

Abstract
The neuro-glial interface extends far beyond mechanical support alone and includes interactions through coagulation cascade proteins. Here, we systematically review the evidence indicating that synaptic and node of Ranvier glia cell components modulate synaptic transmission and axonal conduction by a coagulation cascade protein system, leading us to propose the concept of the neuro-glial coagulonome. In the peripheral nervous system, the main thrombin receptor protease activated receptor 1 (PAR1) is located on the Schwann microvilli at the node of Ranvier and at the neuromuscular junction. PAR1 activation effects can be both neuroprotective or harmful, depending on thrombin activity levels. Low physiological levels of thrombin induce neuroprotective effects in the Schwann cells which are mediated by the endothelial protein C receptor. High levels of thrombin induce conduction deficits, as found in experimental autoimmune neuritis, the animal model for Guillaine-Barre syndrome. In the central nervous system, PAR1 is located on the peri-synaptic astrocyte end-feet. Its activation by high thrombin levels is involved in the pathology of primary inflammatory brain diseases such as multiple sclerosis, as well as in other central nervous system insults, including trauma, neoplasms, epilepsy and vascular injury. Following activation of PAR1 by high thrombin levels the seizure threshold is lowered. On the other hand, PAR1 activation by lower levels of thrombin in the central nervous system protects against a future ischemic insult. This review presents the known structure and function of the neuro-glial coagulonome, focusing on coagulation, thrombin and PAR1 in a pathway which may be either physiological (neuroprotective) or detrimental in peripheral nervous system and central nervous system diseases. Understanding the neuro-glial coagulonome may open opportunities for novel pharmacological interventions in neurological diseases.
Keywords: thrombin, protease activated receptor 1, protease nexin 1, glia, node of Ranvier, synapse, epilepsy, glioblastoma, Guillaine-Barre syndrome
Introduction
It has long been known that glial cells have many functional roles beyond supporting neuronal structure. Many studies indicate that glial cells play an active role in regulation of neural activity, while undergoing changes in intracellular calcium-dependent signaling and self-active secretion of various factors including neurotransmitters and cytokines (Haydon, 2001; Fields, 2010). Thrombin, a serine protease, is a central factor in the coagulation pathway, and mediates its cellular effects through activation of G-protein coupled protease activating receptors (PARs) (Schmidlin and Bunnett, 2001). The known interaction between coagulation and inflammation in a variety of pathologies suggests the importance of the thrombin pathway (Esmon, 2001; Chapman, 2013; Ebrahimi et al., 2017). The thrombin receptor PAR1 is strategically located in both the peripheral and central nervous systems (PNS and CNS, respectively), further supporting its possible role in various neurological disease processes. In the PNS, PAR1 is located at the node of Ranvier (NOR) on a specific glial structure known as the Schwann cell microvilli and its activation causes conduction block (Shavit et al., 2008). The endogenous glial derived thrombin inhibitor protease nexin (PN) 1 plays a central role in synaptic regeneration that follows peripheral nerve injury (Lino et al., 2007). Together this evidence suggests that a fine balance between the coagulation proteases, their inhibitors and the target receptors exists and is necessary for the neural function. Here we will review the current data that indicates the involvement of coagulation system proteins in neuro-glia interaction and define this system as “neuro-glial coagulonome” (NG-coagulonome). Since the NG-coagulonome appears to play a central role in maintaining the physiological function of the peripheral nerve, it is reasonable to speculate that its disruption may be important in several peripheral neurological diseases that involve nerve conduction deficits such as Guillian-Barre syndrome (GBS), diabetic neuropathy and nerve injuries.
In the CNS, PAR1 and thrombin are involved in synaptic transmission and plasticity (Gingrich et al., 2000; Maggio et al., 2008; Ben Shimon et al., 2015). PAR1 is located at the peri-synaptic astrocytic end-feet (Shavit et al., 2011) and its activation causes hyperexcitability and epileptic threshold modification (Maggio et al., 2008). The peri-synaptic astrocyte is a known component of the tripartite synapse and modulates its function (Haydon, 2001). The unique localization of PAR1 on the synaptic adjunct glia, the brain endogenous expression of prothrombin (Riek-Burchardt et al., 2002) and the role of PN1 at the NOR (Dutta et al., 2018) strongly suggest NG-coagulonome involvement in physiological neuronal function. As in the PNS, a disruption of the NG-coagulonome may play a central role in the development of pathologies involving impaired nerve conduction and synaptic transmission, such as multiple sclerosis (MS), brain trauma, ischemia and epilepsy.
In addition to the importance of the NG-coagulonome in neurophysiology and pathophysiology, its role in glial cell proliferation and extension formation links it to brain and nerve neoplastic processes (Stein et al., 2015; Smith and Winkelstein, 2017; Shavit-Stein et al., 2018).
This review systematically presents NG-coagulonome involvement in the physiology of the nervous system and its role in the pathophysiology of a number of neural diseases, modulation of this pathway as a therapeutic target, and potential future research directions.
This review includes the experience of our own laboratory, as well as articles retrieved using electronic search of the PubMed and google scholar databases. Searching criteria included articles describing the following subjects: Thrombin and PAR1 pathways, tripartite synapse, astrocytes, peripheral nerve injury, Guillaine-Barre syndrome, dorsal root ganglion and neuromuscular junction, multiple sclerosis, ischemic and traumatic CNS insults, epilepsy, glioblastoma multiforme. Articles that were published up to April 2019 were included.
The Physiological Role of the Neuro-Glial Coagulonome
The NG-coagulonome is involved in a number of regulatory mechanisms and processes that are necessary for the physiological function of the CNS and PNS. As earlier described, it is localized to two structures which are highly important for nerve function, the NOR and synapse. During neuronal development, coagulation cascade proteases and inhibitors participate in neuronal survival and growth and physiologic elimination of synapses, including the neuromuscular junction (NMJ) (Festoff et al., 2001; Lanuza et al., 2003). The specific effects of neuronal activity were measured on Schwann cell culture using high levels of KCl which is an accepted model for neuronal depolarization in vitro. The Schwann derived PN1 is transiently up regulated in an electrical activity dependent manner in parallel to a corresponding drop in thrombin activity (Gera et al., 2019). This important finding links neuronal activity with glia coagulation factor secretion. It suggests the glial coagulation system acts physiologically as a sensor for axonal activity and a mediator for neuronal-glial crosstalk.
In the mature CNS, thrombin and its receptors are differentially localized to several brain regions including the hippocampus, cerebellum and cortex and are expressed by both neurons and glia. PAR3 and PAR4 are co-localized to different parts of the brain, including the cortex and the hippocampus (Wang and Reiser, 2003). PAR4 is activated by higher levels of thrombin compared to the other PARs, and PAR3 acts as a cofactor for PAR4 activation (Nakanishi-Matsui et al., 2000). PAR1 is the most studied among the CNS PARs, and is the central mediator of thrombin effects on neurons and glia cells. Therefore, this review mainly discusses PAR1 pathway in the CNS.
In the CNS synapse, activation of PAR1 on peri-synaptic astrocytes affects electrophysiological function. Its activation in the solitary tract leads to an astrocytic calcium increase coupled with glutamate secretion, further activating neuronal N-methyl-D-aspartic acid (NMDA) receptors (Vance et al., 2015). In the hippocampus, PAR1 activation potentiates pyramidal neuron NMDA receptors (Gingrich et al., 2000) and at a low concentration its activation shifts the threshold of long term potentiation (LTP) (Maggio et al., 2008), suggesting the important role of the PAR1 pathway in the physiology of synaptic plasticity and memory formation.
The glia cells at the NOR can modulate nerve conduction by several suggested mechanisms, among them, by controlling myelin thickness. Astrocytes exocytosis is necessary for normal myelin thickness and gaps at the NOR. Transgenic mice with reduced astrocytes exocytosis suffer thinner myelin and larger gaps at the NOR in the optic nerve, with reduced visual acuity (Dutta et al., 2018). One of the suggested mechanism by which thrombin is thought to interfere with axonal conduction is by causing proteolysis of neurofascin 155 which acts as a cell adhesion molecule between axons and myelin. The putative mechanism by which perinodal astrocytes regulate this proteolysis is secretion of thrombin inhibitors such as PN1 (Dutta et al., 2018). This allows glia to modulate nerve conduction and serves as a novel pathway for NG-coagulonome involvement in neural physiology.
Neuro-Glial Coagulonome in Diseases of the Peripheral Nervous System
In the PNS, Schwann cells secrete a large number of proteins, which are generally referred to as the Secretome. Among these proteins are several extracellular matrix proteins and neurotrophic factors secreted in response to various neuronal signals (Brown et al., 2013). This new field of Schwann-secretome is highly important for neuronal regeneration, axonal growth, neuronal function and survival.
Peripheral nerve injury
Mechanical injuries to the PNS are extremely common, and are a significant cause of disability. They can be caused by traction of the nerve, laceration, direct compression, or any combination of the three. The nerve injuries can be divided based on severity and involved structure. The Schwann and myelin known to support the axon structure and function and have a role in the regeneration process of damaged nerves. The degree of damage to the glia determines the prognosis.
Numerous studies support the involvement of coagulation factors, and specifically the thrombin pathway, in Schwann cell mediated regeneration and axonal function. In the early 1990’s the presence of the thrombin inhibitor PN1 was demonstrated in the secreted surrounding medium of the Schwann cells (Mulligan et al., 1991). Prothrombin and thrombin elevation is found in the peripheral nerve crush model. This increase is accompanied by PN1 elevation after the injury. The source of this PN1 was traced to the Schwann cells (Smirnova et al., 1996).
Proteomic differential expression analysis of the Schwann cells following injury indicates the involvement of coagulation by a significant upregulation of Factor V levels (Shi et al., 2018). Although thrombin is clearly involved in Schwann-supported neuronal regeneration, the specific pathway by which it mediates this effect remains unclear.
PAR1 activation may have either protective or detrimental effects on nerve conduction and regeneration. As mentioned earlier, raising thrombin levels in the NOR causes a conduction block (Shavit et al., 2008). Levels of thrombin rise in response to crush injury of the sciatic nerve (Gera et al., 2016; Friedmann et al., 2018). These high levels have been linked to a deleterious effect on nerve function. On the other hand, low levels of thrombin generate activated protein C (aPC) which, when coupled with its receptor, endothelial protein C receptor (EPCR), can activate PAR1 (Thiyagarajan et al., 2007). This activation has a neuro-regenerative effect (Thiyagarajan et al., 2008; Festoff and Citron, 2019). PAR1 activation increases neurotrophic and neuroprotective properties of cultured Schwann cells (Pompili et al., 2017). Interestingly, activation of PAR1 by the EPCR pathway inhibits thrombin induced vascular permeability and increase the protective properties of the blood-brain barrier endothelial cells. These effects are mediated by upregulation of sphingosine-phosphate, and thus tie the sphingosine-phosphate pathway to the coagulation system (Mahajan-Thakur et al., 2015). Positive activation of the PAR1 pathway is further supported by the effect of the sphingosine-phosphate receptor modulator fingolimod on Schwann cells. Fingolimod, which is in clinical use for MS treatment, causes a “regenerative phenotype” in Schwann cells, including Schwann cell secretion of proteins known to support neurite growth (Schira et al., 2019). Gera et al. (2016) studied this interplay in response to injury in the sciatic nerve crush model. Thrombin level rise was detected in the first hour after injury, with normalization after a week. EPCR levels were elevated 4 days after injury especially distal to the injury. Furthermore, EPCR was located to the Schwann microvilli in the NOR. The presence of elevated EPCR levels distal to injury site, in which Schwann cells rapidly proliferate, further supports the beneficial effect of PAR1 activation via aPC (Gera et al., 2016).
The mechanism by which active thrombin is generated at injured nerve sites has also been studied. In the blood coagulation system, tissue factor (TF) and factor Xa are two important players in the thrombin generation process, where factor X is cleaved by the TF/factor VIIa complex to create factor Xa that cleaves prothrombin to active thrombin. In addition, FVIIa potentially binds EPCR. Interestingly, all these three important factors are found in the Schwann support system (Gera et al., 2018) where both TF and factor VIIa were clearly located to the NOR. Factor Xa protein activity as well as its coding mRNA are significantly increased after nerve crush. The clinical relevance is further supported by data which indicates that treatment with Apixaban (a selective factor Xa inhibitor) improves motor function following the injury (Gera et al., 2018).
An additional important player is tissue plasminogen activator (tPA), which is secreted from Schwann cells, and clears debris after nerve injury. tPA converts plasminogen to active plasmin, which is another endogenous activator of PAR1. Expression of tPA was found to be regulated by the mircoRNA miR340. This regulation system is found in vivo in the sciatic nerve following nerve crush induction, with direct influence on debris clearance and axonal growth. The microRNA system was suggested as a regulation mediator of the interaction between neurons and glia (Li et al., 2017). Together, this data suggests a highly important role for the NG-coagulonome in processes that occur following peripheral nerve injury. Deficits in nerve conduction are also observed in various inflammatory diseases. The tight link between inflammation and coagulation raises the question regarding NG-coagulonome involvement in such diseases. The GBS and its animal model will be discussed here as a representative of the thrombin pathway in peripheral autoimmune neuropathy.
Guillaine-Barre syndrome
Inflammatory neuropathies are a group of conditions involving damage to myelin, axons, or both. Since the NG-coagulonome is affected by inflammatory processes on one hand and has the potential to modify the Schwann-axon function on the other hand, it is important to evaluate its role in such diseases. The name GBS refers to the acute inflammatory neuropathies, with acute inflammatory demyelinating polyneuropathy being the most common variant (Wijdicks and Klein, 2017). 25% of GBS patients require artificial ventilation, 20% are unable to walk after 6 months and 3-7% dies (van den Berg et al., 2014), raising the need for novel therapy for GBS of which NG-coagulonome modification may be one. The experimental autoimmune neuritis animal model is the accepted murine model for GBS, and shows slowing of nerve conduction velocity, together with destruction of the NOR architecture. These functional and structural abnormalities are accompanied by elevation of thrombin levels in the sciatic nerve itself together with a reduction of nodal PAR1 levels. Treating the animals with thrombin inhibitors normalizes both the conduction velocity and the NOR architecture (Shavit-Stein et al., 2019). Improvement of both function and anatomy of the NOR in this inflammatory neuropathy suggests thrombin pathway inhibition as a possible future treatment in GBS in particular and in inflammatory neuropathies in general.
The neuro-glial coagulonome in the dorsal root ganglion
The rat dorsal root ganglion cells express all four PARs (Zhu et al., 2005). PAR2, which is activated by the proinflammatory agent trypsin rather than thrombin, is related to inflammatory pain (Kawabata et al., 2003; Wang et al., 2012b). Activation of PAR4 leads to dorsal root ganglia (DRG) reduced excitability (Karanjia et al., 2009), similar to the excitability reduction induced by gastrointestinal bacteria taken from healthy human donors. This effect is blocked by PAR4 inhibition (Sessenwein et al., 2017). PAR1 is upregulated in the DRG of individuals infected with viruses from the lentivirus genus such as human immunodeficiency virus. This elevated activation results in neurite retraction and soma atrophy in human DRG (Acharjee et al., 2011), linking the painful neuropathy often seen in these patients with PAR1 activation. PAR1, PAR2, and PAR4 are expressed in human thoracic DRG (in 20%, 40%, and 40% of the sensory neurons, respectively). PAR1 activation (but not the activation of PAR2 and PAR4) by agonist peptide increases intracellular calcium concentration in a dose-dependent manner. Interestingly, PAR4 activation prevents the increased intracellular calcium following PAR1 activation whereas PAR4 blocking further increases it (Desormeaux et al., 2018). The exact localization of PARs to specific cells in the DRG is challenging because of the close approximation of neurons and glia. Further studies are needed in order to define the distribution of these receptors in DRG neural and glial structures.
The closely related thrombolytic protein tPA is involved in the pathogenesis of neuropathic pain in the DRG as well, with inhibition of the anexin 2-tPA complex suppressing neuropathic pain (Yamanaka et al., 2016). Together this suggests that a fine balance between PARs activation and coagulation proteins levels are important to DRG sensory nerve conduction and pain signal transduction.
The neuro-glial coagulonome at the neuro-muscular junction
Thrombin activity and electrical activation are known to participate in synapse reduction in the NMJ since the 1990’s (Jia et al., 1999). Thrombin and electrical activation lead to synaptic reduction in a Schwann cell mediated pathway. This was demonstrated by Gould et al. using the erbB3 mice, which lack Schwann cells, and suffer degeneration of the NMJ. Blocking activation of the NMJ leads to the preservation of the synapse, alongside with reduced production of prothrombin in the muscle. Expression of serpin C1 (anti-thrombin) and serpin D1, both inhibitors of thrombin activity, was significantly lower in the erbB3 mice. These results, together with former studies (Festoff et al., 1991), suggest that the Schwann cells regulate the NMJ synapses by secretion of thrombin inhibitors, which counteract the thrombin secreted from the active muscle fiber (Gould et al., 2019). The involvement of PAR1 in NMJ regulation was studied in the wobbler mice, the animal model for amyotrophic lateral sclerosis. PAR1 expression was higher in motor neurons in the cervical spine of the wobbler mice, which undergoes degeneration (Festoff et al., 2000). The morphology of the NMJ was further studied using plastic-embedded semithin cross-sections. PAR1 was located both to the post synaptic membrane of the muscle fiber and to the membrane of the peri-synaptic Schwann cell (Lanuza et al., 2007). Coagulation cascade proteases and inhibitors participate in pathologic degenerative processes at the NMJ, that lead to unwanted loss of synapses and eventually neuronal apoptosis (Festoff et al., 2001). The strategic localization of PAR1, active thrombin and thrombin inhibitors to the NMJ indicate the involvement of the NG-coagulonome in NMJ pathologies.
The Neuro-Glial Coagulonome in Diseases of the Central Nervous System
A growing body of evidence supports the complex role of thrombin and its linked pathway in CNS physiology and pathology. In the CNS, as in the PNS, many of thrombin’s functions are mediated by glia cells. The thrombin pathway affects many cellular processes that can be protective or detrimental depending on several factors including dose, receptor mode of activation and down-stream signaling. The source of thrombin may be extrinsic, related to inflammatory processes and blood-brain barrier breakdown, or intrinsic, mainly secreted by glial cells. Regardless its source, the effect of thrombin on brain function is highly significant.
As mentioned above, the PAR1 pathway participates in myelin regulation, and is essential for nerve conduction (Yoon et al., 2015; Dutta et al., 2018). The myelin protein expression pattern in the spinal cord was found to be dysregulated in PAR1 knockout (KO) mice. This effect on myelin proteins expression was further supported by an in-vitro study which demonstrated higher levels of proteolipid protein and myelin basic protein in an oligodendrogila culture that lacks PAR1 (Yoon et al., 2015). In cultured microglia, thrombin activation of PAR1 leads to cytosol calcium increase and activation of mitogen activated protein kinases (Suo et al., 2002), as well as tumor necrosis factor-α and nitric oxide production (Ryu et al., 2000). Thrombin via PAR1 activation causes morphological and functional changes in astrocytes leading to extension retraction (Grabham and Cunningham, 1995) and astrogliosis. As described earlier, the NG-coagulonome modulates neuronal electrophysiological functions and nerve conduction in physiology thus strongly implies its potential involvement in pathologies of the CNS.
In the last decade, low thrombin levels were indicated to have protective properties in the CNS, creating the term thrombin preconditioning (TPC). Low concentration of thrombin delivered directly to the rat caudate nucleus prior to ischemic insult reduced ischemic damage and brain edema (Masada et al., 2000).
The dual effect of thrombin raises the question regarding the concentration cut-off between the beneficial and harmful effects (Ben Shimon et al., 2015). In order to trace the concentration at which thrombin ceases to have a protective effect, García et al. (García et al., 2015) studied the effect of different thrombin concentrations on cultured cortical neurons. Indeed, dose-dependent responses were demonstrated where low levels of thrombin (1 nM) encourage neurons viability and mediate neuroprotective effects, while high levels of thrombin (100 nM) are neurotoxic.
The mechanism by which TPC creates its protective effect was further studied both in vitro and in vivo. PAR1 and PN1 were both found to participate in its mechanism. TPC can be achieved by PAR1 agonists and be blocked by PAR1 antagonists. Additional support for the role of PAR1 in mediating TPC beneficial effects is that TPC protects astrocytes from death following oxygen glucose deprivation and this protection was prevented in PAR1 KO mice (Bao et al., 2018). Furthermore, PN1 elevation as well serves as an important mediator for the TPC beneficial effects since TPC is ineffective in PN1 KO hippocampal culture (Mirante et al., 2013). The accumulation of data clearly supports the importance and involvement of the NG-coagulonome in the brain function in health and disease.
Multiple sclerosis
MS is the most common demyelinated immune mediated CNS disease, with a prevalence varying from 2/100,000 to more than 100/100,000 according to geographical location (Leray et al., 2016). The clinical symptoms depend on the involved area, usually presenting as optic neuritis, diplopia, brain stem and cerebellum syndromes, or myelitis (Thompson et al., 2018). Transient inflammation and focal lymphocytic infiltrates causes reversible loss of myelin at first stage, but chronic inflammation leads to microglia activation and neurodegeneration (Dendrou et al., 2015). The brain inflammation and the neural pathology progression raise the question regarding the NG-coagulonome involvement. Current MS treatments include drugs against acute exacerbations and chronic, disease-modifying drugs. Current MS treatments include drugs against acute exacerbations and chronic, disease-modifying drugs (Goldenberg, 2012). Chronic treatments include a number of injectable, infusion, and oral therapies (Rae-Grant et al., 2018). Treatment for acute exacerbation, although effective, include numerous side effects. Chronic treatments have abundant side effects as well, with variable efficiency. This residual significant disease burden calls for further research into new targets.
Experimental autoimmune encephalomyelitis (EAE) is an accepted animal model for MS, induced by immunization with spinal cord homogenates. EAE animals demonstrate high levels of endogenous thrombin inhibition as can be measured in brain homogenate, including anti-thrombin and PN1 (Beilin et al., 2005). Starting at preclinical stages EAE rats also express an elevated level of another endogenous thrombin inhibitor named PN2 which is the secreted form of the amyloid precursor protein that contains a KPI+ domain (Beilin et al., 2007). Rising levels of the thrombin inhibitors can be used as markers for EAE, and may be part of the protective inhibitory counter-reaction of the CNS to elevated thrombin activity. This strongly supports the hypothesis that the brain-NG coagulonome is a major player in MS pathology.
Involvement of coagulation factors in human MS pathophysiology is supported by proteomic analysis of plaques taken from MS patients (Han et al., 2008). The chronic plaques highly express TF and protein C inhibitor. This finding led to further research on the effect of thrombin inhibition in EAE using hirudin and recombinant aPC which both decreased EAE severity (Han et al., 2008).
Brain inflammation may be the primary cause of certain pathologies in diseases such as MS and it can also occur secondary to systemic inflammation as seen in sepsis. The link between systemic inflammation and cognitive deficits (such as confusion and memory impairments) is known (Holmes et al., 2009; Marsland et al., 2015). Systemic injection of lipopolysaccharides serves as an accepted model for systemic inflammation in animals and humans (Andreasen et al., 2008). Interestingly, this systemic injection modifies brain cytokines profiles and inflammatory markers (Cunningham et al., 2009). Inflammation and coagulation in the CNS are hypothesized to share a common pathway (Chapman, 2013). A recent study demonstrates a significant change in hippocampal expression of coagulation proteins and their coding mRNA as well as increased thrombin activity following induction of systemic inflammation by lipopolysaccharides injection. Systemic thrombin inhibition prevents the intrinsic hippocampal increase of the central inflammatory cytokine tumor necrosis factor-α following induction of inflammation (Shavit Stein et al., 2018). These results further support the link between NG-coagulonome and systemic and brain inflammation.
Traumatic neuronal damage
Traumatic brain injury (TBI) is a term used to describe a wide range of conditions involving the application of external force on the brain. Mild TBI (mTBI), as defined by Glasgow Coma Scale of 13–15 after head injury, is relatively benign, but has short and long term implications including headache and cognitive impairment. Since TBI involves a breakage in the blood-brain barrier on either microscopic or macroscopic levels, it may create an opportunity for plasma proteins such as thrombin to contact brain tissue, along with the creation and release of proteases endogenous to the brain.
There are various TBI animal models, mimicking different mechanisms of injury. These include free weight drop devices, fluid percussion injuries and penetrating mechanisms (Xiong et al., 2013).
In spinal cord injuries, PAR1 KO mice show improved motor outcomes, reduced astrogliosis, and reduced production of pro-inflammatory cytokines including interleukin-1β and interleukin-6. In vitro, thrombin causes increased secretion of inflammatory cytokines from astroglia, and astrocytes that are exposed to interleukin-6 create a feedback loop by up-regulation of PAR1 and thrombin (Radulovic et al., 2016). Thrombin was hypothesized to take part in the creation of hyperalgesia following spinal cord injury in rats. Indeed, one day following nerve root compression, fibrin levels are elevated as an indirect measure of thrombin activity. Injection of either the thrombin inhibitor hirudin or PAR1 antagonists protected the animals from hyperalgesia. Furthermore, intra-thecal injection of thrombin caused hyperalgesia without an antecedent trauma. This effect was prevented by blocking spinal PAR1 prior to thrombin injection (Smith and Winkelstein, 2017). These two spinal cord studies highlight the role of thrombin-PAR1 pathway in some of the inflammatory processes and their clinical outcome following CNS trauma, but they do not necessarily explain thrombin’s origin. This will be discussed later on.
Amnesia following mTBI is a well-known phenomenon, studied in an animal model using free weight drop, creating minimal TBI, which parallels the definition of mild TBI. Brain thrombin levels following mTBI are elevated and the animals become amnestic, as assessed by behavior and memory tests. A similar amnesia is also reproduced by intra-ventricular injection of either thrombin or PAR1 agonist. All three interventions impair LTP in hippocampal slices taken from the animals. This amnestic effect is completely blocked by PAR1 antagonists, supporting the PAR1 pathway involvement in the pathogenesis of amnesia following mTBI (Itzekson et al., 2014). Further research conducted on the same animal model reveals that the recovery from trauma induced amnesia correlates with normalization of thrombin activity level in the hippocampus. This strongly suggests a link between cognitive deficits in the context of brain trauma and thrombin level in the brain. (Ben Shimon et al., 2017).
It is tempting to assume that thrombin presence in the CNS following injury is due to plasma leakage following damage to the blood-brain barrier. The origin of this thrombin activity was addressed by following its dynamics post-injury. Two peaks of thrombin levels were measured in the mTBI model. The first elevation was measured immediately after the insult, with normalization after one hour. This elevation probably represents the breakage of the blood-brain barrier. The second elevation peaked 72 hours later, and was accompanied by elevation of PAR1 and interestingly, elevation of the thrombin inhibitor PN1 as well. This late elevation most likely represents an inflammatory process, mediated by astrocytes (Itsekson-Hayosh et al., 2016). The same study found that injection of thrombin to the brain ventricle causes increased sensitivity to seizures 72 hours after the insult, compared to animals which were injected with thrombin but did not experience mTBI. The elevated sensitivity was hypothesized to be due to the elevation of PAR1 following mTBI.
Epilepsy
Brain trauma and seizures commonly present together, leading researchers to question the mechanism relating the two. An important study found that injection of thrombin directly into rat brains caused motor seizures (Lee et al., 1997). This pioneering work was one of many to come, studying the tight link between thrombin, trauma and epileptic activity.
The detrimental effect of thrombin in the CNS was studied in the context of brain injuries, when its level is abnormally high. Mice expressing either excess or lack of PN1 are prone to epileptic activity, supporting the involvement of PAR1 pathway (Lüthi et al., 1997). Thrombin regulates neuronal plasticity in a dose depended matter, resembling its effects in the PNS. High levels evoke a slow, NMDA dependent LTP, whereas low levels induce LTP via the activation of aPC (Maggio et al., 2013c). Both of the effects are PAR1 mediated. In the presence of a short tetanic stimulus, PAR1 and aPC facilitate LTP in hippocampal slices (Maggio et al., 2014) in a mechanism which involves sphingosine-phosphate receptor 1.
Thrombin, via PAR1 activation, affects the electrophysiology of mouse hippocampal brain slices. It creates both enhancement of the CA1 neurons reaction to afferent stimulus (attenuated by NMDA receptor antagonist), and lowered epileptic threshold in CA3 neurons (Maggio et al., 2008). These two well established effects directly link thrombin to memory and learning deficits on the one hand and to hyperexcitability and increase susceptibility to seizures production on the other hand. This last described effect was further studied using whole-cell patch recording of the pyramidal neurons in the hippocampus and found that thrombin causes increased spontaneous action potentials in CA3 neurons which correlates with the high expression of PAR1 in CA3 (Maggio et al., 2013b). The suggested mechanism of seizures according to this finding is the positive feedback loop of depolarization in a PAR1 depended matter, blood-brain barrier damage, and entrance of more thrombin (Maggio et al., 2013a).
Epileptic seizure and status epilepticus with subsequent brain damage can be provoked using organophosphates such as paraoxon. High levels of thrombin, PAR1 and pERK are found in the brain of paraoxon treated mice, coupled with increased electrical activity in the CA1 and CA3 neurons of the hippocampus. Elevated electrical activity is reduced using a PAR1 antagonist (Golderman et al., 2019). These results show the involvement of thrombin in epileptic activity which is not induced by trauma, raising further questions regarding the source of thrombin in the CNS.
Ischemic injury
Ischemic stroke is caused by thrombosis, embolism, or systemic hypoperfusion. Elevation in levels of prothrombin mRNA were noted after induction of global ischemia in rats (Riek-Burchardt et al., 2002), with preserved levels of PN1 and PAR1. This raises questions regarding the role of thrombin in the setting of ischemic insult. In vivo ischemic injury conducted by transient occlusion of the carotid arteries causes increased thrombin activity throughout the ischemic hemisphere, including the peri-infract areas together with increased mRNA levels of prothrombin and factor X in the ischemic core (Bushi et al., 2015). Interestingly, hippocampal slices treated with thrombin concentrations as found in the ischemic hemisphere show altered synaptic responses (Bushi et al., 2015). In a permanent ischemic animal model, a linear significant increase of brain thrombin level is seen over time (up to 24 hours post ischemia) accompanied by a decrease of PAR1 level in the ischemic core (Bushi et al., 2017). Treatment with a specific factor Xa inhibitor (apixaban) given systemically immediately after ischemia induction decreases brain thrombin and reduces infract size (Bushi et al., 2018). PAR1 KO mice show reduced brain edema and neuronal damage, and less prominent behavioral change compare to wild-type mice (Wang et al., 2012a). In vitro ischemia by acute oxygen and glucose deprivation in mice brain slices causes increased hippocampal thrombin presence and activity together with prothrombin mRNA decrease. The functional impairment following thrombin elevation is expressed by the creation of ischemic LTP as measured by in vitro recordings from CA1 neurons of the hippocampus following oxygen and glucose deprivation. The ischemic LTP generation is mediated by thrombin via PAR1 and is blocked following inhibition of thrombin or PAR1 (Stein et al., 2015). These results suggest the NG-coagulonome as a therapeutic target in ischemic stroke treatment.
Neoplasms
The incidence of primary brain neoplasms in the united states is 29.9 per 100,000 persons (Ostrom et al., 2018). Glioblastoma (GBM) is a tumor of glial origin, and is responsible for 15% of the primary CNS neoplasms. It is a highly aggressive tumor with 10–12 months of median overall survival (Ostrom et al., 2016). Current treatments include extensive resection for tumors in accessible locations (Brown et al., 2016). The infiltrative nature of this tumor necessitate adjuvant treatment with radiotherapy (Corso et al., 2017), and the alkylating agent temozolomide (Hart et al., 2013). Despite this combined approach, these therapies rarely achieve extension of more than a few months in the median survival time and are highly aggressive to the patients. More specific interventions in GBM mechanisms are needed.
Thrombin is known to participate in the pathogenesis of GBM (de Almeida and Monteiro, 2016; Xie et al., 2016; Krenzlin et al., 2017), and PAR1 was found to be expressed in glioma cells (Kuhn et al., 2014). Glioma cells produce and secret active thrombin in culture (Itsekson-Hayosh et al., 2015) which in turn causes increased proliferation that is prevented by the thrombin specific inhibitor Dabigatran (Vianello et al., 2016). GBM edema volume as well as glioma progression markers (vascular endothelial growth factor and hypoxia inducible factor-1α) are decreased in PAR1 KO mice (Xie et al., 2016). Furthermore, a differential analysis for the PAR1 gene (F2R) expression in human GBM indicates that it is in the top 2% of genes which are overexpressed in GBM patients (Shavit-Stein et al., 2018). The increased PAR1 levels positively correlate with the expression of TF and negatively correlate with tumor suppressor factors in human GBM patients (Carneiro-Lobo et al., 2014) which further supports this pathway involvement in GBM pathology. Thrombin activity is elevated in-vivo in brains of a rat GBM model and is correlated with brain edema volume. PAR1 elevation is restricted to the tumor itself, without the involvement of adjacent areas (Itsekson-Hayosh et al., 2015). A specific irreversible inhibitor of the proteolytic activation of PAR1 named SIXAC decreases proliferation, invasion and colony formation in glioma cells in vitro. Furthermore, SIXAC was found to reduce brain edema and prolong survival when applied directly into the tumor bed of rat GBM model. (Shavit-Stein et al., 2018). We propose the NG-coagulonome as a possible therapeutic target for this incurable disease.
Conclusions and Future Research Directions
In this review we summarized the current knowledge regarding thrombin pathway in the NG-coagulonome in various physiological and pathological processes. A summary of the involved players can be seen in Figure 1.
Figure 1.
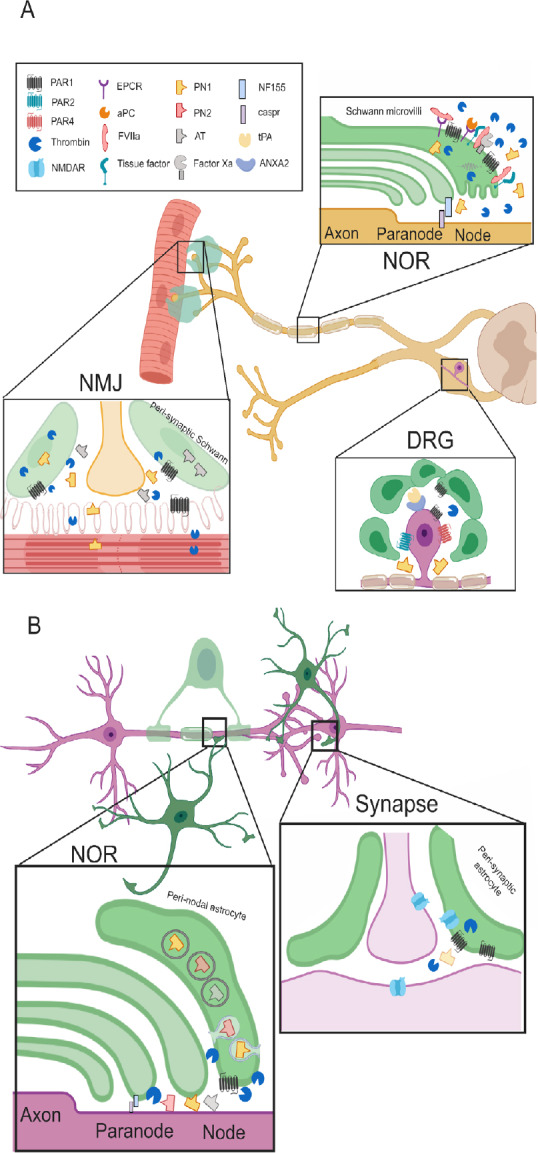
The neuro-glial coagulonome (NG-coagulonome) in the peripheral nervous system (PNS) and central nervous system (CNS).
(A) The NG-coagulonome in the PNS: Schwann cells modulate nerve function in the PNS by secretion of coagulation factors and inhibitors and expression of corresponding receptors. NOR: Schwann cells microvilli at the NOR express protease activating receptor 1 (PAR1). High levels of thrombin activate PAR1 and cause conduction block, whereas low levels activate PAR1 via the endothelial protein C receptor (EPCR) pathway, inducing a neuroprotective phenotype in Schwann cells. Tissue factor binds FVIIa which cleave FX to generate FXa. Levels of FXa and activated protein C (aPC) rises after injury, modulate activation of prothrombin to thrombin. The Schwann secretes PN1 in electrical activity dependent manner which inhibits thrombin. Thrombin inhibition supports neuroregenerative processes. FVIIa potentially binds EPCR as well, suggesting fine balance between the different modes of PAR1 activation. Myelin expresses the adhesion protein neurofascin 155 (NF155) which connects it to the axons via caspr. This structure is disturbed by thrombin. DRG: Satellite cells at the DRG express PAR1 and neurons express PAR1, 2 and 4. PAR1 is elevated in diseases involving peripheral neuropathy such as human immunodeficiency virus (HIV). The complex of tissue plasminogen activator (tPA) and annexin A2 (ANXA2) is expressed on the DRG cells and associated with chronic pain. This suggests a local production of plasmin which is another PAR1 activating protease. Thrombin is inhibited by PN1 which is secreted from glia cells and counteracts the detrimental PAR1 activation. NMJ: Both muscle fibers and Schwann express PAR1 and secrete thrombin. Electrical activity induces thrombin secretion which leads to synaptic elimination. This is prevented by secretion of the thrombin inhibitors such as anti-thrombin (AT) and PN1 from glia cells mainly. (B) The known NG-coagulonome in the CNS: Astrocytes modulate nerve conduction in the CNS at the NOR and at the synapse. NOR: Peri-nodal astrocytes express PAR1, implying its role in nerve conduction. Thrombin cleaves NF155 thus, induces neurodegeneration. Protease nexin (PN)1 is secreted from astrocytes by exocytosis and modulate thrombin depended proteolysis of NF155. PN2 and AT are assumed to be secreted from astrocytes as well. These thrombin inhibitors are part of a feedback loop opposing thrombin damage seen in disease models such as experimental autoimmune encephalomyelitis (EAE). Synapse: Astrocyte’s PAR1 activation by thrombin depolarizes the synapse, whereas depolarization itself releases thrombin from the astrocyte. High levels of thrombin following injury activates PAR1 and lead to the secretion of glutamate, thus activating N-methyl-D-aspartic acid receptor (NMDAR). NMDAR is localized to all of the three partite synapse components. Its activation creates slow long term potentiation (LTP) which changes synaptic plasticity. NOR: node of Ranvier; DRG: dorsal root ganglia; NMJ: neuromuscular junction. Created with BioRender.com.
In the PNS, the PAR1 receptor is located on the Schwann microvilli at the NOR. The Schwann cells endogenously express a significant part of the coagulation cascade proteins (TF, factor X, factor VII, and tPA), enabling them to locally produce active thrombin which serves as a neuro-glial modulator. Thrombin activity is further regulated by the Schwann secretion of PN1, which serves as a protective feedback mechanism. PAR1 located on the glia can have different effect on nerve conduction and regeneration depending on its specific mode of activation. High levels of thrombin activate PAR1 to create conduction deficits. Low levels of thrombin lead to the activation of EPCR, which encourages Schwann cells neuroregeneration phenotype. Although most of these components are localized to the glial cells, their activity is both affected by and influences neuronal activity and thus merits designation as the NG-coagulonome.
PAR1 is located to the NMJ as well, on both the Schwann cells and the muscle fibers. Active muscle fibers secrete thrombin, which in turns lead to the elimination of the NMJ. The Schwann cells participating in the NMJ secrete thrombin inhibitors, protecting the NMJ. The NMJ is rich in interactions between neuronal and glial components of the NG-coagulonome.
PAR1 is expressed on both neurons and glia in the CNS. In astrocytes, it is located to the peri-synaptic astrocyte end-feet indicating its role in synaptic transmission. PAR1 activation by thrombin induces secretion of inflammatory cytokines thus play a central role in brain inflammatory diseases. Astrocytes may regulate nerve conduction in the NOR by secretion of PN1, effecting myelin thickness by inhibition of thrombin induced proteolysis of myelin adhesion molecules.
The NG-coagulonome participates in primary inflammatory disease of the brain and in the brain reaction to general inflammation as showed by the lipopolysaccharides model. In CNS trauma, thrombin increases secretion of inflammatory cytokines from glia, causing amnesia by impairing LTP. High levels of thrombin promote epileptic seizures mediated by the PAR1 pathway. Low levels of thrombin are protective against ischemic insults, an effect which is mediated via the PAR1 pathway as well. PAR1 expression is upregulated in the highly malignant GBM, and its selective inhibition has a positive effect on tumor size and survival in animals.
The details of thrombin generation, mainly on glial cells, and the influence of these changes on neuronal function in these various CNS pathologies are yet to be fully determined. The complete pathways by which it creates its deleterious or protective effect need to be better defined. Future research will characterize these details with the aim of utilizing them for therapeutic purposes.
Additional file: Open peer review report 1 (93.1KB, pdf) .
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Peng Luo, Air Force Medical University, China.
P-Reviewer: Luo P; C-Editors: Zhao M, Yu J; T-Editor: Jia Y
References
- 1.Acharjee S, Zhu Y, Maingat F, Pardo C, Ballanyi K, Hollenberg MD, Power C. Proteinase-activated receptor-1 mediates dorsal root ganglion neuronal degeneration in HIV/AIDS. Brain. 2011;134:3209–3221. doi: 10.1093/brain/awr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreasen A, Krabbe K, Krogh-Madsen R, Taudorf S, Pedersen B, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- 3.Bao X, Hua Y, Keep RF, Xi G. Thrombin-induced tolerance against oxygen-glucose deprivation in astrocytes: role of protease-activated receptor-1. Cond Med. 2018;1:57–63. [PMC free article] [PubMed] [Google Scholar]
- 4.Beilin O, Karussis DM, Korczyn AD, Gurwitz D, Aronovich R, Hantai D, Grigoriadis N, Mizrachi-Kol R, Chapman J. Increased thrombin inhibition in experimental autoimmune encephalomyelitis. J Neurosci Res. 2005;79:351–359. doi: 10.1002/jnr.20270. [DOI] [PubMed] [Google Scholar]
- 5.Beilin O, Karussis DM, Korczyn AD, Gurwitz D, Aronovich R, Mizrachi-Kol R, Chapman J. Increased KPI containing amyloid precursor protein in experimental autoimmune encephalomyelitis brains. Neuroreport. 2007;18:581–584. doi: 10.1097/WNR.0b013e328091c1e6. [DOI] [PubMed] [Google Scholar]
- 6.Ben Shimon M, Lenz M, Ikenberg B, Becker D, Shavit Stein E, Chapman J, Tanne D, Pick CG, Blatt I, Neufeld M, Vlachos A, Maggio N. Thrombin regulation of synaptic transmission and plasticity: implications for health and disease. Front Cell Neurosci. 2015;9:151. doi: 10.3389/fncel.2015.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben Shimon M, Zeimer T, Shavit Stein E, Artan-Furman A, Harnof S, Chapman J, Eisenkraft A, Pick CG, Maggio N. Recovery from trauma induced amnesia correlates with normalization of thrombin activity in the mouse hippocampus. PLoS One. 2017;12:e0188524. doi: 10.1371/journal.pone.0188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown KJ, Seol H, Pillai DK, Sankoorikal BJ, Formolo CA, Mac J, Edwards NJ, Rose MC, Hathout Y. The human secretome atlas initiative: Implications in health and disease conditions. Biochim Biophys Acta. 2013;1834:2454–2461. doi: 10.1016/j.bbapap.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown TJ, Brennan MC, Li M, Church EW, Brandmeir NJ, Rakszawski KL, Patel AS, Rizk EB, Suki D, Sawaya R, Glantz M. Association of the extent of resection with survival in glioblastoma. JAMA Oncol. 2016;2:1460. doi: 10.1001/jamaoncol.2016.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bushi D, Ben Shimon M, Shavit Stein E, Chapman J, Maggio N, Tanne D. Increased thrombin activity following reperfusion after ischemic stroke alters synaptic transmission in the hippocampus. J Neurochem. 2015;135:1140–1148. doi: 10.1111/jnc.13372. [DOI] [PubMed] [Google Scholar]
- 11.Bushi D, Chapman J, Wohl A, Stein ES, Feingold E, Tanne D. Apixaban decreases brain thrombin activity in a male mouse model of acute ischemic stroke. J Neurosci Res. 2018;96:1406–1411. doi: 10.1002/jnr.24253. [DOI] [PubMed] [Google Scholar]
- 12.Bushi D, Stein ES, Golderman V, Feingold E, Gera O, Chapman J, Tanne D. A linear temporal increase in thrombin activity and loss of its receptor in mouse brain following ischemic stroke. Front Neurol. 2017;8:138. doi: 10.3389/fneur.2017.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carneiro-Lobo TC, Lima MT, Mariano-Oliveira A, Dutra-Oliveira A, Oba-Shinjo SM, Marie SKN, Sogayar MC, Monteiro RQ. Expression of tissue factor signaling pathway elements correlates with the production of vascular endothelial growth factor and interleukin-8 in human astrocytoma patients. Oncol Rep. 2014;31:679–686. doi: 10.3892/or.2013.2880. [DOI] [PubMed] [Google Scholar]
- 14.Chapman J. Coagulation in inflammatory diseases of the central nervous system. Semin Thromb Hemost. 2013;39:876–880. doi: 10.1055/s-0033-1357482. [DOI] [PubMed] [Google Scholar]
- 15.Corso CD, Bindra RS, Mehta MP. The role of radiation in treating glioblastoma: here to stay. J Neurooncol. 2017;134:479–485. doi: 10.1007/s11060-016-2348-x. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham C, Campion S, Lunnon K, Murray CL, Woods JFC, Deacon RMJ, Rawlins JNP, Perry VH. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry. 2009;65:304–312. doi: 10.1016/j.biopsych.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Almeida VH, Monteiro RQ. Protease-activated receptor 1 (PAR1): a promising target for the treatment of glioblastoma? Transl Cancer Res. 2016;5:S1274–S1280. [Google Scholar]
- 18.Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545–558. doi: 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- 19.Desormeaux C, Bautzova T, Garcia-Caraballo S, Rolland C, Barbaro MR, Brierley SM, Barbara G, Vergnolle N, Cenac N. Protease-activated receptor 1 is implicated in irritable bowel syndrome mediators-induced signaling to thoracic human sensory neurons. Pain. 2018;159:1257–1267. doi: 10.1097/j.pain.0000000000001208. [DOI] [PubMed] [Google Scholar]
- 20.Dutta DJ, Woo DH, Lee PR, Pajevic S, Bukalo O, Huffman WC, Wake H, Basser PJ, SheikhBahaei S, Lazarevic V, Smith JC, Fields RD. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc Natl Acad Sci U S A. 2018;115:11832–11837. doi: 10.1073/pnas.1811013115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebrahimi S, Jaberi N, Avan A, Ryzhikov M, Keramati MR, Parizadeh MR, Hassanian SM. Role of thrombin in the pathogenesis of central nervous system inflammatory diseases. J Cell Physiol. 2017;232:482–485. doi: 10.1002/jcp.25501. [DOI] [PubMed] [Google Scholar]
- 22.Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemost. 2001;86:51–56. [PubMed] [Google Scholar]
- 23.Festoff BW, Citron BA. Thrombin and the coag-inflammatory nexus in neurotrauma, als, and other neurodegenerative disorders. Front Neurol. 2019;10:59. doi: 10.3389/fneur.2019.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Festoff BW, D’Andrea MR, Citron BA, Salcedo RM, Smirnova I V, Andrade-Gordon P. Motor Neuron cell death in wobbler mutant mice follows overexpression of the g-protein-coupled, protease-activated receptor for thrombin. Mol Med. 2000;9:410–429. [PMC free article] [PubMed] [Google Scholar]
- 25.Festoff BW, Rao JS, Hantaï D. Plasminogen activators and inhibitors in the neuromuscular system: III. The serpin protease nexin I is synthesized by muscle and localized at neuromuscular synapses. J Cell Physiol. 1991;147:76–86. doi: 10.1002/jcp.1041470111. [DOI] [PubMed] [Google Scholar]
- 26.Festoff BW, Suo Z, Citron BA. Plasticity and stabilization of neuromuscular and CNS synapses: interactions between thrombin protease signaling pathways and tissue transglutaminase. Int Rev Cytol. 2001;211:153–177. doi: 10.1016/s0074-7696(01)11018-1. [DOI] [PubMed] [Google Scholar]
- 27.Fields RD. Release of neurotransmitters from glia. Neuron Glia Biol. 2010;6:137–139. doi: 10.1017/S1740925X11000020. [DOI] [PubMed] [Google Scholar]
- 28.Friedmann I, Faber-Elman A, Yoles E, Schwartz M. Injury-induced gelatinase and thrombin-like activities in regenerating and nonregenerating nervous systems. FASEB J. 2018;13:533–543. doi: 10.1096/fasebj.13.3.533. [DOI] [PubMed] [Google Scholar]
- 29.García PS, Ciavatta VT, Fidler JA, Woodbury A, Levy JH, Tyor WR. Concentration-dependent dual role of thrombin in protection of cultured rat cortical neurons. Neurochem Res. 2015;40:2220–2229. doi: 10.1007/s11064-015-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gera O, Bushi D, Ben Shimon M, Artan-Furman A, Harnof S, Maggio N, Dori A, Chapman J, Shavit-Stein E. Local regulation of thrombin activity by factor Xa in peripheral nerve schwann cells. Neuroscience. 2018;371:445–454. doi: 10.1016/j.neuroscience.2017.12.035. [DOI] [PubMed] [Google Scholar]
- 31.Gera O, Shavit-Stein E, Bushi D, Harnof S, Shimon M Ben, Weiss R, Golderman V, Dori A, Maggio N, Finegold K, Chapman J. Thrombin and protein C pathway in peripheral nerve Schwann cells. Neuroscience. 2016;339:587–598. doi: 10.1016/j.neuroscience.2016.10.034. [DOI] [PubMed] [Google Scholar]
- 32.Gera O, Shavit-Stein E, Chapman J. The effect of neuronal activity on glial thrombin generation. J Mol Neurosci. 2019;67:589–594. doi: 10.1007/s12031-019-01265-4. [DOI] [PubMed] [Google Scholar]
- 33.Gingrich MB, Junge CE, Lyuboslavsky P, Traynelis SF. Potentiation of NMDA receptor function by the serine protease thrombin. J Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldenberg MM. Multiple sclerosis review. P T. 2012;37:175–184. [PMC free article] [PubMed] [Google Scholar]
- 35.Golderman V, Shavit-Stein E, Gera O, Chapman J, Eisenkraft A, Maggio N. Thrombin and the protease-activated receptor-1 in organophosphate-induced status epilepticus. J Mol Neurosci. 2019;67:227–234. doi: 10.1007/s12031-018-1228-6. [DOI] [PubMed] [Google Scholar]
- 36.Gould TW, Dominguez B, de Winter F, Yeo GW, Liu P, Sundararaman B, Stark T, Vu A, Degen JL, Lin W, Lee KF. Glial cells maintain synapses by inhibiting an activity-dependent retrograde protease signal. PLoS Genet. 2019;15:e1007948. doi: 10.1371/journal.pgen.1007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grabham P, Cunningham DD. Thrombin receptor activation stimulates astrocyte proliferation and reversal of stellation by distinct pathways: involvement of tyrosine phosphorylation. J Neurochem. 1995;64:583–591. doi: 10.1046/j.1471-4159.1995.64020583.x. [DOI] [PubMed] [Google Scholar]
- 38.Han MH, Hwang S Il, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 39.Hart MG, Garside R, Rogers G, Stein K, Grant R. Temozolomide for high grade glioma. Cochrane Database Syst. 2013:CD007294. doi: 10.1002/14651858.CD007415.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haydon PG, Blendy J, Moss SJ, Rob Jackson F. Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology. 2009;1(56 Suppl):83–90. doi: 10.1016/j.neuropharm.2008.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–774. doi: 10.1212/WNL.0b013e3181b6bb95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Itsekson-Hayosh Z, Shavit-Stein E, Katzav A, Rubovitch V, Maggio N, Chapman J, Harnof S, Pick CG. Minimal traumatic brain injury in mice: protease-activated receptor 1 and thrombin-related changes. J Neurotrauma. 2016;33:1848–1854. doi: 10.1089/neu.2015.4146. [DOI] [PubMed] [Google Scholar]
- 43.Itsekson-Hayosh Z, Shavit-Stein E, Last D, Goez D, Daniels D, Bushi D, Gera O, Zibly Z, Mardor Y, Chapman J, Harnof S. Thrombin activity and thrombin receptor in rat glioblastoma model: possible markers and targets for intervention? J Mol Neurosci. 2015;56:644–651. doi: 10.1007/s12031-015-0512-y. [DOI] [PubMed] [Google Scholar]
- 44.Itzekson Z, Maggio N, Milman A, Shavit E, Pick CG, Chapman J. Reversal of trauma-induced amnesia in mice by a thrombin receptor antagonist. J Mol Neurosci. 2014;53:87–95. doi: 10.1007/s12031-013-0200-8. [DOI] [PubMed] [Google Scholar]
- 45.Jia M, Li M, Dunlap V, Nelson PG. The thrombin receptor mediates functional activity-dependent neuromuscular synapse reduction via protein kinase C activation in vitro. J Neurobiol. 1999;38:369–381. [PubMed] [Google Scholar]
- 46.Karanjia R, Spreadbury I, Bautista-cruz F, Tsang ME, Vanner S. Activation of protease-activated receptor-4 inhibits the intrinsic excitability of colonic dorsal root ganglia neurons. Neurogastroenterol Motil. 2009;21:1218–1221. doi: 10.1111/j.1365-2982.2009.01353.x. [DOI] [PubMed] [Google Scholar]
- 47.Kawabata A, Kuroda R, Nishikawa H, Tanaka A, Kawao N, Itoh H. Peripheral PAR-2 triggers thermal hyperalgesia and nociceptive responses in rats. Neuroreport. 2003;12:715–719. doi: 10.1097/00001756-200103260-00020. [DOI] [PubMed] [Google Scholar]
- 48.Krenzlin H, Lorenz V, Alessandri B. The involvement of thrombin in the pathogenesis of glioblastoma. J Neurosci Res. 2017;95:2080–2085. doi: 10.1002/jnr.24049. [DOI] [PubMed] [Google Scholar]
- 49.Kuhn SA, Martin M, Brodhun M, Kratzsch T, Hanisch UK, Haberl H. Overexpression of protease-activated receptor type 1 (PAR-1) in glioblastoma multiforme WHO IV cells and blood vessels revealed by NCAM-assisted glioblastoma border labeling. Neurol Res. 2014;36:709–721. doi: 10.1179/1743132813Y.0000000303. [DOI] [PubMed] [Google Scholar]
- 50.Lanuza MA, Besalduch N, Garcia N, Sabaté M, Santafé MM, Tomàs J. Plastic-embedded semithin cross-sections as a tool for high-resolution immunofluorescence analysis of the neuromuscular junction molecules: Specific cellular location of protease-activated receptor-1. J Neurosci Res. 2007;85:748–756. doi: 10.1002/jnr.21192. [DOI] [PubMed] [Google Scholar]
- 51.Lanuza MA, Garcia N, González CM, Santafé MM, Nelson PG, Tomas J. Role and expression of thrombin receptor PAR-1 in muscle cells and neuromuscular junctions during the synapse elimination period in the neonatal rat. J Neurosci Res. 2003;73:10–21. doi: 10.1002/jnr.10576. [DOI] [PubMed] [Google Scholar]
- 52.Lee KR, Drury I, Vitarbo E, Hoff JT. Seizures induced by intracerebral injection of thrombin: a model of intracerebral hemorrhage. J Neurosurg. 1997;87:73–78. doi: 10.3171/jns.1997.87.1.0073. [DOI] [PubMed] [Google Scholar]
- 53.Leray E, Moreau T, Fromont A, Edan G. Epidemiology of multiple sclerosis. Rev Neurol (Paris) 2016;172:3–13. doi: 10.1016/j.neurol.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 54.Li S, Zhang R, Yuan Y, Yi S, Chen Q, Gong L, Liu J, Ding F, Cao Z, Gu X. MiR-340 regulates fibrinolysis and axon regrowth following sciatic nerve injury. Mol Neurobiol. 2017;54:4379–4389. doi: 10.1007/s12035-016-9965-4. [DOI] [PubMed] [Google Scholar]
- 55.Lino MM, Atanasoski S, Kvajo M, Fayard B, Moreno E, Brenner HR, Suter U, Monard D. Mice lacking protease nexin-1 show delayed structural and functional recovery after sciatic nerve crush. J Neurosci. 2007;27:3677–3685. doi: 10.1523/JNEUROSCI.0277-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lüthi A, van der Putten H, Botteri FM, Mansuy IM, Meins M, Frey U, Sansig G, Portet C, Schmutz M, Schröder M, Nitsch C, Laurent JP, Denis M. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J Neurosci. 1997;17:4688–4699. doi: 10.1523/JNEUROSCI.17-12-04688.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maggio N, Blatt I, Vlachos A, Tanne D, Chapman J, Segal M. Treating seizures and epilepsy with anticoagulants? Front Cell Neurosci. 2013a;7:19. doi: 10.3389/fncel.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maggio N, Cavaliere C, Papa M, Blatt I, Chapman J, Segal M. Thrombin regulation of synaptic transmission: implications for seizure onset. Neurobiol Dis. 2013b;50:171–178. doi: 10.1016/j.nbd.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 59.Maggio N, Itsekson Z, Dominissini D, Blatt I, Amariglio N, Rechavi G, Tanne D, Chapman J. Thrombin regulation of synaptic plasticity: implications for physiology and pathology. Exp Neurol. 2013c;247:595–604. doi: 10.1016/j.expneurol.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 60.Maggio N, Itsekson Z, Ikenberg B, Strehl A, Vlachos A, Blatt I, Tanne D, Chapman J. The anticoagulant activated protein C (aPC) promotes metaplasticity in the hippocampus through an EPCR-PAR1-S1P1 receptors dependent mechanism. Hippocampus. 2014;24:1030–1038. doi: 10.1002/hipo.22288. [DOI] [PubMed] [Google Scholar]
- 61.Maggio N, Shavit E, Chapman J, Segal M. Thrombin induces long-term potentiation of reactivity to afferent stimulation and facilitates epileptic seizures in rat hippocampal slices: toward understanding the functional consequences of cerebrovascular insults. J Neurosci. 2008;28:732–736. doi: 10.1523/JNEUROSCI.3665-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahajan-Thakur S, Böhm A, Jedlitschky G, Schrör K, Rauch BH. Sphingosine-1-Phosphate and Its Receptors: A Mutual Link between Blood Coagulation and Inflammation. Mediators Inflamm. 2015;2015:1–11. doi: 10.1155/2015/831059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marsland AL, Gianaros PJ, Kuan DC, Sheu LK, Krajina K, Manuck SB. Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain Behav Immun. 2015;48:195–204. doi: 10.1016/j.bbi.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Masada T, Xi G, Hua Y, Keep RF. The effects of thrombin preconditioning on focal cerebral ischemia in rats. Brain Res. 2000;867:173–179. doi: 10.1016/s0006-8993(00)02302-7. [DOI] [PubMed] [Google Scholar]
- 65.Mirante O, Price M, Puentes W, Castillo X, Benakis C, Thevenet J, Monard D, Hirt L. Endogenous protease nexin-1 protects against cerebral ischemia. Int J Mol Sci. 2013;14:16719–16731. doi: 10.3390/ijms140816719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mulligan LP, Rosenblatt DE, Toms R, Johnson D. Protease nexin-1 activity in cultured Schwann cells. Neurosci Lett. 1991;128:42–46. doi: 10.1016/0304-3940(91)90756-j. [DOI] [PubMed] [Google Scholar]
- 67.Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, Coughlin SR. PAR3 is a cofactor for PAR4 activation by thrombin. Nature. 2000;404:609–613. doi: 10.1038/35007085. [DOI] [PubMed] [Google Scholar]
- 68.Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. 2018;20:iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016;18:v1-75. doi: 10.1093/neuonc/now207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pompili E, Fabrizi C, Somma F, Correani V, Maras B, Schininà ME, Ciraci V, Artico M, Fornai F, Fumagalli L. PAR1 activation affects the neurotrophic properties of Schwann cells. Mol Cell Neurosci. 2017;79:23–33. doi: 10.1016/j.mcn.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 71.Radulovic M, Yoon H, Wu J, Mustafa K, Scarisbrick IA. Targeting the thrombin receptor modulates inflammation and astrogliosis to improve recovery after spinal cord injury. Neurobiol Dis. 2016;93:226–242. doi: 10.1016/j.nbd.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rae-Grant A, Day GS, Marrie RA, Rabinstein A, Cree BAC, Gronseth GS, Haboubi M, Halper J, Hosey JP, Jones DE, Lisak R, Pelletier D, Potrebic S, Sitcov C, Sommers R, Stachowiak J, Getchius TSD, Merillat SA, Pringsheim T. Practice guideline recommendations summary: Disease-modifying therapies for adults with multiple sclerosis: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018;90:777–788. doi: 10.1212/WNL.0000000000005347. [DOI] [PubMed] [Google Scholar]
- 73.Riek-Burchardt M, Striggow F, Henrich-Noack P, Reiser G, Reymann K. Increase of prothrombin-mRNA after global cerebral ischemia in rats, with constant expression of protease nexin-1 and protease-activated receptors. Neurosci Lett. 2002;329:181–184. doi: 10.1016/s0304-3940(02)00645-6. [DOI] [PubMed] [Google Scholar]
- 74.Ryu J, Pyo H, Jou I, Joe E. Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-κB. J Biol Chem. 2000;275:29955–29959. doi: 10.1074/jbc.M001220200. [DOI] [PubMed] [Google Scholar]
- 75.Schira J, Heinen A, Poschmann G, Ziegler B, Hartung HP, Stühler K, Küry P. Secretome analysis of nerve repair mediating Schwann cells reveals Smad-dependent trophism. FASEB J. 2019;33:4703–4715. doi: 10.1096/fj.201801799R. [DOI] [PubMed] [Google Scholar]
- 76.Schmidlin F, Bunnett NW. Protease-activated receptors: how proteases signal to cells. Curr Opin Pharmacol. 2001;1:575–582. doi: 10.1016/s1471-4892(01)00099-6. [DOI] [PubMed] [Google Scholar]
- 77.Sessenwein JL, Baker CC, Pradhananga S, Maitland ME, Petrof EO, Allen-Vercoe E, Noordhof C, Reed DE, Vanner SJ, Lomax AE. Protease-mediated suppression of DRG neuron excitability by commensal bacteria. J Neurosci. 2017;37:11758–11768. doi: 10.1523/JNEUROSCI.1672-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shavit-Stein E, Sheinberg E, Golderman V, Sharabi S, Wohl A, Gofrit SG, Zivli Z, Shelestovich N, Last D, Guez D, Daniels D, Gera O, Feingold K, Itsekson-Hayosh Z, Rosenberg N, Tamarin I, Dori A, Maggio N, Mardor Y, Chapman J, et al. A novel compound targeting protease receptor 1 activators for the treatment of glioblastoma. Front Neurol. 2018;9:1087. doi: 10.3389/fneur.2018.01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shavit-Stein E, Aronovich R, Sylantiev C, Gera O, Gofrit SG, Chapman J, Dori A. Blocking thrombin significantly ameliorates experimental autoimmune neuritis. Front Neurol. 2019;9:1139. doi: 10.3389/fneur.2018.01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shavit E, Beilin O, Korczyn AD, Sylantiev C, Aronovich R, Drory VE, Gurwitz D, Horresh I, Bar-Shavit R, Peles E, Chapman J. Thrombin receptor PAR-1 on myelin at the node of Ranvier: a new anatomy and physiology of conduction block. Brain. 2008;131:1113–1122. doi: 10.1093/brain/awn005. [DOI] [PubMed] [Google Scholar]
- 81.Shavit E, Michaelson DM, Chapman J. Anatomical localization of protease-activated receptor-1 and protease-mediated neuroglilal crosstalk on peri-synaptic astrocytic endfeet. J Neurochem. 2011;119:460–473. doi: 10.1111/j.1471-4159.2011.07436.x. [DOI] [PubMed] [Google Scholar]
- 82.Shavit Stein E, Ben Shimon M, Artan Furman A, Golderman V, Chapman J, Maggio N. Thrombin inhibition reduces the expression of brain inflammation markers upon systemic LPS treatment. Neural Plast. 2018;2018:7692182. doi: 10.1155/2018/7692182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shi GD, Shen X, Zhou XH, Fan BY, Ren YM, Lin W, Zhang XL, Liu S, Hao Y, Wei ZJ, Feng SQ. iTRAQ-based proteomics profiling of Schwann cells before and after peripheral nerve injury. Iran J Basic Med Sci. 2018;21:832–841. doi: 10.22038/IJBMS.2018.26944.6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smirnova I V, Ma JY, Citron BA, Ratzlaff KT, Gregory EJ, Akaaboune M, Festoff BW. Neural thrombin and protease nexin I kinetics after murine peripheral nerve injury. J Neurochem. 1996;67:2188–2199. doi: 10.1046/j.1471-4159.1996.67052188.x. [DOI] [PubMed] [Google Scholar]
- 85.Smith JR, Winkelstein BA. The role of spinal thrombin through protease-activated receptor 1 in hyperalgesia after neural injury. J Neurosurg Spine. 2017;26:532–541. doi: 10.3171/2016.9.SPINE16501. [DOI] [PubMed] [Google Scholar]
- 86.Stein ES, Itsekson-Hayosh Z, Aronovich A, Reisner Y, Bushi D, Pick CG, Tanne D, Chapman J, Vlachos A, Maggio N. Thrombin induces ischemic LTP (iLTP): Implications for synaptic plasticity in the acute phase of ischemic stroke. Sci Rep. 2015;5:7912. doi: 10.1038/srep07912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suo Z, Wu M, Ameenuddin S, Anderson HE, Zoloty JE, Citron BA, Andrade-Gordon P, Festoff BW. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J Neurochem. 2002;80:655–666. doi: 10.1046/j.0022-3042.2001.00745.x. [DOI] [PubMed] [Google Scholar]
- 88.Thiyagarajan M, Cheng T, Zlokovic BV. Endothelial cell protein C receptor: Role beyond endothelium? Circ Res. 2007;100:155–157. doi: 10.1161/01.RES.0000258167.48227.84. [DOI] [PubMed] [Google Scholar]
- 89.Thiyagarajan M, Fernández J, Lane S, Griffin G, Zlocovik B. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. 2008;28:12788–12797. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, Correale J, Fazekas F, Filippi M, Freedman MS, Fujihara K, Galetta SL, Hartung HP, Kappos L, Lublin FD, Marrie RA, Miller AE, Miller DH, Montalban X, Mowry EM, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–173. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 91.van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barré syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10:469–482. doi: 10.1038/nrneurol.2014.121. [DOI] [PubMed] [Google Scholar]
- 92.Vance KM, Rogers RC, Hermann GE. PAR1-activated astrocytes in the nucleus of the solitary tract stimulate adjacent neurons via NMDA receptors. J Neurosci. 2015;35:776–785. doi: 10.1523/JNEUROSCI.3105-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vianello F, Sambado L, Goss A, Fabris F, Prandoni P. Dabigatran antagonizes growth cell-cycle progression, migration, and endothelial tube formation induced by thrombin in breast and glioblastoma cell lines. Cancer Med. 2016;5:2886–2898. doi: 10.1002/cam4.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang H, Reiser G. Thrombin signaling in the brain: The role of protease-activated receptors. Biol Chem. 2003;384:193–202. doi: 10.1515/BC.2003.021. [DOI] [PubMed] [Google Scholar]
- 95.Wang J, Jin H, Hua Y, Keep RF, Xi G. Role of protease-activated receptor-1 in brain injury after experimental global cerebral ischemia. Stroke. 2012a;43:2476–2482. doi: 10.1161/STROKEAHA.112.661819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang S, Dai Y, Kobayashi K, Zhu W, Kogure Y, Yamanaka H, Wan Y, Zhang W, Noguchi K. Potentiation of the P2X3 ATP receptor by PAR-2 in rat dorsal root ganglia neurons, through protein kinase-dependent mechanisms, contributes to inflammatory pain. Eur J Neurosci. 2012b;36:2293–2301. doi: 10.1111/j.1460-9568.2012.08142.x. [DOI] [PubMed] [Google Scholar]
- 97.Wijdicks EF, Klein CJ. Guillain-Barré Syndrome. Mayo Clin Proc. 2017;92:467–479. doi: 10.1016/j.mayocp.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 98.Xie Q, Bao X, Chen ZH, Xu Y, Keep RF, Muraszko KM, Xi G, Hua Y. Role of protease-activated receptor-1 in glioma growth. Acta Neurochir Suppl. 2016;121:355–360. doi: 10.1007/978-3-319-18497-5_61. [DOI] [PubMed] [Google Scholar]
- 99.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yamanaka H, Kobayashi K, Okubo M, Noguchi K. Annexin A2 in primary afferents contributes to neuropathic pain associated with tissue type plasminogen activator. Neuroscience. 2016;314:189–199. doi: 10.1016/j.neuroscience.2015.11.058. [DOI] [PubMed] [Google Scholar]
- 101.Yoon H, Radulovic M, Drucker KL, Wu J, Scarisbrick IA. The thrombin receptor is a critical extracellular switch controlling myelination. Glia. 2015;63:846–859. doi: 10.1002/glia.22788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu WJ, Yamanaka H, Obata K, Dai Y, Kobayashi K, Kozai T, Tokunaga A, Noguchi K. Expression of mRNA for four subtypes of the proteinase-activated receptor in rat dorsal root ganglia. Brain Res. 2005;1041:205–211. doi: 10.1016/j.brainres.2005.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.