing protein folding and conformational transitions with fluorescence. Chem. Rev. 2006, 106, 1769–1784. 10.1021/cr0404390. [DOI] [PubMed] [Google Scholar]
- Royer C. A.; Mann C. J.; Matthews C. R. Resolution of the fluorescence equilibrium unfolding profile of trp apo repressor using single tryptophan mutants. Protein Sci. 1993, 2, 1844–1852. 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumestand C.; Boyer M.; Guignard M.; Barthe P.; Royer C. A. Characterization of the folding and unfolding reactions of a small beta-barrel protein of novel topology, the MTCP1 oncogene product P13. J. Mol. Biol. 2001, 312, 247–259. 10.1006/jmbi.2001.4928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.