

Abstract
Placenta-specific protein 8 (PLAC8) is a conserved protein with a molecular weight of 12.5 kDa. The specific function of this protein has not been fully elucidated, however, PLAC8 has been found to play an important tumor regulatory role in certain types of cancer, including colon, pancreatic and liver cancer. PLAC8 also participates in the regulation of the cell cycle, autophagy, epithelial-mesenchymal transition and other cellular functions, indicating its potential as a molecular target worth further investigation. The present study investigated the effect of PLAC8 on the proliferation of lung adenocarcinoma PC-9 cells and their sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI). It was found that the inhibition of PLAC8 expression in PC-9 cells resulted in significantly decreased proliferation, whereas overexpression of PLAC8 significantly increased the proliferation (P<0.05) of PC-9 cells. Furthermore, inhibition of PLAC8 expression resulted in decreased activity of the ERK signaling pathway, while PLAC8 overexpression increased activity of this pathway. Inhibition of the ERK signaling pathway with U0126 reversed the effects induced by inhibiting or overexpressing PLAC8 on cell proliferation. In addition, overexpression of PLAC8 significantly decreased the sensitivity of PC-9 cells to gefitinib, and this effect was reversed by U0126. Overall, these results suggest that PLAC8 is involved in the regulation of proliferation of lung adenocarcinoma PC-9 cells and impacts their sensitivity to an EGFR-TKI. Thus, PLAC8 is a potential novel target in lung adenocarcinoma for future studies.
Keywords: lung adenocarcinoma, placenta-specific protein 8, proliferation, Erk1/2, epidermal growth factor receptor tyrosine kinase inhibitor
Introduction
Lung adenocarcinoma is one of the most common malignant tumors in China (1). It has a high incidence rate, progresses rapidly and poses a threat to human health (2). Lung adenocarcinoma is prone to metastasis at an early stage and is likely to develop resistance to standard treatments such as radiotherapy and chemotherapy, resulting in poor clinical efficacy and poor prognosis (3). A number of studies (4–6) have been conducted to explore the molecular mechanism underlying proliferation and drug sensitivity of lung adenocarcinoma cells, in order to enhance the understanding of the disease pathogenesis and for the identification of potential therapeutic targets.
Among these studies, ERK has been frequently studied as a molecular target (7,8). ERK has several regulatory roles in various cellular functions, including cell proliferation and differentiation (9). Moreover, ERK is a member of the mitogen-activated protein kinase (MAPK) signaling cascade, which includes three classes of protein kinases: MAPK kinase kinase, MAPK kinase, MAPK/ERK kinase (MEK) and MAPK ERK. The ERK signaling pathway is activated by a combination of growth factors, extracellular matrix, receptor tyrosine kinases and integrins (10). Phosphorylated (p)-ERK, the active form of ERK, is an important indicator of ERK signaling activity (11–13). p-ERK translocates to the nucleus and regulates gene expression by directly phosphorylating various nuclear proteins, including transcription factors, and thereby regulates cell proliferation (14,15). However, the upstream regulatory mechanism of the ERK signaling pathway in tumor progression requires further investigation.
Placenta-specific protein 8 (PLAC8) is a protein with a molecular weight of 12.5 kDa, and its molecular structure is highly conserved from amphibians to humans (16). PLAC8 was originally discovered in mice and was considered to be a placental-specific transcriptional gene (17). It was eventually found to play an important role in malignant tumor progression, including in leukemia, pancreatic cancer, colon cancer and osteosarcoma (18–21), by regulating various cellular functions, including apoptosis, differentiation and autophagy (20–22). Recently, a study by Jin et al (23) found that PLAC8 expression levels were significantly higher in lung adenocarcinoma cells that were tolerant to radiotherapy compared with that in lung adenocarcinoma cells that were sensitive to radiotherapy. Moreover, it was demonstrated that overexpression of PLAC8 enhanced the stemness and tolerance to radiotherapy of lung adenocarcinoma cells, indicating a role for PLAC8 in the regulation of sensitivity to radiotherapy (23). A study by Zhang et al (24) revealed that expression of PLAC8 was high in osteosarcoma cells with a p53 gene mutation, and this was found to be associated with promotion of osteosarcoma metastasis through the MAPK signaling pathway. Considering the diversity of PLAC8 function and its regulatory role in lung adenocarcinoma, it was hypothesized that PLAC8 may be involved in lung adenocarcinoma cell proliferation and the regulation of drug sensitivity to gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), via the ERK signaling pathway. This study aimed to determine whether PLAC8 may enhance the effect of gefitinib and represent a potential novel target in lung adenocarcinoma.
Materials and methods
Cell culture
PC-9 cells were purchased from the American Type Culture Collection. The cells were maintained in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.) containing 10% fetal calf serum (Gibco; Thermo Fisher Scientific, Inc.) and cultured at 37°C in 5% carbon dioxide.
Lentiviral transfections. In the present study, two short hairpin (sh) RNA interference sequences for PLAC8, sh1# and sh2#, were designed as follows: Sh1, 5′-CCGGCAATGAGGACTCTCTACAGGACTCGAGTCCTGTAGAGAGTCCTCATTGTTTTTTG-3′; and Sh2, 5′-CCGGCTTTGCCAAATCAAGAGATGTCTGGAGATCTCTCTTGATTTGGCAAAGTTTTTTG-3′ (RuboBio Biotechnology Co., Ltd.). PC-9 cells (50% confluence) were transfected with 2 µg plasmid for 72 h. The DNA sequence of human PLAC8 (NM_001130715.1) was obtained from human monocytes by PCR and inserted into the vector using Cloning Kit (Promega Corporation) (25). The primers for PLAC8 used for the PCR were as follows: PLAC8 forward, 5′-GAACTCAGATCTCGAAAAATGCAAGCTCAGGCGC-3′, and reverse 5′-CATGACCGGTGGATCGAAAGTACGCATGGCTCTC-3′. The pLKO.1 puro lentiviral vector (Chengdu Biomart Biotechnology Co., Ltd.) was used as the interference vector and a scrambled shRNA sequence 5′-CCGGGAGCAATCGCACGAGCTAATTCTCGAGAATTAGCTCGTGCGATTGCTCTTTTTTG-3′ was used as a knockdown negative control (KDNC). The pLVX–IRES-puro lentiviral vector (Chengdu Biomart Biotechnology Co., Ltd.) was used as the overexpression vector and an empty vector was used as an overexpression negative control (OENC). After 72 h of lentivirus transfection, positive cells were selected with puromycin. To do so, cells were incubated with 2 µg/ml puromycin for 5 days at 37°C. Cells that were resistant to puromycin represented cells that were efficiently transfected.
Western blot analysis
Protein extraction and western blot analyses were performed as previously described (26). Briefly, IP lysis buffer (Beyotime Institute of Biotechnology) and protease inhibitor (complete ULTRA tablets; Roche Diagnostics GmbH) were used for protein extraction at 4°C for 30 min. Cell lysate was then centrifuged at 12,000 × g for 5 minutes and the supernatant was discarded. A BCA protein assay kit (Beyotime Institute of Biotechnology) was used to detect protein concentrations. Proteins (30 µg) were separated by 10% SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes. Membranes were blocked with 5% skimmed milk for 1 h at room temperature, and incubated with primary antibodies against PLAC8 (1:1,000; cat. no. 13885), ERK1/2 (1:2,000; cat. no. 4695), p-ERK1/2 (1:2,000; cat. no. 9101), EGFR (1:3,000; cat. no. 4267), p-EGFR (1:800; cat. no. 3777), cleaved caspase 3 (1:1,500; cat. no. 9661), cleaved poly (ADP-ribose) polymerase 1 (PARP) (1:3,000; cat. no. 9532) and GAPDH (1:5,000; cat. no. 5174) overnight at 4°C. All primary antibodies were purchased from Cell Signaling Technology. Membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies (cat. nos. ZB-2301 and ZB-2305, respectively; 1:10,000; OriGene Technologies, Inc.). Bands were detected using enhanced chemiluminescence substrate (Bio-Rad Laboratories, Inc.)
Detection of cell viability and proliferation by Cell Counting Kit-8 (CCK-8) assay
Cell viability was measured using the CCK-8 assay kit (Beyotime Institute of Biotechnology). Following 72 h of cell transfection with lentivirus, cells were seeded in 96-well plates at 5,000 cells/well. At the time of detection, 10 µl CCK-8 solution was added to each well, and the absorbance was measured at 450 nm using a microplate reader (Thermo Fisher Scientific, Inc.) after 90 min. To measure the relative proliferative rate of cells, PC-9 OENC and PC-9 PLAC8 cells were seeded in 96-well plates, and each group was administered DMSO or 0.001, 0.01, 0.05, 0.1, 0.5, 1 and 5 µM gefitinib (Selleck Chemicals; cat. no. S1025). For U0216 (MedChemExpress; cat. no. HY-12031) administration, only PC-9 OENC and PC-9 PLAC8 groups treated with DMSO were added as controls. After 72 h, medium was changed and cells were incubated with 100 µl medium containing 10% CCK-8 for 90 min. Absorbance was then measured at 450 nm using a microplate reader, and the relative proliferative rate of the cells was calculated as the ratio of the optical density (OD) value of the treatment group at 72 h to the OD value of the control group at 72 h. Eight replicate wells were tested for each set of samples, and the average absorbance was calculated. GraphPad Prism software v7.0 (GraphPad Software, Inc.) was used to plot the proliferation curves and to calculate the IC50 values.
Statistical analysis
Each experiment was repeated 3 times and representative results were selected. Data analysis was performed using SPSS version 18.0 statistical software (IBM Corp.). The data are expressed as the mean ± SD. The CCK-8 assay was analyzed using one-way ANOVA, followed by the least significant difference test. P<0.05 was considered to indicate a statistically significant difference.
Results
Stable infection of the PC-9 cell line with PLAC8-silencing and PLAC8-overexpressing lentiviruses
Lung adenocarcinoma PC-9 cells were infected with lentiviruses carrying PLAC8-targeting shRNAs (sh1#, sh2#), a knockdown control (KDNC), PLAC8 or an empty vector control (OENC). Western blot analysis demonstrated reduced protein expression of PLAC8 in the sh1# and sh2# groups compared with that in the KDNC group (Fig. 1A). PLAC8 protein expression was increased in the PLAC8 group compared with that in the OENC group (Fig. 1B). Thus, the western blot analysis confirmed the successful transfection of PLAC8 shRNAs and overexpression vector.
Figure 1.

Western blot analysis of PLAC8 protein expression in PC-9 cells. (A) PLAC8 protein expression was detected following transfection of PC-9 cells with PLAC8-shRNA and control lentiviruses. (B) PLAC8 protein expression following transfection of PC-9 cells with PLAC8 overexpression and control lentiviruses. PLAC8, placenta-specific protein 8; KDNC, knockdown negative control; OENC, overexpression negative control; sh, short hairpin.
PLAC8 enhances PC-9 cell proliferation
To study the effect of PLAC8 on the proliferation of lung adenocarcinoma PC-9 cells, the CCK-8 assay was used. Proliferation was detected at 0, 24, 48 and 72 h (Fig. 2). As presented in Fig. 2A, the proliferative capacity of PC-9 cells was significantly decreased following PLAC8 knockdown. At 24 h, there was no significant difference between the three groups (P>0.05). At 48 h, the proliferation was significantly reduced in sh1# and sh2# groups compared with that in the KDNC group (P<0.05). At 72 h, the difference between the KDNC group and the sh1# and sh2# groups was greater (P<0.05). As presented in Fig. 2B, the proliferation of PC-9 cell was significantly increased following PLAC8 overexpression. There was no significant difference between the PLAC8 and OENC groups at 0 and 24 h. However, by 48 h, the proliferation was higher in the PLAC8 overexpression group compared with the OENC group and at 72 h, the difference between the two groups was further increased (P<0.05). These results indicated that PLAC8 influenced PC-9 cell proliferation, as it was inhibited following knockdown and enhanced following overexpression of PLAC8.
Figure 2.

PC-9 cell proliferation curves. (A) CCK-8 assay was used to determine proliferation following transfection of PC-9 cells with PLAC8-shRNA and control lentiviruses. (B) Proliferation of PC-9 cells following transfection of PC-9 cells with PLAC8 overexpression and control lentiviruses. *P<0.05 vs. control group. PLAC8, placenta-specific protein 8; KDNC, knockdown negative control; OENC, overexpression negative control; sh, short hairpin.
PLAC8 enhances ERK signaling pathway activity
Western blot analysis was performed to study the mechanism underlying the impact of PLAC8 on proliferation of PC-9 cells. The ERK signaling pathway was investigated as it plays an important regulatory role in tumor cell viability (27,28). As presented in Fig. 3A, there was no change in ERK1/2 total protein expression after knockdown of PLAC8, however p-ERK1/2 expression was decreased, indicating decreased ERK signaling activity. As shown in Fig. 3B, protein expression of ERK1/2 did not change after PLAC8 overexpression, however p-ERK1/2 protein levels were increased in cells with overexpression of PLAC8. These results indicate a regulatory role for PLAC8 in the activity of the ERK signaling pathway.
Figure 3.

Western blot analysis of p-ERK1/2 and ERK1/2 expression in PC-9 cells. Protein expression of p-ERK1/2 and ERK1/2 in (A) PLAC8-shRNA and control PC-9 cells and in (B) PLAC8-overexpressing and control PC-9 cells. PLAC8, placenta-specific protein 8; KDNC, knockdown negative control; OENC, overexpression negative control; sh, short hairpin.
PLAC8 promotes PC-9 cell proliferation through the ERK signaling pathway
To investigate whether PLAC8 regulates the proliferation of PC-9 cells via the ERK signaling pathway, PC-9 cells with PLAC8 overexpression and knockdown were treated with the ERK signaling pathway inhibitor U0126 (5 µM). The CCK-8 assay was used to detect cell proliferation with and without the addition of the ERK signaling pathway inhibitor U0126 at 0, 24, 48 and 72 h (Fig. 4). As presented in Fig. 4A, when U0126 was not added, cell proliferation was significantly lower in cells transfected with two PLAC8-targeting shRNAs compared to control KDNC cells. However, there were no significant differences in the proliferation rate between KDNC cells and those with PLAC8 knocked down that had been treated with the ERK signaling pathway inhibitor U0126. Similarly, the proliferation of PC-9 PLAC8 cells was significantly higher compared with that of the PC-9 OENC group in untreated cells, but there was no significant difference in the proliferation ability of the two groups after the addition of U0126 (Fig. 4B). These results demonstrated that altering the expression of PLAC8 while inhibiting the ERK signaling activity did not affect the proliferation of PC-9 cells.
Figure 4.

Proliferation in PC-9 cells with PLAC8 knockdown and overexpression following U0126 treatment. Cell proliferation in (A) PLAC8-shRNA and control groups and (B) PLAC8-overexpressing and control groups of PC-9 cells, following treatment with 5 µM U0126 (red) or untreated (black). *P<0.05. PLAC8, placenta-specific protein 8; KDNC, knockdown negative control; OENC, overexpression negative control; sh, short hairpin.
PLAC8 decreases the sensitivity of PC-9 cells to gefitinib
PC-9 cells carry EGFR-sensitive mutations and can be treated with gefitinib, an EGFR-TKI. OENC and PLAC8-overexpressing cells were treated with 0.001, 0.01, 0.05, 0.1, 0.5, 1 and 5 µM gefitinib. Cells treated with DMSO alone were used as controls. Following 72 h, the effect of gefitinib was detected by CCK-8 assay and the cell viability of OENC and PLAC8-overexpressing cells was determined. Fig. 5A shows that the PC-9 cells with overexpression of PLAC8 were more tolerant to gefitinib than PC-9 OENC cells; the PLAC8 group had an IC50 value of 0.1526 µM, which was higher than that of the PC-9 OENC group (0.0177 µM). Subsequently, OENC and PLAC8 groups were treated with 0.5 µM gefitinib for 24 h to detect the expression of apoptosis-associated proteins. As shown in Fig. 5B, apoptosis-associated protein levels were lower in PC-9 PLAC8 cells compared with those in PLAC8 OENC cells, further indicating that the sensitivity of PC-9 cells to gefitinib was decreased following PLAC8 overexpression.
Figure 5.
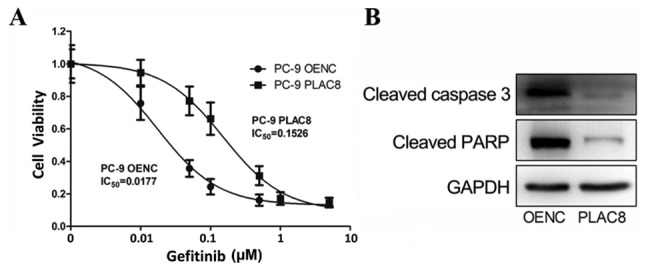
Cell viability in PLAC8-overexpressing PC-9 cells treated with gefitinib. (A) Cell viability at 72 h following treatment with gefitinib in PLAC8-overexpressing and control PC-9 cells. (B) The expression of apoptotic proteins 24 h following treatment with 0.5 µM gefitinib was determined by western blot analysis in PC-9 OENC and PC-9 PLAC8 cells. PLAC8, placenta-specific protein 8; OENC, overexpression negative control; IC, inhibitory concentration.
PLAC8 decreases the sensitivity of PC-9 cells to gefitinib by activating the ERK pathway
The role of ERK in the PLAC8-mediated decrease in sensitivity to gefitinib was investigated. OENC and PLAC8-overexpressing cells were treated with 0.05 µM gefitinib alone or in combination with 5 µM U0126. Cells treated with DMSO were considered as the control groups. The expression of ERK proteins and the relative cell viability was detected by western blot and CCK-8 assays, respectively.
As presented in Fig. 6, western blot analysis revealed a decrease in the protein expression of p-EGFR in PC-9 OENC and PC-9 PLAC8 cells following 72 h of gefitinib treatment, but EGFR expression level was not changed. The anticancer effect exerted by gefitinib is mediated by the inhibition of EGFR phosphorylation (29,30). Following PLAC8 overexpression, ERK was phosphorylated in PC-9 cells, but ERK protein expression was not significantly changed. Following treatment with U0126 and gefitinib, ERK phosphorylation was inhibited in PLAC8 overexpressing cells. The CCK-8 results presented in Fig. 7 revealed that the relative survival rates of OENC cells and PLAC8-overexpressing cells were significantly different following 72 h of gefitinib treatment. The survival rate of PLAC8-overexpressiong cells was significantly higher compared with that of OENC cells (P<0.05). By contrast, when U0126 treatment was used to inhibit the ERK signaling pathway, the relative cell survival rate was not significantly different between OENC and PLAC8 cells following 72 h of gefitinib treatment, indicating that PLAC8 could not induce resistance in PC-9 cells to gefitinib following inhibition of the ERK signaling pathway. These results suggested that the ERK signaling pathway was involved in the regulation of PC-9 cell sensitivity to gefitinib by PLAC8.
Figure 6.

Expression of ERK pathway proteins following treatment of PLAC8-overexpressing PC-9 cells with gefitinib and the ERK inhibitor, U0126. Cells were treated with DMSO (control treatment group) or with gefitinib alone or in combination with ERK inhibitor U0126. PLAC8, placenta-specific protein 8; OENC, overexpression negative control; p, phosphorylated; EGFR, epidermal growth factor receptor; DMSO, dimethyl sulfoxide.
Figure 7.
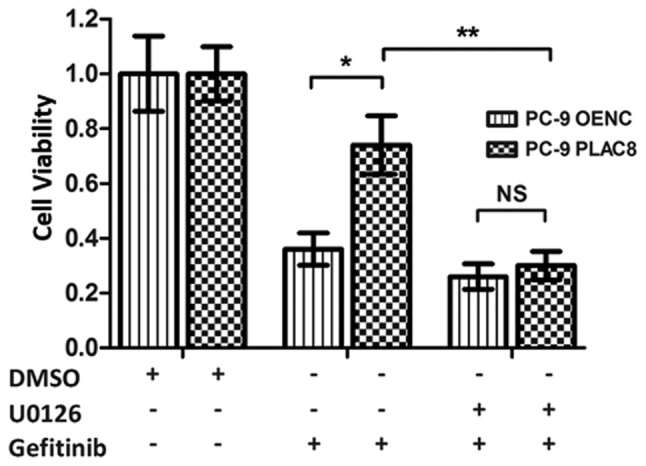
Relative cell viability rate of PLAC8-overexpressing PC-9 cells treated with gefitinib and U0126. CCK-8 assays were used to detect the proliferation of OENC and PLAC8-overexpressing cells treated with DMSO (control treatment group) or with the gefitinib alone or in combination with ERK inhibitor, U0126. NS, non-significant; PLAC8, placenta-specific protein 8; OENC, overexpression negative control. *P<0.05 and **P<0.01.
PLAC8 decreases the expression of apoptosis-associated proteins via the ERK pathway in PC-9 cells treated with gefitinib
OENC and PLAC8-overexpressing cells were treated with 0.5 µM gefitinib alone or in combination with 5 µM U0126 and the expression levels of apoptosis-associated proteins, cleaved caspase 3 and cleaved PARP, were detected by western blot analysis after 24 h. As presented in Fig. 8, the expression of cleaved caspase 3 and cleaved PARP was reduced in PLAC8-overexpressing cells compared with that in OENC cells following 24 h of gefitinib treatment. This suggested that PLAC8 decreased the sensitivity of PC-9 cells to gefitinib-induced apoptosis. However, inhibition of the ERK signaling pathway by treatment with U0126 reversed the effect of PLAC8 overexpression on the expression of cleaved caspase 3 and cleaved PARP in cells treated with gefitinib. This suggested that PLAC8-mediated resistance to gefitinib-induced apoptosis may be dependent on the ERK signaling pathway.
Figure 8.
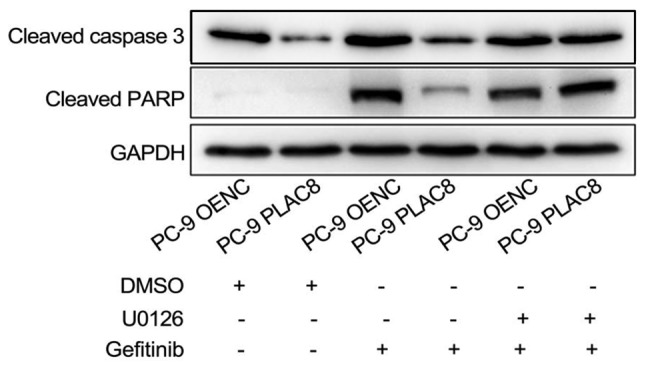
Expression of apoptotic proteins in PLAC8-overexpressing PC-9 cells at 24 h following treatment with gefitinib and U0126. The expression levels of cleaved caspase 3 and cleaved PARP was detected by western blot analysis. PLAC8, placenta-specific protein 8; OENC, overexpression negative control; PARP, poly (ADP-ribose) polymerase 1.
Discussion
In recent years, the role of PLAC8 in tumor progression has begun to be uncovered (18,31). However, to the best of our knowledge, the regulatory role and mechanism of PLAC8 in lung adenocarcinoma have not yet been reported. Recently, Jin et al (23) reported that PLAC8 is involved in the resistance of lung adenocarcinoma to radiotherapy. No studies have been performed on the regulation of lung adenocarcinoma cell proliferation by PLAC8.
Firstly, the present study demonstrated a role for PLAC8 in regulating the proliferation of lung adenocarcinoma PC-9 cells. Cell proliferation was decreased following the knockdown of PLAC8 and significantly increased with overexpression of PLAC8 in PC-9 cells. These results indicate that PLAC8 can enhance the proliferation ability of PC-9 cells. Subsequently, it was revealed that PLAC8 plays a regulatory role in the ERK signaling pathway, as the inhibition of PLAC8 resulted in decreased p-ERK expression. Finally, it was found that PLAC8 was unable to regulate PC-9 cell proliferation following the inhibition of the ERK signaling pathway. This indicates that the regulation of PC-9 cell proliferation by PLAC8 is mediated via the ERK signaling pathway.
U0126 is an effective compound that inhibits ERK1/2 activation by specifically inhibiting the activity of the ERK1/2 upstream kinase MEK1/2 (32). In the present study, cells were treated with U0126, which can induce apoptosis rapidly via inhibiting the ERK signaling pathway activity, and the impact on PC-9 cell proliferation was investigated using the CCK-8 assay. This revealed that the inhibition of ERK activity had no effect on cell proliferation. The proliferation of cells at 72 h after U0126 administration was the latest time point detected; therefore, a low concentration of U0126 (5 µM) was used to prevent the drug from directly causing apoptosis (33). The results indicated that inhibition of ERK signaling pathway activity resulted in the loss of PLAC8-mediated regulation of PC-9 cell proliferation. These findings demonstrate that PLAC8 regulates PC-9 cell proliferation in a manner dependent on ERK signaling pathway activation. In addition, these results confirm that the ERK signaling pathway activation may be critical for promoting lung adenocarcinoma cell proliferation, and this observation is consistent with previous studies showing that ERK signaling pathway activation promotes tumor proliferation (34–36).
Certain subtypes of lung cancer have EGFR mutations. Approximately 10–50% of patients with non-small cell lung cancer have EGFR-activating mutations (37,38). These patients are sensitive to EGFR-TKIs such as gefitinib and erlotinib, and benefit from better therapeutic effects. However, such patients will eventually develop EGFR-TKI resistance (39). Although the number of studies on the mechanism of EGFR-TKI resistance and resistance reversal has increased, current, alternative treatments are limited, and there is still no effective way to reverse EGFR-TKI resistance. The present study found that PC-9 cells are sensitive to gefitinib. A low concentration of gefitinib significantly inhibited cell proliferation, and overexpression of PLAC8 significantly enhanced the resistance of PC-9 cells to gefitinib, demonstrating that PLAC8 may play an important role in mediating resistance to EGFR-TKIs in lung adenocarcinoma. Furthermore, it was found that PLAC8 activated the ERK signaling pathway and that ERK signaling was required for the PLAC8-mediated resistance of PC-9 cells to gefitinib. In addition, overexpression of PLAC8 decreased expression of gefitinib-induced apoptosis-associated proteins in PC-9 cells, and inhibition of ERK signaling pathway blocked this effect. These results indicated that PLAC8 induced resistance of PC-9 cells to gefitinib via the ERK signaling pathway. The present study revealed the mechanism underlying the resistance of lung adenocarcinoma cell to EGFR-TKIs, thus providing a novel approach for reversing EGFR-TKI resistance.
One of the limitations of the present study was that the mechanistic studies were not comprehensive. The mechanism by which PLAC8 activates the ERK signaling pathway remains to be determined. Furthermore, in addition to its regulatory role in lung adenocarcinoma cells, the role of PLAC8 in other diseases remains to be further explored.
In conclusion, the present study demonstrated that PLAC8 enhanced proliferation and resistance to EGFR-TKIs in lung adenocarcinoma PC-9 cells by activating the ERK signaling pathway. These findings demonstrate the functional diversity of the role of PLAC8 in lung adenocarcinoma and provide a novel therapeutic target for future studies.
Acknowledgements
Not applicable.
Funding
This study was supported by the Scientific Research Topics of Sichuan Education Department (grant no. 18ZA0165).
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Authors' contributions
XZ and QL analyzed the data and wrote the paper. YY and SL performed the CCK-8 and western blotting assays. DH, WJ and RM designed the experiment and revised the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Cao M, Chen W. Epidemiology of lung cancer in China. Thorac Cancer. 2019;10:3–7. doi: 10.1111/1759-7714.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Liu X, Xu Y, Zhang K, Huang J, Pan B, Chen D, Cui S, Song H, Wang R, et al. TFAP2C-activated MALAT1 modulates the chemoresistance of docetaxel-resistant lung adenocarcinoma cells. Mol Ther Nucleic Acids. 2019;14:567–582. doi: 10.1016/j.omtn.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai B, Kong DL, Tian J, Liu TW, Zhou H, Wang ZF. microRNA-1205 promotes cell growth by targeting APC2 in lung adenocarcinoma. Eur Rev Med Pharmacol Sci. 2019;23:1125–1133. doi: 10.26355/eurrev_201902_17003. [DOI] [PubMed] [Google Scholar]
- 6.Song J, Wang W, Wang Y, Qin Y, Wang Y, Zhou J, Wang X, Zhang Y, Wang Q. Epithelial-mesenchymal transition markers screened in a cell-based model and validated in lung adenocarcinoma. BMC Cancer. 2019;19:680. doi: 10.1186/s12885-019-5885-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cong Z, Diao Y, Xu Y, Li X, Jiang Z, Shao C, Ji S, Shen Y, De W, Qiang Y. Long non-coding RNA linc00665 promotes lung adenocarcinoma progression and functions as ceRNA to regulate AKR1B10-ERK signaling by sponging miR-98. Cell Death Dis. 2019;10:84. doi: 10.1038/s41419-019-1361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu C, Li H, Jia J, Ruan X, Liu Y, Zhang X. High metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) expression promotes proliferation, migration, and invasion of non-small cell lung cancer via ERK/mitogen-activated protein kinase (MAPK) signaling pathway. Med Sci Monit. 2019;25:5143–5149. doi: 10.12659/MSM.913308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 10.Tanimura S, Takeda K. ERK signalling as a regulator of cell motility. J Biochem. 2017;162:145–154. doi: 10.1093/jb/mvx048. [DOI] [PubMed] [Google Scholar]
- 11.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang LF, Li X, Gao YB, Wang SM, Zhao L, Dong J, Yao BW, Xu XP, Chang GM, Zhou HM, et al. Activation of VEGF/Flk-1-ERK pathway induced blood-brain barrier injury after microwave exposure. Mol Neurobiol. 2015;52:478–491. doi: 10.1007/s12035-014-8848-9. [DOI] [PubMed] [Google Scholar]
- 13.Garces S, Yin CC, Patel KP, Khoury JD, Manning JT, Jr, Li S, Xu J, Pina-Oviedo S, Johnson MR, González S, et al. Focal Rosai-Dorfman disease coexisting with lymphoma in the same anatomic site: A localized histiocytic proliferation associated with MAPK/ERK pathway activation. Mod Pathol. 2019;32:16–26. doi: 10.1038/s41379-018-0152-1. [DOI] [PubMed] [Google Scholar]
- 14.Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/MCB.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenormand P, Sardet C, Pages G, L'Allemain G, Brunet A, Pouyssegur J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang WL, Liu YW, Dang YL, Jiang XX, Xu H, Huang X, Wang YL, Wang H, Zhu C, Xue LQ, et al. PLAC8, a new marker for human interstitial extravillous trophoblast cells, promotes their invasion and migration. 2018;145(pii) doi: 10.1242/dev.148932. dev148932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galaviz-Hernandez C, Stagg C, de Ridder G, Tanaka TS, Ko MS, Schlessinger D, Nagaraja R. Plac8 and Plac9, novel placental-enriched genes identified through microarray analysis. Gene. 2003;309:81–89. doi: 10.1016/S0378-1119(03)00508-0. [DOI] [PubMed] [Google Scholar]
- 18.Kaistha BP, Lorenz H, Schmidt H, Sipos B, Pawlak M, Gierke B, Kreider R, Lankat-Buttgereit B, Sauer M, Fiedler L, et al. PLAC8 localizes to the inner plasma membrane of pancreatic cancer cells and regulates cell growth and disease progression through critical cell-cycle regulatory pathways. Cancer Res. 2016;76:96–107. doi: 10.1158/0008-5472.CAN-15-0216. [DOI] [PubMed] [Google Scholar]
- 19.Li C, Ma H, Wang Y, Cao Z, Graves-Deal R, Powell AE, Starchenko A, Ayers GD, Washington MK, Kamath V, et al. Excess PLAC8 promotes an unconventional ERK2-dependent EMT in colon cancer. J Clin Invest. 2014;124:2172–2187. doi: 10.1172/JCI71103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu SF, Huang Y, Hou JK, Yuan TT, Zhou CX, Zhang J, Chen GQ. The downregulation of onzin expression by PKCepsilon-ERK2 signaling and its potential role in AML cell differentiation. Leukemia. 2010;24:544–551. doi: 10.1038/leu.2009.280. [DOI] [PubMed] [Google Scholar]
- 21.Kinsey C, Balakrishnan V, O'Dell MR, Huang JL, Newman L, Whitney-Miller CL, Hezel AF, Land H. Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep. 2014;7:1143–1155. doi: 10.1016/j.celrep.2014.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mourtada-Maarabouni M, Watson D, Munir M, Farzaneh F, Williams GT. Apoptosis suppression by candidate oncogene PLAC8 is reversed in other cell types. Curr Cancer Drug Targets. 2013;13:80–91. doi: 10.2174/1568009611309010080. [DOI] [PubMed] [Google Scholar]
- 23.Jin Z, Guan L, Xiang GM, Gao BA. Radiation resistance of the lung adenocarcinoma is related to the AKT-Onzin-POU5F1 axis. Biochem Biophys Res Commun. 2018;499:538–543. doi: 10.1016/j.bbrc.2018.03.185. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Hu Q, Li G, Li L, Liang S, Zhang Y, Liu J, Fan Z, Li L, Zhou B, et al. ONZIN upregulation by mutant p53 contributes to osteosarcoma metastasis through the CXCL5-MAPK signaling pathway. Cell Physiol Biochem. 2018;48:1099–1111. doi: 10.1159/000491976. [DOI] [PubMed] [Google Scholar]
- 25.Segawa S, Kondo Y, Nakai Y, Iizuka A, Kaneko S, Yokosawa M, Furuyama K, Tsuboi H, Goto D, Matsumoto I, Sumida T. Placenta specific 8 suppresses IL-18 production through regulation of autophagy and is associated with adult still disease. J Immunol. 2018;201:3534–3545. doi: 10.4049/jimmunol.1800667. [DOI] [PubMed] [Google Scholar]
- 26.Zhao L, Liu S, Xu J, Li W, Duan G, Wang H, Yang H, Yang Z, Zhou R. A new molecular mechanism underlying the EGCG-mediated autophagic modulation of AFP in HepG2 cells. Cell Death Dis. 2017;8:e3160. doi: 10.1038/cddis.2017.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong C, Fang J, Li G, Liu HH, Liu ZS. Effects of microRNA-126 on cell proliferation, apoptosis and tumor angiogenesis via the down-regulating ERK signaling pathway by targeting EGFL7 in hepatocellular carcinoma. Oncotarget. 2017;8:52527–52542. doi: 10.18632/oncotarget.17283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G, Sun J, Liu G, Fu Y, Zhang X. Bradykinin promotes cell proliferation, migration, invasion, and tumor growth of gastric cancer through ERK signaling pathway. J Cell Biochem. 2017;118:4444–4453. doi: 10.1002/jcb.26100. [DOI] [PubMed] [Google Scholar]
- 29.Verma N, Muller AK, Kothari C, Panayotopoulou E, Kedan A, Selitrennik M, Mills GB, Nguyen LK, Shin S, Karn T, et al. Targeting of PYK2 Synergizes with EGFR Antagonists in Basal-like TNBC and Circumvents HER3-Associated Resistance via the NEDD4-NDRG1 Axis. Cancer Res. 2010;77:86–99. doi: 10.1158/0008-5472.CAN-16-1797. [DOI] [PubMed] [Google Scholar]
- 30.Tanimoto A, Takeuchi S, Arai S, Fukuda K, Yamada T, Roca X, Ong ST, Yano S. Histone deacetylase 3 inhibition overcomes BIM deletion polymorphism-mediated osimertinib resistance in EGFR-mutant lung cancer. Clin Cancer Res. 2017;23:3139–3149. doi: 10.1158/1078-0432.CCR-16-2271. [DOI] [PubMed] [Google Scholar]
- 31.Jia Y, Ying X, Zhou J, Chen Y, Luo X, Xie S, Wang QC, Hu W, Wang L. The novel KLF4/PLAC8 signaling pathway regulates lung cancer growth. Cell Death Dis. 2018;9:603. doi: 10.1038/s41419-018-0580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou ZQ, Zhang LN, Wang F, Bellenger J, Shen YZ, Zhang XH. The novel dual PI3K/mTOR inhibitor GDC-0941 synergizes with the MEK inhibitor U0126 in non-small cell lung cancer cells. Mol Med Rep. 2012;5:503–508. doi: 10.3892/mmr.2011.682. [DOI] [PubMed] [Google Scholar]
- 33.Ochi N, Takigawa N, Harada D, Yasugi M, Ichihara E, Hotta K, Tabata M, Tanimoto M, Kiura K. Src mediates ERK reactivation in gefitinib resistance in non-small cell lung cancer. Exp Cell Res. 2014;322:168–177. doi: 10.1016/j.yexcr.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Ito Y, Sasaki Y, Horimoto M, Wada S, Tanaka Y, Kasahara A, Ueki T, Hirano T, Yamamoto H, Fujimoto J, et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27:951–958. doi: 10.1002/hep.510270409. [DOI] [PubMed] [Google Scholar]
- 35.Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, Ding L, Mardis ER, Wilson RK, Solit D, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008;68:5524–5528. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao X, Chen G, Gao C, Zhang DH, Kuan SF, Stabile LP, Liu G, Hu J. MAP4K4 is a novel MAPK/ERK pathway regulator required for lung adenocarcinoma maintenance. Mol Oncol. 2017;11:628–639. doi: 10.1002/1878-0261.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 38.Shu Y, Wu X, Tong X, Wang X, Chang Z, Mao Y, Chen X, Sun J, Wang Z, Hong Z, et al. Circulating tumor DNA mutation profiling by targeted next generation sequencing provides guidance for personalized treatments in multiple cancer types. Sci Rep. 2017;7:583. doi: 10.1038/s41598-017-00520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato Y, Hosomi Y, Watanabe K, Yomota M, Kawai S, Okuma Y, Kubota K, Seike M, Gemma A, Okamura T. Impact of clinical features on the efficacy of osimertinib therapy in patients with T790M-positive non-small cell lung cancer and acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors. J Thorac Dis. 2019;11:2350–2360. doi: 10.21037/jtd.2019.06.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.