

Abstract
Recent interest in high sensitivity multiple myeloma (MM) measurable residual disease (MRD) testing is a direct consequence of the high-quality responses achieved using novel therapeutic agents and better treatment strategies. Traditional diagnostic measures such as immunohistochemistry and morphology have detection sensitivities of only 10−2 – 10−3, which do not reliably predict progression free survival (PFS) or overall survival (OS) after these treatments. Contemporary monitoring of MM MRD has switched to more sensitive platforms such as quantitative allele-specific oligonucleotide polymerase chain reaction (ASO-qPCR), next-generation sequencing (NGS), and multiparametric flow cytometry (MFC). Though both ASO-qPCR and NGS have excellent detection sensitivities (10−5 – 10−6), both technologies have lower applicability when compared to MFC. Conventional MFC can easily reach a detection sensitivity of 10−4 and when optimized can achieve a sensitivity of 10−5 – 10−6. Current consensus guidelines require a minimum of 2 million and recommend 5 million events be acquired to reach a minimum sensitivity of 10-5. As conventional immunophenotyping protocols are unable to attain these numbers, alternative MFC staining procedures are required. This manuscript describes two high-sensitivity MFC approaches that can be used for MM MRD testing.
Keywords: Measurable Residual Disease, Multiple Myeloma, Flow Cytometry, Plasma Cell, Bulk-lysis, Pooled Tube
INTRODUCTION
Multiple myeloma (MM) is an irremediable B-cell malignancy characterized by the neoplastic expansion of clonal plasma cells (PCs) in the bone marrow, which is often typified by elevated calcium concentration, renal failure, anemia, increased infection, and osteolytic bony lesions (Kumar et al., 2017). In the United States, MM represents approximately 1% of all cancers and 10% of all hematological malignancies (Siegel et al., 2018). Improvement in standards of care and the emergence of novel therapeutics over the last 2 decades have resulted in substantial increase in median survival for both elderly and young patients, with greater than 70% of these patients achieve complete response (CR) post-treatment (Kristinsson et al., 2014; Kumar et al., 2014; Landgren et al., 2017). Despite these advances MM remains an incurable disease, as most patients eventually relapse, develop resistance to their treatments, and succumb to the disease, indirectly suggesting that the traditional method defining a CR (e.g. measurement of monoclonal proteins in the serum and urine by immunofixation and protein electrophoresis) lacks the sensitivity to evaluate a patients’ long-term response to therapy. Thus, there is a need to refine response criteria and alternative detection methods that are more sensitive for evaluating patients’ long-term outcomes are necessary to predict disease relapse risk so that patient survival can be improved.
The ability to detect and quantify small number of residual tumor cells (e.g. measurable residual disease) that have not been eliminated by the therapy can provide indispensable information pertaining to patient response to therapy. The presence of MRD in MM patients has been associated with worse PFS and OS regardless of the detection methods used (Avet-Loiseau et al., 2015; Korde et al., 2014; Martinez-Lopez et al., 2014; Paiva et al., 2008; Rawstron et al., 2013). Consequently, MRD monitoring has been seen as a potential strategy to assess a patient’s long-term clinical outcome and to reduce the long latency between drug development and approval to be considered as a therapeutic intervention. In this regard, several high sensitivity detection platforms have been investigated and described for MRD monitoring in the bone marrow of patients with MM and these techniques include MFC, quantitative allele-specific oligonucleotide quantitative PCR (ASO-qPCR), and next-generation sequencing (NGS) (Biran et al., 2014). While each of these methods has its own advantages and disadvantages, MFC has consistently demonstrated to provide higher applicability (>95% patients), similar sensitivities (10-5 – 10−6), broader availability, shorter turnaround time (<6 hours), and better reproducibility when compared to the other methods (Ladetto et al., 2014; Logan et al., 2013; Paiva et al., 2012; Puig et al., 2014; Soh et al., 2017; van der Velden et al., 2003). Additionally, MFC is an excellent tool for MRD evaluation because it allows simultaneous characterization of the composition of PCs, both normal PCs and myeloma cells, at the single cell level.
To date, all studies employing MFC for MM MRD monitoring have correlated MRD-negativity with longer PFS and/or OS (Gormley et al., 2016; Paiva et al., 2008). These finding prompted the International Myeloma Working Group (IMWG) to incorporate MM MRD assessment by MFC into their response criteria (Kumar et al., 2016) and for the FDA to conclude it could be used as a surrogate end-point biomarker in clinical trials when properly validated and standardized. As growing evidence supports a strong correlation for MRD status and PFS/OS, MFC faces the challenges of widespread adoption. One such challenge is the highly varying criteria used for MRD determination (Flanders et al., 2013; Paiva et al., 2008; Rawstron et al., 2013). Indeed, a recent survey of 30 major medical institutes in the United States reported methodological differences in MM MRD testing by MFC (Flanders et al., 2013) and initial effort to harmonize sample processing and data analysis have resulted in some improvement (Salem et al., 2016).
Earlier generations of MM MFC assays simultaneously assessed 4 – 5 fluorochromes and collected 500,000 or fewer events. This resulted in sensitivities ranging from 10−3 to 10−4 (e.g. detection of 1 PC in 1,000 – 10,000 analyzed cells). Improvements of MFC technologies have translated into faster acquisition speeds and the ability to simultaneously analyze a larger number of fluorophores. In turn, this has allowed the clinical MFC field to develop panels with 8 – 10 antibodies per tube and to record a larger number of events, improving the sensitivity to as high as 10−6 (e.g. 1 PC in 1,000,000 events) (Flores-Montero et al., 2017; Royston et al., 2016). However, to achieve detection sensitivities of 10−5 − 10−6, millions of cells have to be acquired and conventional MFC staining procedure needs to be modified to achieve these numbers.
The following protocols provide detailed instructions for performing and validating high sensitivity MFC assays for detecting MRD in MM patients. The BASIC PROTOCOL 1 describes a staining procedure known as the ‘Bulk-lysis’ or ‘Pre-lysis’ method designed to obtain sufficient bone marrow cells for the assay. This procedure is adopted from the next-generation flow method recommended by the NCI/ICCS consensus group and developed in partnership with the EuroFlow consortium (Burgos et al., 2018). ALTERNATE PROTOCOL 1 provides an alternative processing method known as the ‘Pooled-tube’ method that is a slight variant of the ‘Pre-lysis’ method. Detailed guidelines for evaluating the performance of fluorochrome-conjugated antibodies used for detecting and distinguishing normal vs. abnormal PCs are described in SUPPROT PROTOCOL 1. Finally, a complete workflow for establishing the sensitivity of MFC assays for detecting rare events are described in SUPPORT PROTOCOL 2. It should be noted that for detailed recommendations and descriptions on validating clinical flow cytometric methods the reader is encouraged to consult the International Council for Standardization of Haematology (ICSH) and the International Clinical Cytometry Society (ICCS) consensus guidelines published in 2013 (Barnett et al., 2013; Davis, Dasgupta, et al., 2013; Davis, Wood, et al., 2013; Tanqri et al., 2013; Wood et al., 2013).
BASIC PROTOCOL 1
PROCESSING OF BONE MARROW SAMPLES USING ‘BULK-LYSIS’ METHOD
Current consensus guidelines require a minimum of 2 million and recommend 5 million events be acquired per tube for a sensitivity of 10−5 (Flores-Montero et al., 2016; Flores-Montero et al., 2017). Conventional MFC staining methods do not normally process sufficient number of cells to satisfy this requirement. Accordingly, the NCI/ICCS initiative resulted in the creation of a consensus 8-color, two-tube panel that is consistent with the methodology simultaneously developed by the EuroFlow group (Flores-Montero et al., 2016). This panel consists of six backbone antibodies targeting CD45, CD38, CD138, CD19, CD56, and CD27, with antibodies to CD81 and CD117 in one tube and cytoplasmic kappa (cKappa) and cytoplasmic lambda (cLambda) light chains in the other tube. Recently, a 10-color assay for MRD evaluation that incorporates into one tube all of these monoclonal antibodies (mAbs) was described which was found to have near concordance with the 8-color method detailed here (Royston et al., 2016).
In this BASIC PROTOCOL, we describe the ‘Pre-lysis’ (or ‘Bulk-lysis’) staining procedure. Briefly, this method lyses a sufficient volume of bone marrow sample upfront, stains the cells with the desired combination of fluorochrome-conjugated antibodies, and then lyses the cells a second time to remove residual erythrocytes. The analysis approach recommended by the NCI/ICCS Consensus Group utilizes the expression of CD38, CD138, CD45, and light scatter characteristics to define PCs, while excluding contaminant lymphocytes, doublets, and debris. Such ‘fit-for-purpose’ gating can be easily adapted to any MM analysis, regardless of the instrument, panel, or type of flow cytometric analysis software package used.
Materials
Specimen:
EDTA or sodium heparin anti-coagulated human bone marrow aspirate should be used in this protocol. EDTA samples should be stored at room temperature for no more than 24 hours prior to use, whereas sodium heparin samples should be processed within 48 hours (CLSI, 2007). Samples with less than 85% viability are deemed suboptimal for flow cytometry testing. Bone marrow is an irreplaceable sample and should not be rejected if it exceeds these specimen age and viability requirements. Instead, it should be processed, and its age and viability noted on the final report.
Reagents:
Refer to Table 1 for a comprehensive list of fluorochrome-conjugated monoclonal antibodies used in this protocol
LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific, Catalog #L34957); after suspending in DMSO as recommended by the manufacturer, a working stock is prepared by diluting 1:10 in PBS; store at 4 – 8˚C for no longer than 24 hours; LIVE/DEAD Fixable Aqua is used as if it was a surface mAb; samples are stained with 5 μL of the working stock
12 × 75 mm Polystyrene Tubes (BD Falcon, Catalog #352052)
10-mL and 50-mL conical centrifuge tubes
Ammonium-Chloride (ACK) Lysing Buffer (Thermo Fisher Scientific, Catalog #A1049201); store at room temperature (RT)
10X BD FACS™ Lysing Solution (BD, Catalog #349202); store at RT; dilute in purified water prior to use
10% Methanol-free Formaldehyde, Ultra-Pure (Polysciences, Catalog #04018–1); store at RT; dilute in Phosphate Buffered Saline (PBS)
10X Flow Cytometry Stain (FCM) Buffer (Leinco Technologies, Catalog #F1175) containing 0.5% Bovine Serum Albumin (BSA), 0.1% Sodium Azide (NaN3), and 0.04 g/L disodium ethylenediaminetetraacetic acid (NA2EDTA) in PBS; store at 4oC; dilute 1:10 with deionized water
Permeabilization Medium B (Thermo Fisher Scientific, Catalog #GAS002S-100); store at RT; dilute 1:4 with PBS to prepare a working solution
Table 1.
Fluorochrome-conjugated antibodies used in the study.
| Antibody Specificity | Format | Clone | Source | Catalog Number |
|---|---|---|---|---|
| (a) Tube 1 | ||||
| CD138 | BV421 | MI15 | Becton Dickinson | 562935 |
| CD27 | BV510 | O323 | BioLegend | 302835 |
| CD38 | FITC | T16 | Beckman Coulter | IM0775U |
| CD56 | PE | C5.9 | Cytognos | CYT-56PE |
| CD45 | PCPCy5.5 | HI30 | BioLegend | 304027 |
| CD19 | PECy7 | J3–119 | Beckman Coulter | IM3628U |
| CD117 | APC | 104D2 | Becton Dickinson | 341106 |
| CD81 | APCH7 | JS-81 | Becton Dickinson | 656154 |
| (B) Tube 2 | ||||
| CD138 | BV421 | MI15 | Becton Dickinson | 562935 |
| CD27 | BV510 | O323 | BioLegend | 302835 |
| CD38 | FITC | T16 | Beckman Coulter | IM0775U |
| CD56 | PE | C5.9 | Cytognos | CYT-56PE |
| CD45 | PCPCy5.5 | HI30 | BioLegend | 304027 |
| CD19 | PECy7 | J3–119 | Beckman Coulter | IM3628U |
| cKappa | APC | Poly | Dako | C0222 |
| cLambda | APC-C750 | Poly | Cytognos | CYT-LAC750 |
| (C) Tube 3 | ||||
| CD138 | BV421 | MI15 | Becton Dickinson | 562935 |
| Live Dead Aqua | - | - | Thermo Fisher Scientific | L34957 |
| CD38 | FITC | T16 | Beckman Coulter | IM0775U |
| CD45 | PCPCy5.5 | HI30 | BioLegend | 304027 |
| (D) Tube 4 | ||||
| CD27 | BV510 | O323 | BioLegend | 302835 |
| CD56 | PE | C5.9 | Cytognos | CYT-56PE |
| CD45 | PCPCy5.5 | HI30 | BioLegend | 304027 |
| CD19 | PECy7 | J3–119 | Beckman Coulter | IM3628U |
| CD117 | APC | 104D2 | Becton Dickinson | 341106 |
| CD81 | APCH7 | JS-81 | Becton Dickinson | 656154 |
BV421: Brilliant Violet 421; BV510: Brilliant Violet 510; FITC: Fluorescein Isothiocyanate; PE: Phycoerythrin; PerCPCy5.5: Peridinin Chlorophyll Cyanine 5.5; PECy7: Phycoerythrin Cyanine 7; APC: Allophycocyanin; APCH7: Allophycocyanin Hilite 7; APC-C750: Allophycyanin Tandem 750
Equipment:
Clinically-validated flow cytometer equipped with suitable excitation lasers and detectors for measuring fluorescence generated from BV421, BV510, FITC, PE, PCPCy5.5, PECy7, APC, and APCH7
Automated cell counter
Centrifuge capable of at least 520 × g
Falcon® 70 μm cell strainer (Corning, Catalog #352350)
Protocols Steps:
Filter bone marrow sample through a 70 μm cell strainer to exclude spicules from the sample.
- Perform cell count and transfer at least 30 × 106 white blood cells (in a volume of less than 2 mL) to a 50-mL Falcon tube. Fill the tube to 50 mL mark using ACK Lysing Buffer.
- If the bone marrow volume exceeds 2 mL, use multiple 50-mL Falcon tubes.
Let sample lyse at RT for 10 minutes.
Centrifuge at 520 × g for 5 minutes and wash the sample once with 50 mL of FCM buffer to remove residual ACK Lysing Buffer solution.
Perform absolute cell count and adjust cell concentration to 5 × 107 cells/mL using FCM buffer.
- Separately label sufficient 12 × 75 mm polystyrene tubes (e.g. Tube 1, Tube 2, and Tube 3). To Tube 1 and Tube 2, transfer 100 – 200 μL (5 – 10 × 106 cells). To determine sample viability, transfer 10 – 20 μL (5 – 10 × 105 cells) to Tube 3.
- If the sample is limiting, priority should be given to collecting a sufficient number of cells in Tube 1. Any remaining sample can be used for Tube 2 and Tube 3.
- While it is recommended that sample viability be obtained for each sample, Tube 3 has the lowest priority.
- Add mAbs for surface labeling and incubate for 30 minutes at RT in the dark.
- Information about the mAbs used for labeling Tube 1, Tube 2, and Tube 3 can be found in Table 1. Each of the mAb used should be titrated individually and use at saturation for optimal results.
Add 2 mL of BD FACS™ Lysing Solution to each tube. Let sit for 10 minutes at RT in the dark.
Centrifuge at 520 × g for 5 minutes and wash once with FCM buffer to remove residual lysis solution.
For tubes to be labeled with surface antibodies only (e.g. Tube 1 and Tube 3), resuspend the cells in 500 μL of 0.5% methanol-free formaldehyde and proceed to Step 15 for MFC data acquisition. For tubes to be labeled with intracellular antibodies to immunoglobulin light chains (e.g. Tube 2), proceed to Step 11.
For intracellular staining (e.g. Tube 2), resuspend the cells in 100 μL of 2% formaldehyde and incubate for 10 minutes at RT in the dark.
Wash the cells once with 3 mL of FCM buffer and centrifuge at 520 × g for 5 minutes. Resuspend the residual volume with 100 μL of diluted Permeabilization Medium B.
Add saturating concentrations of anti-Kappa and anti-Lambda antibodies and incubate the cells for 30 minutes at RT in the dark.
- Add 3 mL of FCM buffer and let sit for 10 minutes at RT in the dark. Centrifuge the cells at 520 × g for 5 minutes and resuspend residual volume using 500 μL of FCM buffer.
- If storage of samples is desired before data acquisition, resuspend the samples using 0.5% methanol-free formaldehyde and store at 4oC for no longer than 3 days.
Setup and optimize the flow cytometer’s voltages and compensation for data acquisition using standardized methods as described by Wang et al. (Wang et al., 2017). Adjust threshold setting based on the forward scatter light characteristics to include hematogones while judiciously avoiding the recording of unwanted background noise.
Analysis and Gating Strategy:
-
16.Identification of Total Plasma Cells:
- The recommended gating strategies employed for the phenotypic identification, enumeration, and characterization of normal and malignant PCs, as well as the identification of mast cells, hematogones, and erythroid precursors to assess the quality of the bone marrow samples are described below.
- Analyses performed in this section are not software-specific, as any commercially available software capable of generating MFC plots and associated results from millions of events can be used.
- On a bivariate plot of Time vs. FSC-A (Figure 1A), create and place a rectangular region (R1) to include all valid events acquired in chronologic homogeneity.
- On a bivariate plot of FSC-A vs. FSC-H (Figure 1B), draw a rectangular region (R2) to circumscribe single cell events. Gate this bivariate plot on (R1).
- When defining the singlet cells, be careful not to exclude PCs as they can be large and may fall slightly outside a tightly defined singlet region. Back-gating the FSC-A vs. FSC-H bivariate plot on CD38+ and CD138+ events will help inform how this region is placed especially when a small number of PCs are present.
- On a bivariate plot of FCS-A vs. SSC-A (Figure 1C), construct a generous region (R3) to include all the cellular events that are of interest, while excluding aggregate, debris, and dead cells. Gate this bivariate plot on (R1 & R2). Events that are gated on (R1 & R2 & R3) are defined as the ‘Total Cells’.
- PCs may exhibit aberrantly high light scatter characteristics. Caution should be exercised not to exclude these cell populations of interest.
- Generate 3 additional bivariate plots of CD138 BV421 vs. CD38 FITC (Figure 1D), CD45 PCPCy5.5 vs. CD38 FITC (Figure 1E), and CD45 PCPCy5.5 vs. CD138 BV421 (Figure 1F). Gate these 3 dot plots on ‘Total Cells’. On Figure 1D, create an irregular region (R4) to identify CD138+, CD38+ events. On Figure 1E, create an irregular region (R5) to identify CD45het, CD38+ events. On Figure 1F, create an irregular region (R6) to identify CD45het, CD138+ events. The combined Boolean gate definition of (R1 & R2 & R3 & R4 & R5 & R6) represents the ‘Total Plasma Cells’.
- Create a bivariate plot of CD38 FITC vs. CD45 PCPCy5.5 gated on ‘Total Cells’ (Figure 1G), draw an irregular region (R7) to include all CD45+ leukocytes and CD38+ PCs. Gate this bivariate on (R1 & R2 & R3). Cells that are gated on (R1 & R2 & R3 & R7) are defined as the ‘Total Leukocytes’ and used as the denominator for reporting percentages. The final patient report should clearly state the calculated percentages are based on the total leukocyte population.
-
17.Immunophenotypic differences between normal and abnormal plasma cells:
- Multiple myeloma is a heterogeneous disease where the myeloma cells can upregulate or down-modulate a broad number of antigens at the same time. Therefore, the identification of abnormal PCs can be challenging.
- Draw all permutations of bivariate plots gated on ‘Total Plasma Cells’.
-
The antigen expression profiles of normal vs. abnormal PCs can be found in Table 2. An example representing some of the expected results from these analyses can be found in Figure 2.
- Non-neoplastic PCs exhibit heterogeneous expression levels of CD45 and CD19, are mostly negative for CD56 and CD117, and invariably show homogeneous bright expression of CD81 and CD27. Dim or absent CD81 and/or CD27 expression is only observed in abnormal PCs. The expression of CD56 and the lack of CD19 are observed in a small subset of non-neoplastic PCs (between 5 – 20% of all PCs) with higher percentages reported in post-treatment bone marrow samples. Due to the clonal nature of neoplastic PCs, their antigen expression is often more uniform rather than present across a heterogeneous spectrum as seen in normal bone marrow. Additionally, increased CD117 expression and diminished expression of CD27 are often observed in neoplastic PCs. The myeloma cells are often slightly dimmer for CD38 than normal PCs. These aberrancies will be co-expressed within the same cell population, whereas immunophenotypic variations in normal PCs tend to be heterogeneous and/or distributed among different subsets. Lastly, provided a sufficient number of PCs are acquired, analysis of cytoplasmic light chain expression within subsets showing myeloma-like aberrant phenotypes (e.g. CD19- or CD56+ PCs), while not routinely required, can be a valuable supplementary verification step to confirm the clonal nature of a suspicious population. Reliance solely on cytoplasmic light chain clonality can be misleading when minor populations of abnormal PCs become obscured within the polyclonal non-neoplastic population (Figure 2C).
-
Determine which combination of markers best separate normal from abnormal plasma cells and draw regions around each. Assign distinct colors to each population.
- Contemporary standards require at least 2 atypically expressed markers be used to reliably distinguish myeloma cells from normal PCs.
-
18.Results Reporting:
- Create a final report to include all the following information (Arroz et al., 2016):
-
(i)Number and percentage of mast cells, hematogones, and erythroid precursors for quality assessment of the bone marrow specimen;
-
(ii)Total number of CD45+ leukocytes evaluated after excluding doublets and debris;
-
(iii)Number and percentage of normal and abnormal PCs;
-
(iv)the estimated Limit of Detection (LOD) and/or Lower Limit of Quantification (LLOQ) of the sample;
-
(v)Immunophenotypic profiles of abnormal PCs; and
-
(vi)Categorical assessment of MRD status (e.g. MRD positive, MRD negative, or Equivocal).
-
(i)
- Use the following equation to calculate sample specific LOD and LLOQ:
- It is important that the MRD report also include information about the estimated detection limits for each individual sample. This is based on (i) the number of leukocytes acquired and (ii) the number of cells required to reproducibly detect a neoplastic population. These values cannot be lower than the LOD and LLOQ as established in SUPPORT PROTOCOL 2 but may be higher if the sample is limiting and an insufficient number of cells were acquired.
- In our validation and that of others, 20 clustered cells is generally the smallest number of events that can conservatively define an abnormal PC population and 50 events is the smallest number that can be reproducibly quantified, nevertheless, these cutoffs should be independently determined by individual laboratories (Arroz et al., 2016; Hedley et al., 2013; Selliah et al., 2019; Subira et al., 2002).
-
19.Quality assessment of bone marrow aspirate:
- The detection of mast cells, hematogones, and erythroid precursors is a convenient method to assess the quality of bone marrow samples since they are rarely found in peripheral blood.
- Samples that have ≥20 abnormal PCs are by definition considered adequate. Otherwise, samples are considered adequate if mast cells are present and/or hematogones with erythroid precursors. Samples lacking these constitutes are judged to be blood diluted and should be indicated as such on the final report.
- For mast cells, place an elliptic region (R8) on a bivariate plot of CD27 BV510 vs. CD117 APC gated on ‘Total Cells’ (defined in Step 16c) to circumscribe CD27-, CD117br events (Figure 3A).
- For erythroid precursors, create a bivariate plot of SSC-A vs. CD45 PCPCy5.5 gated on ‘Total Cells’ and place an irregular region (R9) to circumscribe events defined as CD45-/lo, SSClo (Figure 3B).
- For hematogones, first define CD19+ B cells by constructing a CD56 PE vs. CD19 PECy7 plot gated on ‘Total Cells’ and placing a rectangular region (R10) around the CD56-, CD19+ events (Figure 3C). Sequentially apply this region (R10) to a bivariate plot of CD45 PCPCy5.5 vs. CD81 APCH7. Create a region (R11) to include cells that are CD45dim, CD81br to identify the hematogones (Figure 3D).
Figure 1: Detection of Total Leukocytes and Total Plasma Cells by high sensitivity MFC.
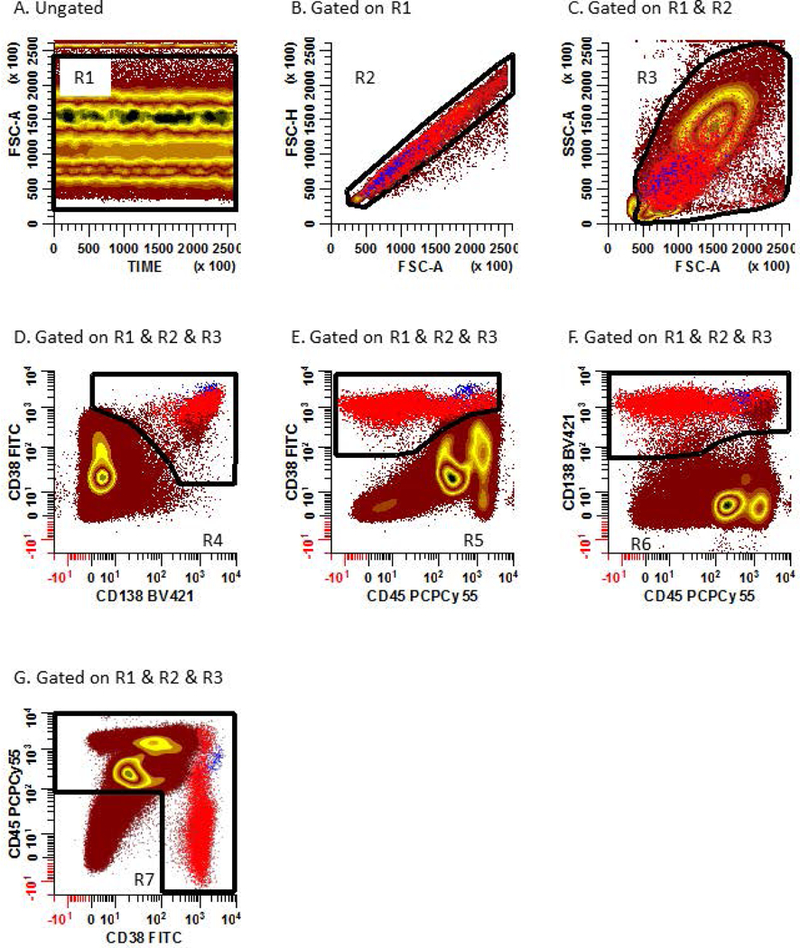
(A) A rectangular region (R1) is created on a bivariate plot of Time (event chronology) vs. FSC-A to assess the compositional homogeneity of collected events. Disinterested and invalid events such as air bubbles collected during sample acquisition can be excluded using this plot. (B) A rectangular region (R2) is created on a gated (R1) bivariate plot of FSC-A vs. FSC-H to include singlet cells event. (C) An irregular region (R3) is created on a gated (R1 and R2) bivariate plot of FSC-A vs. SSC-A to circumscribe all viable cells while excluding debris and large aggregated events. Three separate bivariate plots, each gated on (R1, R2, and R3), are created: (D) CD138 BV421 vs. CD38 FITC, (E) CD45 PCPCy5.5 vs. CD38 FITC, and (F) CD45 PCPCy5.5 vs. CD138 BV421. On dot plot D, create an irregular region (R4) to identify CD138+, CD38+ events. On dot plot E, create an irregular region (R5) to identify CD45het, CD38+ events. On dot plot F, create an irregular region (R6) to identify CD45het, CD138+ events. Normal (blue) and abnormal (red) PCs are defined by the Boolean gate definition (R1 & R2 & R3 & R4 & R5 & R6). (G) An irregular region (R7) is created on a gated (R1, R2, and R3) bivariate plot of CD45 PCPCy5.5 vs. CD38 FITC to circumscribe CD45+ leukocytes and CD38br PCs. ‘Total Leukocytes’ are defined as events falling within (R1 & R2 & R3 & R7).
Table 2.
Antigen expression patterns of normal plasma cells vs. myeloma cells.
| Antigen(s) | Normal Plasma Cells | Myeloma Cells | ||
|---|---|---|---|---|
| Expression Level |
Percentage | Expression Level |
Percentage | |
| CD45 | +++ | 94% | - | 73 – 80% |
| CD38 | +++ | 100% | + | 80% |
| CD138 | +++ | 98% | +++ | 98% |
| CD19 | ++ | >70% | - | 95% |
| CD56 | - | >85% | +++ | 60 – 75% |
| CD117 | - | 100% | ++ | 30 – 32% |
| CD27 | +++ | 100% | - to + | 40 – 68% |
| CD81 | ++ | 100% | - to + | 55% |
Dimly Positive
Moderately Positive
Strongly Positive
Negative
Figure 2: Immunophenotypic profiles of normal and abnormal PCs.
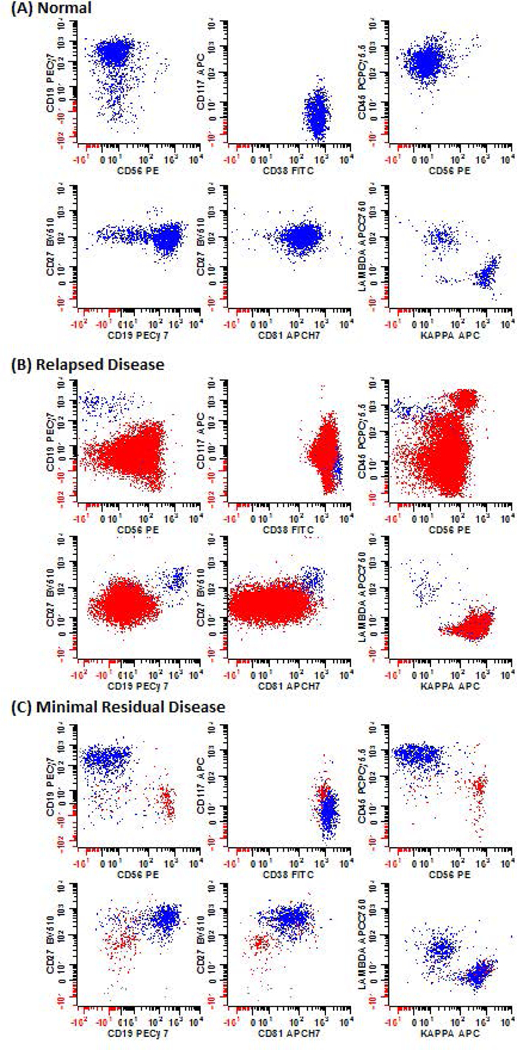
Bone marrow samples from patients with (A) no obvious hematological disease, (B) relapsed disease, and (C) MRD. Samples were stained using the high sensitivity MFC assay. As MM is a heterogeneous disease, different permutations of markers were utilized to distinguish myeloma cells from the normal PCs. Blue: normal PCs; Red: neoplastic PCs.
Figure 3: Quality assessment of bone marrow samples based on the recovery of mast cells, hematogones, and erythroid precursors.
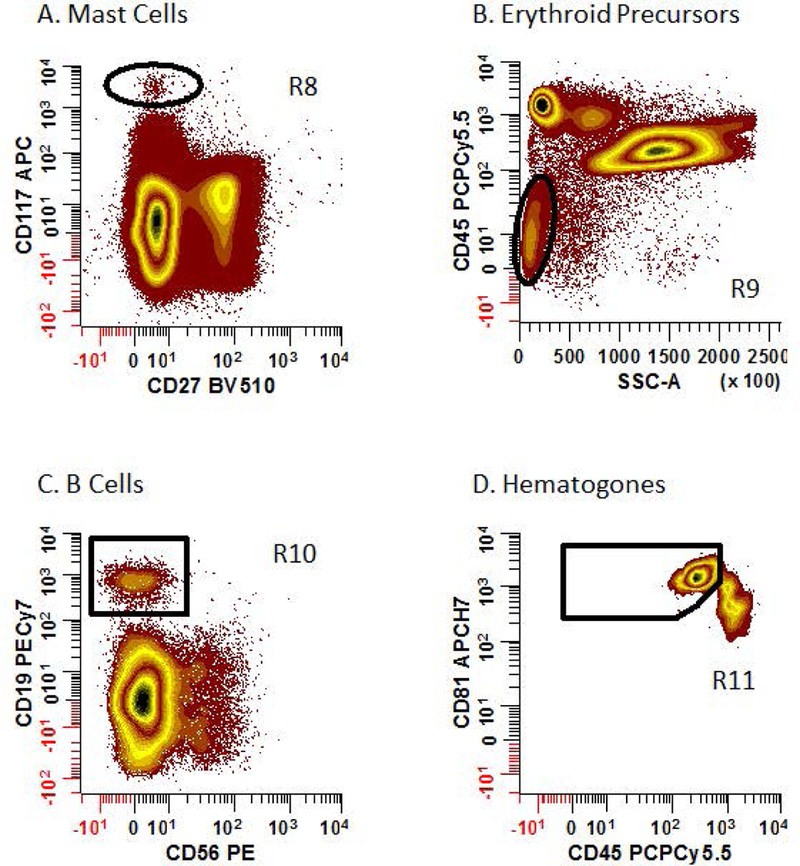
All plots shown are gated on [R1 & R2 & R3 & NOT (R4 & R5 &R6)]. (A) Mast cells (defined as CD27-, CD117br) are identified by drawing an elliptical region (R8) on a bivariate plot of CD27 BV510 vs. CD117 APC. (B) An elliptical region (R9) is placed on a bivariate plot of SSC-A vs. CD45 PCPCy5.5 to identify erythroid precursors (defined as CD45-, SSClo). (C & D) B cells (defined as CD56-, CD19+) are identified by placing a rectangular region (R10) on a bivariate plot of CD56 PE vs. CD19 PECy7. B cell are then delineated using a bivariate plot of CD45 PCPCy5.5 vs. CD81 APCH7. A region (R11) is then drawn to identify hematogones (defined as CD45dim, CD81br).
ALTERNATE PROTOCOL 1
PROCESSING OF BONE MARROW SAMPLES USING THE ‘POOLED-TUBE’ METHOD
An alternative sample staining method which may better fit into the routine sample processing workflow of some laboratories, but will require greater amounts of mAb, is termed the ‘Pooled-tube’ method. This alternative staining method requires only one lysis step, is deemed less stringent and preserves most mAb epitopes better than the ‘Bulk-lysis’ method. The ‘Pooled-tube’ method was devised to maximize cell recovery during processing while maintaining the assay’s high sensitivity to detect MRD. Both protocols are acceptable for MM MRD testing by MFC as they yield comparable results. It is up to the laboratory’s discretion as to which procedures is adopted.
Materials
Specimens:
See BASIC PROTOCOL 1
Reagent:
See BASIC PROTOCOL 1
Equipment:
See BASIC PROTOCOL 1
Protocol Steps:
Filter bone marrow specimen through a 70 μm cell strainer.
Perform cell count and adjust the concentration to 10 × 106 cells/mL using FCM Buffer. For the MM MRD Tube 1 and 2 (Table 1), 4 – 10 × 106 cells/tube is required; for MM MRD Tube 3 a minimum of 5 × 105 cells is required.
Separately label sufficient 12 × 75 mm polystyrene tubes (e.g. Tube 1, Tube 2, and Tube 3). Transfer 200 μL of washed bone marrow samples into a minimum of 2 and, if sample permitting, 6 separate 12 × 75 mm polystyrene tubes for Tube 1. Separately transfer 200 μL of washed bone marrow samples into 2 – 6 separate 12 × 75 mm polystyrene tubes for Tube 2 and 50 – 100 μL to one 12 × 75 mm polystyrene tube for Tube 3.
- Add mAbs used for a surface staining and incubate the bone marrow cells for 30 minutes at RT in the dark.
- Information about the mAbs used for labeling Tube 1, Tube 2, and Tube 3 can be found in Table 1. Each of the mAb used should be titrated individually and used at saturation for optimal results.
Add 2 mL of BD FACS™ Lysing Solution to Tube 1, Tube 2, and Tube 3. Let sit for 10 minutes at RT in the dark.
Centrifuge the cells at 520 × g for 5 minutes, decant and wash each tube with 3 mL of FCM buffer to remove residual lysis solution.
After centrifugation, decant each tube, resuspend the cell pellet in 0.5 mL of FCM buffer, and combine similarly stained tubes into a labeled 12 × 75 mm tube.
Centrifuge the cells at 520 × g for 5 minutes and decant. For tubes to be labeled with surface antibodies only (e.g. Tube 1 and Tube 3), resuspend the cells in 500 μL of 0.5% methanol-free formaldehyde and proceed to Step 13 for flow cytometric data acquisition. For tubes to be labeled with intracellular antibodies (e.g. Tube 2), proceed to Step 9.
For intracellular staining, resuspend the cells in 100 μL of 2% methanol-free formaldehyde. Incubate for 10 minutes at RT in the dark.
Wash the cells with 3 mL of FCM buffer and centrifuge at 520 × g for 5 minutes. Resuspend the residual volume with 100 μL of diluted Permeabilization Medium B.
Add saturating concentrations of anti-cKappa and anti-cLambda antibodies and incubate the cells for 30 minutes at RT in the dark.
- Add 3 mL of FCM buffer and let sit for 10 minutes at RT in the dark. Centrifuge the cells at 520 × g for 5 minutes and resuspend residual volume using 500 μL of FCM buffer.
- If storage of samples is desired before data acquisition, resuspend the samples using 0.5% methanol-free formaldehyde and store at 4oC for no longer than 3 days.
Setup and optimize the flow cytometer for data acquisition using standardized methods as described by Wang et al. (Wang et al., 2017). Adjust the threshold based on forward scatter light characteristics to include hematogones while judiciously avoiding the recording of unwanted background noise. Export all acquired data in listmode FCS format (Version 3.0 or higher).
Analysis and Gating Strategy:
See BASIC PROTOCOL 1.
SUPPORT PROTOCOL 1
VALIDATION OF FLUROROCHROME-CONJUGATED MONOCLONAL ANTIBODIES
MFC immunophenotyping can effectively distinguish normal vs. abnormal PCs when the right combinations of fluorochrome-antibodies are used. Careless selection of staining reagents, however, can result in reduced fluorescence intensity, increased non-specific or background fluorescence, excessive fluorescence crosstalk between channels, and difficulty in circumscribing certain cell populations due to signal broadening. The mAb, clone, and fluorochrome combinations used in this MM MRD panel have been optimized and validated by several independent laboratories. While it is recommended that the standardized combination for Tube 1 and Tube 2 described in Table 1 be used, modifications can be made if the laboratory established that their changes are at least as good as or better than the recommended combinations.
An effective strategy to determine the performance of an mAb is to evaluate its staining resolution in conjunction with all the other mAbs used in the tube, rather than testing the mAb alone. This is best accomplished by defining a negative and positively stained population and calculating their stain indices as first described by Rawstron and colleagues (Rawstron et al., 2016). Since the negative and positive populations used for calculating stain indices can be extracted from within the sample, isotype controls are not required for this evaluation.
Materials
Specimen:
Fresh, remnant EDTA or sodium heparin anti-coagulated human bone marrow aspirates with a viability >85% should be used in this support protocol.
After processing for routine testing, remnant bone marrow samples can be diluted 50:50 in RPMI 1640 to help maintain stability and viability.
Reagents:
See BASIC PROTOCOL 1
CD-Chex CD103™ Plus (Streck, Catalog #213567)
RPMI 1640 with L-glutamine (Corning, Catalog #10–040-CV)
Equipment:
See BASIC PROTOCOL 1
Protocol Steps:
-
1.
Stain the bone marrow cells and acquire the data according to Steps 1 – 15 of BASIC PROTOCOL 1 or Steps 1– 13 for ALTERNATE PROTOCOL 1.
-
2.
Calculate the stain index for each mAb using the formula provided below. Use the gating strategies detailed in Step 3 – 12 below to create a series of bivariate plots for identifying appropriate negative and positive populations used for calculating stain indices for each mAb.
- Stain index is a well-accepted and reliable measure that can be used to evaluate the performance of individual mAb employed in a multi-color tube; it can be calculated using the following formula:
A summary of the gating definitions used to determine negative and positive populations for analysis of this equation in a one parameter histogram can be found in Table 3 and the minimum recommended Stain Index for each fluorochrome-conjugated mAb can be found in Table 4.
-
3.
CD45 (Figure 4A)
Negative population: On a bivariate plot of SSC-A vs. CD45 PCPCy5.5, create an oval region (r1) to circumscribe erythroid precursors (blue events) that are defined as SSClo, CD45-events.
Positive population: On a bivariate plot of SSC-A vs. CD45 PCPCy5.5, create an oval region (r2) to circumscribe CD45+ cells with lymphocytic scatter characteristic. Sequentially apply the region r2 onto a new bivariate plot of CD56 PE vs. CD19 PECy7 and draw a region (r3) to identify T cells that are defined as CD19-, CD56-, CD45br, SSClo (red events).
-
4.
CD19 (Figure 4B)
Negative population: On a bivariate plot of CD45 PCPCy5.5 vs. CD56 PE, create an oval region (r4) to circumscribe NK cells (blue events) that are defined as CD45br, CD56+ events.
Positive population: Create a bivariate plot of CD19 PECy7 vs. CD45 PCPCy5.5 gate it on (r2). Draw an oval region (r5) to circumscribe the majority of B cells (red events) that are defined as CD19+, CD45br, SSClo events.
-
5.CD27 (Figure 4C)
- Negative population: On a bivariate plot of CD45 PCPCy5.5 vs. CD117 APC, create an oval region (r6) to circumscribe mast cells (blue events) that are defined as CD45+, CD117br events.
- Positive population: Create a bivariate plot of CD56 PE vs. CD19 PECy7 and gate it on (r2). Draw an oval region (r7) to identify T cells that are defined as CD19-, CD56-, CD45br, SSClo (red events).
-
6.
CD81 (Figure 4D)
Negative population: On a bivariate plot of SSC-A vs. CD45 PCPCy5.5, create an oval region (r8) to circumscribe granulocytes (blue events) that are defined as CD45dim, SSChi events.
Positive population: On a bivariate plot of SSC-A vs. CD45 PCPCy5.5, create an oval region (r9) to circumscribe CD45+ cells with lymphocytic scatter characteristic. Sequentially apply the region (r9) onto a new bivariate plot of CD56 PE vs. CD19 PECy7 and draw a region (r10) to identify T cells that are defined as CD19-, CD56-, CD45br, SSClo (red events).
-
7.
CD56 (Figure 4E)
Negative population: Create a new bivariate plot of CD56 PE vs. CD19 PECy7 gated on (r2). Draw a region (r11) to identify B cells that are defined as CD19+, CD56-, CD45br, SSClo (blue events).
Positive population: On the bivariate plot of CD56 PE vs. CD19 PECy7 gated on (r2) draw a region (r12) to identify NK cells that are defined as CD19-, CD56+, CD45br, SSClo (red events).
-
8.
CD117 (Figure 4F)
Negative population: Create a bivariate plot of CD56 PE vs. CD19 PECy7 and gate it on (r2). Draw an oval region (r13) to identify T cells that are defined as CD19-, CD56-, CD45br, SSClo (blue events).
Positive population: Draw an oval region (r14) to identify mast cells that are defined as CD117br, CD27- (red events).
-
9.
CD138 (Figure 4G)
For CD138 testing bone marrow samples are spiked with 15 μL of CD-Chex CD103™ Plus which also express CD138.
-
a.
Negative population: Create a bivariate plot of CD56 PE vs. CD19 PECy7 and gate it on (r2). Draw an oval region (r15) to circumscribe B cells (blue events) that are defined as CD19+, CD45br, CD56-, SSClo events.
-
b.
Positive population: On a bivariate plot of CD81 APCH7 vs. CD45 PCPCy5.5, create an elliptical region (r16) to circumscribe CD81+, CD45dim CD-Chex CD103™ Plus cells (red events) that are defined as CD81+, CD45dim events.
-
10.
CD38 (Figure 4H)
Negative population: On a bivariate plot of CD81 APCH7 vs. CD19 PECy7, draw an oval region (r17) to circumscribe mature B cells (blue events) that are defined as CD19+, CD81dim events.
Positive population: On a bivariate plot of CD81 APCH7 vs. CD19 PECy7, draw an oval region (r18) to circumscribe B cell progenitors (red events) that are defined as CD19+, CD81br events.
-
11.
cKappa (Figure 4I)
Negative population: Create a bivariate plot of cLambda APC-C750 vs. CD19 PECy7 and gate it on (r2). Draw a region (r19) to identify cLambda+ B cells that are defined as CD19+, CD45br, cLambda+, SSClo (blue events).
Positive population: On the bivariate plot of cLambda APC-C750 vs. CD19 PECy7 gated on (r2) draw a region (r20) to identify cLambda- B cells that are defined as CD19+, CD45br, cLambda-, SSClo (red events).
-
12.
cLambda (Figure 4J)
Negative population: Create a bivariate plot of cKappa APC vs. CD19 PECy7 gated on (r2). Draw a region (r21) to identify cKappa+ B cells that are defined as CD19+, CD45br, cKappa+, SSClo (blue events).
Positive population: On the bivariate plot of cKappa APC vs. CD19 PECy7 gated (r2) and draw a region (r22) to identify cKappa- B cells that are defined as CD19+, CD45br, cKappa-, SSClo (red events).
Table 3.
Immunophenotypic definition of the negative and positive populations used for calculating Stain Index.
| Tested Markers |
Negative Population |
Positive Population |
||
|---|---|---|---|---|
| Phenotype | Generic Name | Phenotype | Generic Name | |
| CD45 | CD45-, SSClo | Erythroid Precursors | CD19-, CD56-, CD45br, SSClo | T Cells |
| CD19 | CD45br, CD56+, SSClo | NK Cells | CD19+, CD45br, SSClo | B Cells |
| CD27 | CD45+, CD117br | Mast Cells | CD19-, CD56-, CD45br, SSClo | T Cells |
| CD81 | CD45dim,SSChi | Granulocytes | CD19-, CD56-, CD45br, SSClo | T Cells |
| CD56 | CD19+, CD56-, CD45br, SSClo | B Cells | CD19-, CD56+, CD45br, SSClo | NK Cells |
| CD117 | CD19-, CD56-, CD45br, SSClo | T Cells | CD27, CD117br | Mast Cells |
| CD138 | CD19+, CD56-, CD45br, SSClo | B Cells | CD45dim, CD81+ | Spiked Controla |
| CD38 | CD19+, CD81dim | Mature B | CD19+, CD81+ | B-progenitors |
| cKappa | CD19+, CD45br, cLambda+, SSClo | cLambda+ B Cells | CD19+, CD45br, cLambda-, SSClo | cLambda- B Cells |
| cLambda | CD19+, CD45br, cKappa+, SSClo | cKappa+ B cells | CD19+, CD45br, cKappa-, SSClo | cKappa- B cells |
A procedural control for immunophenotyping, CD-Chex CD103™ Plus (Streck, Omaha, NE), which also expresses CD138 is spiked into the bone marrow sample and used as the positive population. Modified from (Soh et al., 2017)
Table 4.
Evaluation of staining performance of monoclonal antibody in the context of multicolor analysis
| Tested Markers | Evaluated Sample, | Stain Index |
||
|---|---|---|---|---|
| na | Mean ± SD | Range | Recommended | |
| CD45 PCP-Cy5.5 | 10 | 170.2 ± 55.7 | 98.8 – 306.3 | >80 |
| CD19 PECy7 | 10 | 7.1 ± 1.7 | 3.9 – 10 | >3 |
| CD27 BV510 | 10 | 27.7 ± 12 | 14.4 – 52.4 | >10 |
| CD81 APCH7 | 10 | 9.8 ± 1.8 | 7.9 – 14.3 | >5 |
| CD56 PE | 10 | 16.5 ± 4.5 | 12.3 – 25.5 | >10 |
| CD117 APC | 10 | 7.7 ± 1.3 | 5.6 – 10.1 | >5 |
| CD138 BV421 | 10 | 60 ± 33.9 | 26.9 – 123.2 | >20 |
| CD38 FITC | 10 | 13.2 ± 7.1 | 3.9 – 26.6 | >3 |
| cKappa APC | 10 | 10.2 ± 5.4 | 3.5 – 18.1 | >3 |
| cLambda APC-C750 | 10 | 8.2 ± 4.1 | 3.4 – 16.5 | >3 |
Ten random patient bone marrow samples received for lymphoma evaluation with no flow cytometric or morphological evidence of hematological disease
Figure 4: Identification of negative and positive populations for calculating stain indices to evaluate antibodies performance.
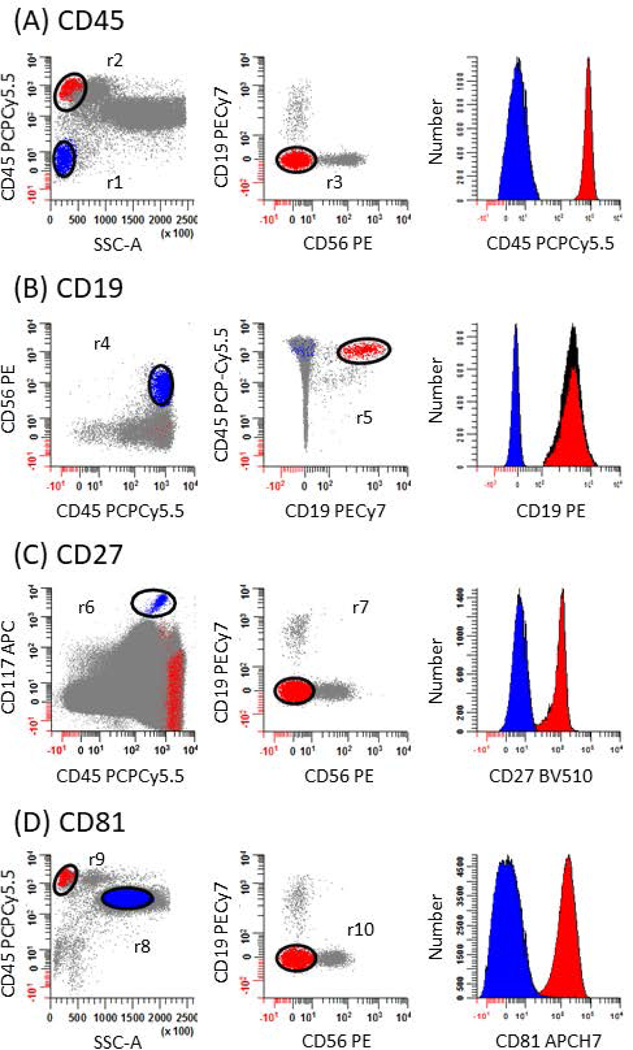
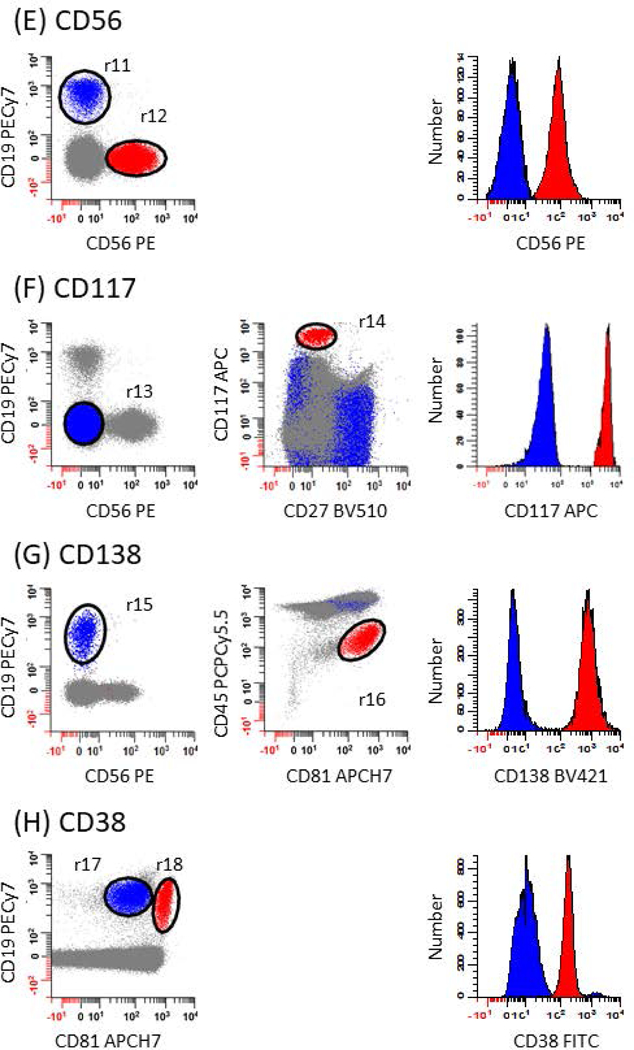
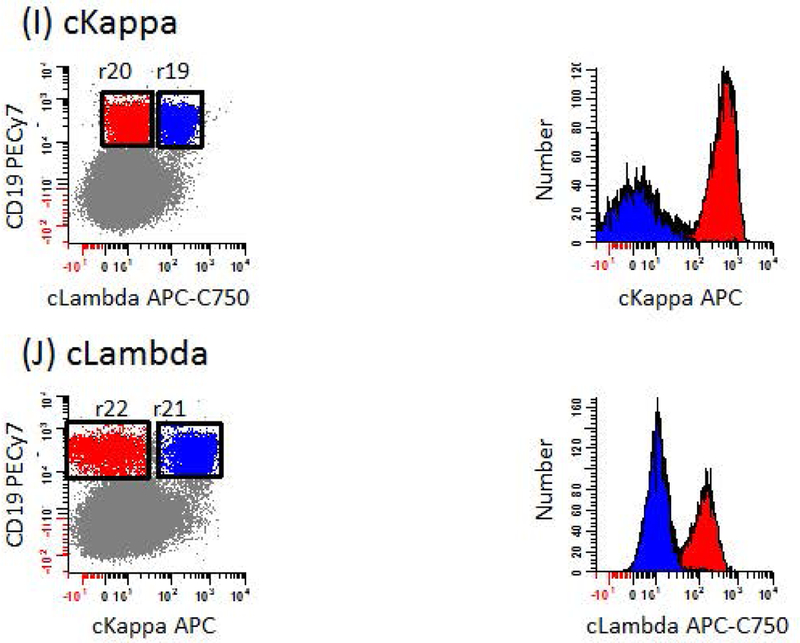
The strategy used to assess the performance of each mAb in the panel relies on defining negative (blue events) and positive (red events) populations for calculating stain indices. This is accomplished by first subtracting the median intensity of negative population from the median intensity of the positive population and dividing the number by 2 times the robust standard deviation of the negative population. A list of gating definitions can be found in Table 2. (A) For CD45, erythroid precursors are used as the negative population defined as SSClo, CD45- events (r1; blue dots) and T cells as the positive population defined as CD19-, CD56-, CD45br, SSClo events (r2 & r3; red dots). (B) For CD19, NK cells are used as the negative population defined as CD45br, CD56+ events (r4: blue dots) and B cells as the positive population defined as CD19+, CD45br events (r5; red dots). (C) For CD27, mast cells are used as the negative population defined as CD45dim, CD117br events (r6; blue dots) and T cells as the positive population defined as CD19-, CD56-, CD45br, SSClo events (r2 & r7; red dots). (D) For CD81, granulocytes are used as the negative population defined as CD45dim, SSChi events (r8; blue dots) and T cells as the positive population defined as CD19-, CD56-, CD45br, SSClo events (r9 & r10; red dots). (E) For CD56, B cells are used as the negative population defined as CD19+, CD56-, CD45br, SSClo events (r2 & r11; blue dots) and NK cells as the positive population defined as CD19-, CD56+, CD45br, SSClo events (r2 & r12; red dots). (F) For CD117, T cells are used as the negative population defined as CD19-, CD56-, CD45br, SSClo events (r2 & r13; blue dots) and mast cells as the positive population defined as CD27-, CD117br events (r14; red dots). (G) For CD138, B cells are used as the negative population defined as CD19+, CD56-, SSClo events (r2 & r15; blue dots) and CD-Chex CD103™ Plus cells as the positive population defined as CD81+, CD45dim events (r16; red dots). (H) For CD38, mature B cells are used as the negative population defined as CD19+, CD81dim events (r17; blue dots) and B progenitors as the positive population defined as CD19+, CD81br events (r18; red dots). (I) For cKappa, cLambda+ B cells are used as the negative population defined as CD19+, cLambda+, CD45br, SSClo events (r2 & r19; blue dots) and cLambda- B cells as the positive population defined as CD19+, cLambda-, CD45br, SSClo (r2 & r20; red dots). (J) For cLambda, cKappa+ B cells are used as the negative population defined as CD19+, cKappa+, CD45br, SSClo events (r2 & r21; blue dots) and cKappa- B cells as the positive population defined as CD19+, cKappa-, CD45br, SSClo (r2 & r22; red dots).
SUPPORT PROTOCOL 2
ESTABLISHING ASSAY SENSITIVITY, LIMIT OF DETECTION, LIMIT OF BLANK, AND LOWER LIMIT OF QUANTIFICATION
Sensitivity of an assay, as defined by Clinical Laboratory Standards Institute (CLSI) is the “measured quantity value for which the probability of falsely claiming the absence of a measurand is β, given a probability α of falsely claiming its presence” (CLSI, 2012). In another words, it describes the probability of an assay to correctly and reliably distinguish the presence of detected signals from background. For the assessment of a low number of events, there is a need to establish limit of blank (LOB), limit of detection (LOD), and the lower limit of quantification (LLOQ) (Armbruster et al., 2008; Barnett et al., 2013; O’Hara et al., 2011). The LOB is defined as the lowest apparent signal (e.g. number or percentage of events) in the absence of measurand falling into the regions used to define PCs. This can be measured in the absence of measurand (e.g. with a bone marrow sample known to lack PCs) or in a bone marrow sample stained without the specific antibodies used to define PCs (e.g. CD38 and CD138). Since bone marrow would normally contain some level of PCs, the latter approach is more practical. The LOD is defined as the ability of an assay to detect the measurand (e.g. number or percentage of cells in a fully stained panel) at a level that can reliably be distinguished from the LOB, such that >95% of samples with low levels of measurand (e.g. PCs) will be detected above the LOB.
The LLOQ is defined as the lowest number or percentage of cells that can be reliably detected above background noise at some predefined criteria for bias and imprecision. Generally, in clinical assays a coefficient of variance (CV) of ≤20% is considered as the acceptable cut-off but in assays evaluating cellular subsets with a frequency or percentage approaching that of the LOD, a CV of ≤30% can be used (Lee et al., 2006). The LLOQ can be validated by enumerating samples with levels of measurand approaching the LOD using a minimum of 5 levels (e.g. different concentrations) in 3 – 5 replicates. A practical way to determine the LLOQ is to serially dilute a known positive sample into a negative sample. The same sample matrix (e.g. bone marrow) should be used for both and the defined number of events for which the assay is being validated collected.
Materials
Specimens:
See SUPPORT PROTOCOL 1. For establishing LLOQ, pooled bone marrow samples that are negative for hematological disease as evidenced by MFC and morphological findings are suggested for use as the dilution matrix.
Reagent:
See BASIC PROTOCOL 1
Equipment:
See BASIC PROTOCOL 1
Protocol Steps:
-
Establishing LOB and LOD:
Follow Steps 1 – 5 of the BASIC PROTOCOL 1.
Separately label 5 – 10, 12 × 75 mm polystyrene tubes. Transfer 100 – 200 μL (5 – 10 × 106 cells) of bone marrow sample without PC dyscrasia into each tube.
Add all the mAbs routinely used for surface labeling except CD38 and CD138 (e.g. Tube 4 as shown in Table 1) and incubate the samples for 30 minutes at RT in the dark.
Proceed to Step 8 of BASIC PROTOCOL 1 and follow through to the end.
Acquire and analyze the data as described in the BASIC PROTOCOL 1.
See Step 16 of BASIC PROTOCOL 1 for the Boolean definition of events falling into the regions used to define PCs.
-
The equations used for the calculation of LOB and LOD are shown below:
Determine the number of PCs identified in Step 6.
Ideally, use a minimum of 5 – 10 replicates in the absence of measurand for these calculations (Armbruster et al., 2008; Barnett et al., 2013; Wood et al., 2013).
-
a.
LOB = Mean of Blank + 1.645 × Standard Deviation of Blank
-
b.
LOD = Mean of Blank + 3 × Standard Deviation of Blank
-
Interpret the calculated LOB and LOD as below:
The LOD can be equal to or greater than the LOB.
The LOB as calculated above is usually lower than 10 events and as a community, Cytometrists will generally concur a LOD of 20 events is a conservative value for the smallest homogeneous population that can be reliably detected by qualified personnel (Arroz et al., 2016; Hedley et al., 2013; Subira et al., 2002). See ANTICIPATED RESULTS for an example.
-
Establishing LLOQ:
-
9.
Follow Steps 1 – 5 of the BASIC PROTOCOL 1.
-
10.
In at least triplicate, prepare 1:10 serial dilutions of a known positive sample into a bone marrow sample know to be free of hematological disease to reach a theoretical abnormal PC dilution of 1:1,000,000 or greater.
-
11.
Transfer 100 – 200 μL (5 – 10 × 106 cells) of each diluted sample into appropriately labeled tubes.
-
12.
Add mAbs employed for surface labeling according to Tube 1 as shown in Table 1. Incubate the cells for 30 minutes at RT in the dark.
-
13.
Proceed to Step 8 of BASIC PROTOCOL 1 and follow through to the end.
-
14.
Acquire and analyze the data as described in the BASIC PROTOCOL 1.
-
15.
See Step 16 of BASIC PROTOCOL 1 for Boolean definition of events falling into regions used to define PCs.
-
16.
Calculate the CV of each dilution using the following equation:
-
a.
CV = [(Standard deviation of number of PCs) / (Mean of Number of PCs)] x 100%
CV should be calculated such that LLOQ can be determined experimentally. Alternatively, CV can be estimated as shown in Step 17.
An example illustrating establishment of the assay sensitivity can be found in section ANCITIPATED RESULTS.
-
a.
-
17.
Alternatively, use Poisson statistics to estimate the number of events required to achieve a defined LLOQ. The relationship between the standard deviation of a distribution, the number of events counted (N), and its CV is given by the following equation:
-
a.
CV = SQRT (N) / N
-
Counting 50 events yields an estimated CV of 14%; therefore, for an assay sensitivity of 0.001%, a theoretical minimum of 50 abnormal events is required and 5,000,000 events would have to be collected for a detection sensitivity of 10-5.
-
–
Assay Sensitivity = (50 / 5,000,000) x 100% = 0.001%
-
–
This alternative method should serve as a guideline for determining the LLOQ, but should not replace experimental validation of the LLOQ
-
a.
-
18. Interpretation the calculated LLOQ as below:
The LLOQ should be greater than or equal to LOD
The calculated CV for the determination of LLOQ should be ≤30% and demonstrate a titratable effect.
-
9.
COMMENTARY
Multiparametric flow cytometry has long been employed in the clinical setting to characterize, diagnose, and monitor hematological malignancies (Foon et al., 1986; Ocqueteau et al., 1998; Orfao et al., 2004; Sarasquete et al., 2005; van Dongen et al., 1988; van Dongen et al., 2012; Vidriales et al., 2003). Although the technology has been used for assessing the bone marrow compartment of patients with MM since the 1990s, widespread adoption has only begun to take place recently (Terstappen et al., 1990). One of the major limiting factors is the lack of suitable markers that can be used to identify PCs. Originally identified as the T10 antigen in the early 1980s by the pioneering work of Reinherz and Schlossman (through the use of OKT10 antibody), CD38 has been recognized as a prognostic marker in many leukemic diseases. Expression of CD38 is widely distributed in myeloid and lymphoid cells; it’s very bright expression on PCs, however, allows this marker to discriminate PCs from other leukocytes (Kumar et al., 2010; Vences-Catalan et al., 2011). To increase the specificity of PC identification, the coincidental expression of CD138 on PCs has been utilized in conjunction with CD38 (Flores-Montero et al., 2016; Kumar et al., 2010). Another antigen routinely used in clinical practice to refine PCs identification is CD45, where moderate to bright expression is found on >90% of normal PCs and it is dim or absent in >70% of aberrant PCs (Soh et al., 2017). The recognition and utilization of CD38, CD138, and CD45 as gating markers for PCs transformed the characterization of PCs disease by MFC. It allows for the identification of relevant markers (e.g. CD19, CD20, CD27, CD28, CD56, CD81, CD117, and CD200) used for the discrimination between normal/reactive and abnormal PCs (Kumar et al., 2010; Soh et al., 2017).
Conventionally measured CR in post-treatment patients no longer satisfies the response criteria for long-term assessment of patient outcomes. Therefore, higher sensitivity modalities for detecting residual tumor cells are required. MFC has been considered a suitable monitoring tool for MM MRD assessment. The earliest studies conducted to explore the applicability and sensitivity of MFC to immunophenotype residual myeloma cells in MM patients was performed by Almeida et al. in 1999 (Almeida et al., 1999). This Spanish group determined that MFC was applicable to >95% of the patient with a detection limit of 10−4 to 10-5. Using a panel consisting of 21 mAbs, the group examined the phenotype of 61 untreated MM patients. PCs with aberrant phenotypes were detected in 87% of their cases, with CD56 (62%), CD117 (28%), sIg (21%), CD28 (16%), CD20 (10%), and CD33 (6%) being the most commonly deregulated markers. Three months following transplantation, a low level of myeloma cells (0.28% ± 0.14%) could still be found in 61% of patient and those with lower percentages of myeloma cells did better (Almeida et al., 1999; San Miguel et al., 2002). In a subsequent and larger clinical trial, this group used 4-color flow cytometry to evaluate 295 patients who achieved a partial response or better after high dose chemotherapy and autologous stem cell transplantation (Paiva et al., 2008). Bone marrow was collected on Day 100 post treatment and the results revealed that MRD-positive vs. MRD-negative patients had shorter PFS (median 37 vs. 71 months; p < 0.001) and OS (89 month vs. median not reached; p = 0.002).
A second pioneering study was reported by Rawstron et al. in 2002 which also demonstrated the presence of MM MRD detected by a 6-color MFC assay correlated with shorter PFS and OS (Rawstron et al., 2002). Bone marrow was collected 3 months after autologous stem cell transplantation from 24 male and 21 female patients enrolled in the Medical Research Council Myeloma VII trial. Neoplastic PCs were detected in 42% of the patients with a PFS of 20 months compared to 35 months (p = 0.003) in patients with no detectable disease. Their study expanded over the next decade to analyze the MRD status of 397 patients who underwent autologous stem cell transplantation after high-dose therapy in the phase 3 MRC Myeloma IX Trial (Rawstron et al., 2013). Consistent with their hypothesis that the presence of MRD was associated with overall outcome, they found that MRD-positive patients when compared to MRD-negative patients had shorter PFS (median 15.5 months vs. 28.6 months; p < 0.001) and OS (median 59.0 months vs 80.6 months; p = 0.0183). Collectively, these studies highlighted the relevance and importance of using MFC for MM MRD assessment and prompted the IMWG to incorporate immunophenotypic response into their response criteria (Kumar et al., 2016). To improve sensitivity, the number of markers acquired simultaneously has increased and the number of events collected from 50,000 – 500,000 to ≥2 million. In 2014, a Phase II study conducted by the Intergroupe Francophone du Myélome incorporated a 7-color panel analyzing a minimum of 2,000,000 cells into their response criteria for a sensitivity of 1 neoplastic cell in 40,000 cells (0.0025%). They reported 21 out of 31 patients became MRD-negative after treatment and none relapsed after a follow-up of 39 months, in contrast 70% of the MRD-positive patients progressed (Roussel et al., 2014). Therefore, it can be inferred from these studies that MRD testing by MFC represents a sensitive surrogate method to predict PFS and OS within months of therapy.
Few studies relating to MM MRD testing have presented all the recommended metrics for establishing and validating an MFC assay, which includes analytical precision, minimal limit of detection, cell recovery, and sample stability (Barnett et al., 2013; Davis, Dasgupta, et al., 2013; Davis, Wood, et al., 2013; Tanqri et al., 2013; Wood et al., 2013). We present a comprehensive workflow to guide Cytometrists on processing bone marrow samples for this assay, their data analysis, and the result interpretation. In light of supporting alternative antibodies clones and markers that may be suitable for replacing current markers used for assessing MM MRD by MFC, we described a systematic approach to critically evaluate the performance of each mAb using Stain Indices as the metric for their assessment. Finally, since the capability of the MFC assay to reliably detect and quantify rare events plays a central role in MM MRD monitoring, we include a structured and complete standard operating procedure for establishing the detection sensitivity of MFC assay for enumerating rare cell events.
In summary, MFC is a powerful and reliable tool that can be used for MM MRD testing. Compared to other existing technologies, MFC is relatively inexpensive, has broader availability, with higher applicability, and a fast turnaround time. The Spanish (Paiva et al., 2008), British (Gormley et al., 2016) and French studies have clearly established that MM MRD status at Day 100 post-transplant is a relevant prognostic tool which correlates with PFS and OS. These finding prompted the IMWG to incorporate immunophenotypic MRD response into the response criteria for MM (Kumar et al., 2016). Moreover, the FDA concluded in 2014 that MM MRD testing by MFC could be used as a surrogate end point biomarker in clinical trials evaluating novel therapies when properly validated and standardized (Landgren et al., 2014). The procedures detailed in this protocol are based on the current International consensus recommendations for performance and validation of the MM MRD assay (Arroz et al., 2016).
CRITICAL PARAMETERS AND TROUBLESHOOTING
Following are some of the consideration that should be taken into account when performing MM MRD testing by MFC.
1. Collection and quality control of bone marrow specimen.
One of the advantages of MFC lies in its capability to identify and enumerate populations of cells in heterogeneous mixtures. It is well-accepted that the percentage of PCs from a bone marrow aspirate is usually underrepresented by MFC as compared to other cytologic methods (Nadav et al., 2006; Smock et al., 2007). One possible reason is that PCs are associated with lipid enriched bone marrow spicules seen by morphology, as opposed to the lipid-depleted fluid analyzed by MFC. The focal nature of MM, hemodilution, and/or selective PC loss during processing and analysis are typically what most experts attribute to PC underestimation by MFC (Al-Quran et al., 2007). Highly representative and non-diluted bone marrow samples are crucial for valid and precise results particularly during MRD evaluation and it is highly recommended that the first bone marrow pull is used for MFC. Unfortunately, the first pull of bone marrow aspirate is frequently reserved for pathology testing in the US, thus it is not uncommon for a flow cytometry laboratory to receive suboptimal specimens for immunophenotyping. Therefore, it is very important that the MFC panel is capable of optimally assessing the overall quality of the bone marrow aspirate and that this result be included on the patient report.
2. Storage
CD138 is considered a labile marker because prolonged storage of bone marrow specimens at cold temperature may downregulate the expression of CD138 (Dorwal et al., 2014; Lin et al., 2004). Various reports by independent investigators have shown that Ficoll Hypaque enrichment and cryopreservation may significantly reduce PC number and may accelerate CD138 antigen loss on PCs (Dorwal et al., 2014; Lin et al., 2004; Reid et al., 2010). For these reasons, bone marrow samples should be processed as soon as possible and Ficoll Hypaque enrichment should be avoided for the optimal detection of PCs.
ANTICIPATED RESULTS
Typical results for the assessment of MM MRD by MFC can be found in Figure 1, Figure 2, and Figure 3. One challenge when interpreting the results is when the samples have been pre-treated using antibody-based immunotherapeutic intervention. As shown in Figure 5, the administration of daratumumab can block the detection of CD38 on PCs. Therefore, the use of antibody-based immunotherapy can impede the detection of PCs by MFC and this creates a need for alternative markers to identify PCs. Prior to the incorporation of alternative markers into the staining panel defined in Table 1, caution has to be taken when analyzing samples from patients on immunotherapy. For example, a CD138 vs. CD45 bivariate plot is useful for evaluating PCs in samples from patients treated with daratumumab.
Figure 5: The detection of CD38 by fluorochrome-conjugated mAb is blocked in cells after treatment with daratumumab.
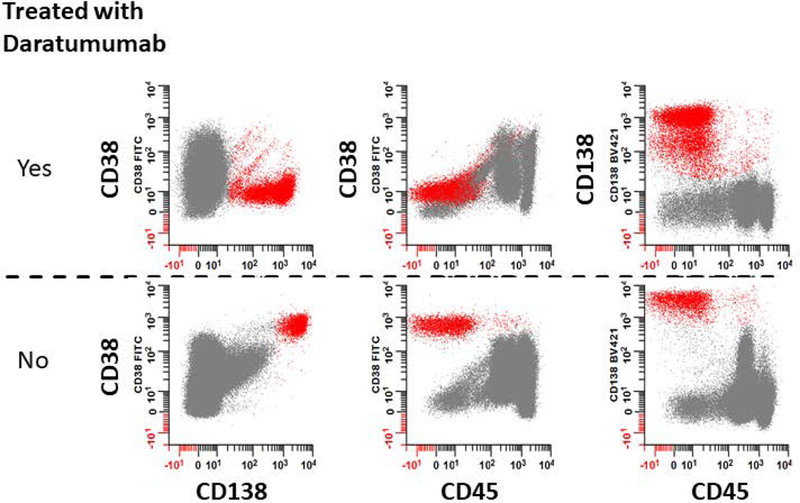
Fresh bone marrow samples from patients with overt disease were processed for MM testing. (Top row) Patient treated with daratumumab, a CD38 targeting immunotherapeutic drug. CD138+ PCs were detected in the absence of CD38 (red dots). (Bottom row) Patient not treated with daratumumab. Populations of PCs that are CD38br, CD138+ (red dots) were clearly seen.
In our LOB validation assay, replicate experiments detected an average of 5.50 ± 2.12 events falling into the gating region defined as PCs using samples not stained with CD38 and CD138. Using the equations provided in SUPPORT PROTOCOL 2, the LOB and LOD were calculated as 9 and 12 events, respectively.
-
(a)
LOB = Mean of Blank + 1.645 × Standard Deviation of Blank
LOB = 5.5 events + 1.645 × 2.121 events
LOB = 9 events
-
(b)
LOD = Mean of Blank + 3 × Standard Deviation of Blank
LOD = 5.5 events + 3 × 2.121 events
LOD = 12 events
As a conservative value, we recommend the use of 20 homogeneously stained abnormal PCs as the minimum number for satisfying the criteria for LOD.
For results describing the estimation and experimental determination of LLOQ, see Table 5 and Figure 6. Briefly, experimentally at the 0.001% level of sensitivity we collected 47.75 ± 2.47 abnormal PCs for a CV of 9.69%. At the next dilution, the CV was 40.85%, which was above our acceptable maximum of 30%. Therefore, in our hands, a minimum of 48 PCs is required for quantitative purposes. For a conservative value, we recommend a minimum of 50 events when considering LLOQ (Arroz et al., 2016).
Table 5.
Estimation and experimental determination of coefficient of variance to establish LLOQ.
| Tube # | Dilution | Number of PCsa | Percentage of PCs, %b | Experimentally Determined CV, %c |
|---|---|---|---|---|
| 1 | Neat | 30781 | 1.08328 | 7.46 |
| 2 | 1:101 | 3028 | 0.07164 | 6.74 |
| 3 | 1:102 | 403 | 0.00927 | 0.00 |
| 4 | 1:103 | 48 | 0.00117 | 9.69 |
| 5 | 1:104 | 17 | 0.00041 | 40.85 |
| 6 | 1:105 | 11 | 0.00026 | 12.12 |
| 7 | 1:106 | 7 | 0.00017 | 33.59 |
| 8 | 1:107 | 6 | 0.00013 | 42.76 |
Average total number of PCs detected based on an acquisition of 5 million total events
Total number of detected PCs divided by the number of Total Leukocytes times 100%
Calculated by dividing the average percentage of PCs by its standard deviation
Figure 6: Establishing the detection sensitivity of MFC for MM MRD monitoring.
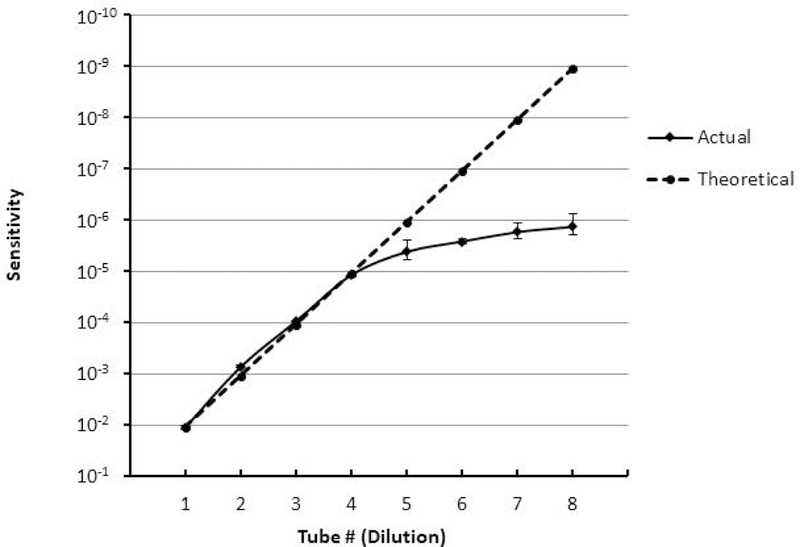
MFC is a relevant tool that can be used for MM MRD monitoring. Prior to the implementation of the assay for clinical utility, the sensitivity of the assay needs to be established. For the determination of LLOQ, a bone marrow sample from a MM patient with a known level of PCs was serially dilution and processed as per procedural steps detailed in SUPPORT PROTOCOL 2. The resulting theoretical sensitivity (represented by dashed line) is established and the actual percentage of detected PCs (represented by solid line) is calculated for each dilution tested in the experiment. The experiment was repeated twice, error bars represent the standard deviation of these measurements.
TIME CONSIDERATIONS
A typical full-length experiment involving processing, acquisition, and analysis of one sample will require a minimum of 3 to 5 hours using the ‘Pre-lysis’ technique. If the ‘Pooled-tube’ method is employed, the time required to complete the sample task will be reduced by approximately 30 minutes due to the removal of bulk lysis step.
ACKNOWLEDGEMENTS
Flow cytometry was performed at Roswell Park Comprehensive Cancer Center’s Department of Flow and Image Cytometry, which was established in part by equipment grants from the NIH Shared Instrument Program and receives support from the Core Grant (5 P30 CA016056–29) from the National Cancer Institute to the Roswell Park Comprehensive Cancer Center. We thank Neil Came (Peter MacCallum Cancer Centre, Sydney) for the insightful discussions and the Total Leukocyte definition used in this manuscript.
Footnotes
Disclosure Statement:
The authors have no commercial or financial conflicts of interest to disclose.
LITERATURE CITED
- Al-Quran SZ, Yang L, Magill JM, Braylan RC, & Douglas-Nikitin VK (2007). Assessment of bone marrow plasma cell infiltrates in multiple myeloma: the added value of CD138 immunohistochemistry. Human pathology, 38(12), 1779–1787. doi: 10.1016/j.humpath.2007.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida J, Orfao A, Ocqueteau M, Mateo G, Corral M, Caballero MD, Blade J, Moro MJ, Hernandez J, & San Miguel JF (1999). High-sensitive immunophenotyping and DNA ploidy studies for the investigation of minimal residual disease in multiple myeloma. Br J Haematol, 107(1), 121–131. [DOI] [PubMed] [Google Scholar]
- Armbruster DA, & Pry T (2008). Limit of blank, limit of detection and limit of quantitation. The Clinical biochemist. Reviews, 29 Suppl 1(Suppl 1), S49–S52. [PMC free article] [PubMed] [Google Scholar]
- Arroz M, Came N, Lin P, Chen W, Yuan C, Lagoo A, Monreal M, de Tute R, Vergilio JA, Rawstron AC, & Paiva B (2016). Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytometry B Clin Cytom, 90(1), 31–39. doi: 10.1002/cyto.b.21228 [DOI] [PubMed] [Google Scholar]
- Avet-Loiseau H, Corre J, Lauwers-Cances V, Chretien M-L, Robillard N, Leleu X, Hulin C, Gentil C, Arnulf B, Belhadj K, Brechignac S, Garderet L, Karlin L, Marit G, Benboubker L, Orsini-Piocelle F, Royer B, Drenou B, Tiab M, Lamy T, Macro M, Richardson PG, Anderson KC, Faham M, Facon T, Moreau P, Attal M, & Munshi NC (2015). Evaluation of minimal residual disease (MRD) by next Generation Sequencing (NGS) Is highly predictive of progression free survival in the IFM/DFCI 2009 trial. Blood, 126(23), 191. [Google Scholar]
- Barnett D, Louzao R, Gambell P, De J, Oldaker T, & Hanson CA (2013). Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part IV - postanalytic considerations. Cytometry B Clin Cytom, 84(5), 309–314. doi: 10.1002/cyto.b.21107 [DOI] [PubMed] [Google Scholar]
- Biran N, Ely S, & Chari A (2014). Controversies in the assessment of minimal residual disease in multiple myeloma: clinical significance of minimal residual disease negativity using highly sensitive techniques. Curr Hematol Malig Rep, 9(4), 368–378. doi: 10.1007/s11899-014-0237-y [DOI] [PubMed] [Google Scholar]
- Burgos L, & Paiva B (2018). EuroFlow-Based Next-Generation Flow Cytometry for Detection of Circulating Tumor Cells and Minimal Residual Disease in Multiple Myeloma In Heuck C & Weinhold N (Eds.), Multiple Myeloma: Methods and Protocols (Vol. 1792, pp. 15–34). Totowa: Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- CLSI. (2007). Clinical Flow Cytometryic Analysis of Neoplastic Hematolymphoid Cells; Approved Guideline - Second Edition. Clinical and Laboratory Standards Institute Document H43-A2, 27(11). [Google Scholar]
- CLSI. (2012). Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline—Second Edition Clinical and Laboratory Standards Institute Document EP17-A2, 32(8). [Google Scholar]
- Davis BH, Dasgupta A, Kussick S, Han JY, & Estrellado A (2013). Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part II - preanalytical issues. Cytometry B Clin Cytom, 84(5), 286–290. doi: 10.1002/cyto.b.21105 [DOI] [PubMed] [Google Scholar]
- Davis BH, Wood B, Oldaker T, & Barnett D (2013). Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part I - rationale and aims. Cytometry B Clin Cytom, 84(5), 282–285. doi: 10.1002/cyto.b.21104 [DOI] [PubMed] [Google Scholar]
- Dorwal P, Thakur R, & Rawat S (2014). CD138 expression in plasma cells is volatile and time-lag dependent. The Egyptian Journal of Haematology, 39(4), 258–259. doi: 10.4103/1110-1067.153978 [DOI] [Google Scholar]
- Flanders A, Stetler-Stevenson M, & Landgren O (2013). Minimal residual disease testing in multiple myeloma by flow cytometry: major heterogeneity. Blood, 122(6), 1088–1089. doi: 10.1182/blood-2013-05-506170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Montero J, de Tute R, Paiva B, Perez JJ, Bottcher S, Wind H, Sanoja L, Puig N, Lecrevisse Q, Vidriales MB, van Dongen JJ, & Orfao A (2016). Immunophenotype of normal vs. myeloma plasma cells: Toward antibody panel specifications for MRD detection in multiple myeloma. Cytometry B Clin Cytom, 90(1), 61–72. doi: 10.1002/cyto.b.21265 [DOI] [PubMed] [Google Scholar]
- Flores-Montero J, Sanoja-Flores L, Paiva B, Puig N, Garcia-Sanchez O, Bottcher S, van der Velden VH, Perez-Moran JJ, Vidriales MB, Garcia-Sanz R, Jimenez C, Gonzalez M, Martinez-Lopez J, Corral-Mateos A, Grigore GE, Fluxa R, Pontes R, Caetano J, Sedek L, Del Canizo MC, Blade J, Lahuerta JJ, Aguilar C, Barez A, Garcia-Mateo A, Labrador J, Leoz P, Aguilera-Sanz C, San-Miguel J, Mateos MV, Durie B, van Dongen JJ, & Orfao A (2017). Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia. doi: 10.1038/leu.2017.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foon KA, & Todd RF 3rd. (1986). Immunologic classification of leukemia and lymphoma. Blood, 68(1), 1–31. [PubMed] [Google Scholar]
- Gormley NJ, Turley DM, Dickey JS, Farrell AT, Reaman GH, Stafford E, Carrington L, & Marti GE (2016). Regulatory perspective on minimal residual disease flow cytometry testing in multiple myeloma. Cytometry B Clin Cytom, 90(1), 73–80. doi: 10.1002/cyto.b.21268 [DOI] [PubMed] [Google Scholar]
- Hedley BD, & Keeney M (2013). Technical issues: flow cytometry and rare event analysis. Int J Lab Hematol, 35(3), 344–350. doi: 10.1111/ijlh.12068 [DOI] [PubMed] [Google Scholar]
- Korde N, Mailankody S, Roschewski M, Faham M, Kotwaliwale C, Moorhead M, Kwok ML, Manasanch EE, Bhutani M, Tageja N, Kazandjian D, Costello R, Zhang Y, Zingone A, Burton D, Mulquin M, Carpenter A, Zuchlinski D, Lamping E, Carter G, Morrison C, Kurdziel K, Lindenberg M, Kurlander R, Maric I, Calvo KR, Braylan RC, Yuan C, Stetler-Stevenson M, Arthur DC, Steinberg SM, Figg WD, Choyke P, & Landgren O (2014). Minimal Residual Disease (MRD) Testing in Newly Diagnosed Multiple myeloma (MM) Patients: A Prospective Head-to-Head Assessment of Cell-Based, Molecular, and Molecular-Imaging Modalities. Blood, 124(21), 2105. [Google Scholar]
- Kristinsson SY, Anderson WF, & Landgren O (2014). Improved long-term survival in multiple myeloma up to the age of 80 years. Leukemia, 28(6), 1346–1348. doi: 10.1038/leu.2014.23 [DOI] [PubMed] [Google Scholar]
- Kumar S, Kimlinger T, & Morice W (2010). Immunophenotyping in multiple myeloma and related plasma cell disorders. Best practice & research. Clinical haematology, 23(3), 433–451. doi: 10.1016/j.beha.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P, Munshi N, Lonial S, Blade J, Mateos MV, Dimopoulos M, Kastritis E, Boccadoro M, Orlowski R, Goldschmidt H, Spencer A, Hou J, Chng WJ, Usmani SZ, Zamagni E, Shimizu K, Jagannath S, Johnsen HE, Terpos E, Reiman A, Kyle RA, Sonneveld P, Richardson PG, McCarthy P, Ludwig H, Chen W, Cavo M, Harousseau JL, Lentzsch S, Hillengass J, Palumbo A, Orfao A, Rajkumar SV, San Miguel J, & Avet-Loiseau H (2016). International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol, 17(8), e328–346. doi: 10.1016/s1470-2045(16)30206-6 [DOI] [PubMed] [Google Scholar]
- Kumar SK, Dispenzieri A, Lacy MQ, Gertz MA, Buadi FK, Pandey S, Kapoor P, Dingli D, Hayman SR, Leung N, Lust J, McCurdy A, Russell SJ, Zeldenrust SR, Kyle RA, & Rajkumar SV (2014). Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia, 28(5), 1122–1128. doi: 10.1038/leu.2013.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos MV, Gay F, & Anderson KC (2017). Multiple myeloma. Nat Rev Dis Primers, 3, 17046. doi: 10.1038/nrdp.2017.46 [DOI] [PubMed] [Google Scholar]
- Ladetto M, Bruggemann M, Monitillo L, Ferrero S, Pepin F, Drandi D, Barbero D, Palumbo A, Passera R, Boccadoro M, Ritgen M, Gokbuget N, Zheng J, Carlton V, Trautmann H, Faham M, & Pott C (2014). Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia, 28(6), 1299–1307. doi: 10.1038/leu.2013.375 [DOI] [PubMed] [Google Scholar]
- Landgren O, Gormley N, Turley D, Owen RG, Rawstron A, Paiva B, Barnett D, Arroz M, Wallace P, Durie B, Yuan C, Dogan A, Stetler-Stevenson M, & Marti GE (2014). Flow cytometry detection of minimal residual disease in multiple myeloma: Lessons learned at FDA-NCI roundtable symposium. Am J Hematol, 89(12), 1159–1160. doi: 10.1002/ajh.23831 [DOI] [PubMed] [Google Scholar]
- Landgren O, & Iskander K (2017). Modern multiple myeloma therapy: deep, sustained treatment response and good clinical outcomes. Journal of Internal Medicine, 281(4), 365–382. doi: 10.1111/joim.12590 [DOI] [PubMed] [Google Scholar]
- Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, Keller S, Weinryb I, Green M, Duan L, Rogers JA, Millham R, O’Brien PJ, Sailstad J, Khan M, Ray C, & Wagner JA (2006). Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res, 23(2), 312–328. doi: 10.1007/s11095-005-9045-3 [DOI] [PubMed] [Google Scholar]
- Lin P, Owens R, Tricot G, & Wilson CS (2004). Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am J Clin Pathol, 121(4), 482–488. doi: 10.1309/74r4-tb90-buwh-27jx [DOI] [PubMed] [Google Scholar]
- Logan AC, Zhang B, Narasimhan B, Carlton V, Zheng J, Moorhead M, Krampf MR, Jones CD, Waqar AN, Faham M, Zehnder JL, & Miklos DB (2013). Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia, 27(8), 1659–1665. doi: 10.1038/leu.2013.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez J, Lahuerta JJ, Pepin F, Gonzalez M, Barrio S, Ayala R, Puig N, Montalban MA, Paiva B, Weng L, Jimenez C, Sopena M, Moorhead M, Cedena T, Rapado I, Mateos MV, Rosinol L, Oriol A, Blanchard MJ, Martinez R, Blade J, San Miguel J, Faham M, & Garcia-Sanz R (2014). Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood, 123(20), 3073–3079. doi: 10.1182/blood-2014-01-550020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadav L, Katz BZ, Baron S, Yossipov L, Polliack A, Deutsch V, Geiger B, & Naparstek E (2006). Diverse niches within multiple myeloma bone marrow aspirates affect plasma cell enumeration. Br J Haematol, 133(5), 530–532. doi: 10.1111/j.1365-2141.2006.06068.x [DOI] [PubMed] [Google Scholar]
- O’Hara DM, Xu Y, Liang Z, Reddy MP, Wu DY, & Litwin V (2011). Recommendations for the validation of flow cytometric testing during drug development: II assays. J Immunol Methods, 363(2), 120–134. doi: 10.1016/j.jim.2010.09.036 [DOI] [PubMed] [Google Scholar]
- Ocqueteau M, Orfao A, Almeida J, Blade J, Gonzalez M, Garcia-Sanz R, Lopez-Berges C, Moro MJ, Hernandez J, Escribano L, Caballero D, Rozman M, & San Miguel JF (1998). Immunophenotypic characterization of plasma cells from monoclonal gammopathy of undetermined significance patients. Implications for the differential diagnosis between MGUS and multiple myeloma. Am J Pathol, 152(6), 1655–1665. [PMC free article] [PubMed] [Google Scholar]
- Orfao A, Ortuno F, de Santiago M, Lopez A, & San Miguel J (2004). Immunophenotyping of acute leukemias and myelodysplastic syndromes. Cytometry A, 58(1), 62–71. doi: 10.1002/cyto.a.10104 [DOI] [PubMed] [Google Scholar]
- Paiva B, Gutierrez NC, Rosinol L, Vidriales MB, Montalban MA, Martinez-Lopez J, Mateos MV, Cibeira MT, Cordon L, Oriol A, Terol MJ, Echeveste MA, de Paz R, de Arriba F, Palomera L, de la Rubia J, Diaz-Mediavilla J, Sureda A, Gorosquieta A, Alegre A, Martin A, Hernandez MT, Lahuerta JJ, Blade J, & San Miguel JF (2012). High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood, 119(3), 687–691. doi: 10.1182/blood-2011-07-370460 [DOI] [PubMed] [Google Scholar]
- Paiva B, Vidriales MB, Cervero J, Mateo G, Perez JJ, Montalban MA, Sureda A, Montejano L, Gutierrez NC, Garcia de Coca A, de Las Heras N, Mateos MV, Lopez-Berges MC, Garcia-Boyero R, Galende J, Hernandez J, Palomera L, Carrera D, Martinez R, de la Rubia J, Martin A, Blade J, Lahuerta JJ, Orfao A, & San Miguel JF (2008). Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood, 112(10), 4017–4023. doi: 10.1182/blood-2008-05-159624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig N, Sarasquete ME, Balanzategui A, Martinez J, Paiva B, Garcia H, Fumero S, Jimenez C, Alcoceba M, Chillon MC, Sebastian E, Marin L, Montalban MA, Mateos MV, Oriol A, Palomera L, de la Rubia J, Vidriales MB, Blade J, Lahuerta JJ, Gonzalez M, Miguel JF, & Garcia-Sanz R (2014). Critical evaluation of ASO RQ-PCR for minimal residual disease evaluation in multiple myeloma. A comparative analysis with flow cytometry. Leukemia, 28(2), 391–397. doi: 10.1038/leu.2013.217 [DOI] [PubMed] [Google Scholar]
- Rawstron AC, Child JA, de Tute RM, Davies FE, Gregory WM, Bell SE, Szubert AJ, Navarro-Coy N, Drayson MT, Feyler S, Ross FM, Cook G, Jackson GH, Morgan GJ, & Owen RG (2013). Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: impact on outcome in the Medical Research Council Myeloma IX Study. J Clin Oncol, 31(20), 2540–2547. doi: 10.1200/jco.2012.46.2119 [DOI] [PubMed] [Google Scholar]
- Rawstron AC, Davies FE, DasGupta R, Ashcroft AJ, Patmore R, Drayson MT, Owen RG, Jack AS, Child JA, & Morgan GJ (2002). Flow cytometric disease monitoring in multiple myeloma: the relationship between normal and neoplastic plasma cells predicts outcome after transplantation. Blood, 100(9), 3095–3100. doi: 10.1182/blood-2001-12-0297 [DOI] [PubMed] [Google Scholar]
- Rawstron AC, Fazi C, Agathangelidis A, Villamor N, Letestu R, Nomdedeu J, Palacio C, Stehlikova O, Kreuzer KA, Liptrot S, O’Brien D, de Tute RM, Marinov I, Hauwel M, Spacek M, Dobber J, Kater AP, Gambell P, Soosapilla A, Lozanski G, Brachtl G, Lin K, Boysen J, Hanson C, Jorgensen JL, Stetler-Stevenson M, Yuan C, Broome HE, Rassenti L, Craig F, Delgado J, Moreno C, Bosch F, Egle A, Doubek M, Pospisilova S, Mulligan S, Westerman D, Sanders CM, Emerson R, Robins HS, Kirsch I, Shanafelt T, Pettitt A, Kipps TJ, Wierda WG, Cymbalista F, Hallek M, Hillmen P, Montserrat E, & Ghia P (2016). A complementary role of multiparameter flow cytometry and high-throughput sequencing for minimal residual disease detection in chronic lymphocytic leukemia: an European Research Initiative on CLL study. Leukemia, 30(4), 929–936. doi: 10.1038/leu.2015.313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S, Yang S, Brown R, Kabani K, Aklilu E, Ho PJ, Woodland N, & Joshua D (2010). Characterisation and relevance of CD138-negative plasma cells in plasma cell myeloma. Int J Lab Hematol, 32(6 Pt 1), e190–196. doi: 10.1111/j.1751-553X.2010.01222.x [DOI] [PubMed] [Google Scholar]
- Roussel M, Lauwers-Cances V, Robillard N, Hulin C, Leleu X, Benboubker L, Marit G, Moreau P, Pegourie B, Caillot D, Fruchart C, Stoppa AM, Gentil C, Wuilleme S, Huynh A, Hebraud B, Corre J, Chretien ML, Facon T, Avet-Loiseau H, & Attal M (2014). Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: a phase II study by the Intergroupe Francophone du Myelome. J Clin Oncol, 32(25), 2712–2717. doi: 10.1200/jco.2013.54.8164 [DOI] [PubMed] [Google Scholar]
- Royston DJ, Gao Q, Nguyen N, Maslak P, Dogan A, & Roshal M (2016). Single-tube 10-fluorochrome analysis for efficient flow cytometric evaluation of minimal residual disease in plasma cell myeloma. Am J Clin Pathol, 146(1), 41–49. doi: 10.1093/ajcp/aqw052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem D, Stetler-Stevenson M, Yuan C, & Landgren O (2016). Myeloma minimal residual disease testing in the United States: Evidence of improved standardization. American Journal of Hematology, 91(12), E502–E503. doi: 10.1002/ajh.24540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Miguel JF, Almeida J, Mateo G, Blade J, Lopez-Berges C, Caballero D, Hernandez J, Moro MJ, Fernandez-Calvo J, Diaz-Mediavilla J, Palomera L, & Orfao A (2002). Immunophenotypic evaluation of the plasma cell compartment in multiple myeloma: a tool for comparing the efficacy of different treatment strategies and predicting outcome. Blood, 99(5), 1853–1856. [DOI] [PubMed] [Google Scholar]
- Sarasquete ME, Garcia-Sanz R, Gonzalez D, Martinez J, Mateo G, Martinez P, Ribera JM, Hernandez JM, Lahuerta JJ, Orfao A, Gonzalez M, & San Miguel JF (2005). Minimal residual disease monitoring in multiple myeloma: a comparison between allelic-specific oligonucleotide real-time quantitative polymerase chain reaction and flow cytometry. Haematologica, 90(10), 1365–1372. [PubMed] [Google Scholar]
- Selliah N, Eck S, Green C, Oldaker T, Stewart J, Vitaliti A, & Litwin V (2019). Flow Cytometry Method Validation Protocols. Curr Protoc Cytom, 87(1), e53. doi: 10.1002/cpcy.53 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, & Jemal A (2018). Cancer statistics, 2018. CA Cancer J Clin, 68(1), 7–30. doi: 10.3322/caac.21442 [DOI] [PubMed] [Google Scholar]
- Smock KJ, Perkins SL, & Bahler DW (2007). Quantitation of plasma cells in bone marrow aspirates by flow cytometric analysis compared with morphologic assessment. Arch Pathol Lab Med, 131(6), 951–955. doi: 10.1043/1543-2165(2007)131[951:qopcib]2.0.co;2 [DOI] [PubMed] [Google Scholar]
- Soh KT, Tario JD Jr., & Wallace PK (2017). Diagnosis of Plasma Cell Dyscrasias and Monitoring of Minimal Residual Disease by Multiparametric Flow Cytometry. Clin Lab Med, 37(4), 821–853. doi: 10.1016/j.cll.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subira D, Castanon S, Aceituno E, Hernandez J, Jimenez-Garofano C, Jimenez A, Jimenez AM, Roman A, & Orfao A (2002). Flow cytometric analysis of cerebrospinal fluid samples and its usefulness in routine clinical practice. Am J Clin Pathol, 117(6), 952–958. doi: 10.1309/123p-ce6v-wyak-bb1f [DOI] [PubMed] [Google Scholar]
- Tanqri S, Vall H, Kaplan D, Hoffman B, Purvis N, Porwit A, Hunsberger B, & Shankey TV (2013). Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part III - analytical issues. Cytometry B Clin Cytom, 84(5), 291–308. doi: 10.1002/cyto.b.21106 [DOI] [PubMed] [Google Scholar]
- Terstappen LW, Johnsen S, Segers-Nolten IM, & Loken MR (1990). Identification and characterization of plasma cells in normal human bone marrow by high-resolution flow cytometry. Blood, 76(9), 1739–1747. [PubMed] [Google Scholar]
- van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, & van Dongen JJ (2003). Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia, 17(6), 1013–1034. doi: 10.1038/sj.leu.2402922 [DOI] [PubMed] [Google Scholar]
- van Dongen JJ, Adriaansen HJ, & Hooijkaas H (1988). Immunophenotyping of leukaemias and non-Hodgkin’s lymphomas. Immunological markers and their CD codes. Neth J Med, 33(5–6), 298–314. [PubMed] [Google Scholar]
- van Dongen JJ, Lhermitte L, Bottcher S, Almeida J, van der Velden VH, Flores-Montero J, Rawstron A, Asnafi V, Lecrevisse Q, Lucio P, Mejstrikova E, Szczepanski T, Kalina T, de Tute R, Bruggemann M, Sedek L, Cullen M, Langerak AW, Mendonca A, Macintyre E, Martin-Ayuso M, Hrusak O, Vidriales MB, & Orfao A (2012). EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia, 26(9), 1908–1975. doi: 10.1038/leu.2012.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vences-Catalan F, & Santos-Argumedo L (2011). CD38 through the life of a murine B lymphocyte. IUBMB Life, 63(10), 840–846. doi: 10.1002/iub.549 [DOI] [PubMed] [Google Scholar]
- Vidriales MB, San-Miguel JF, Orfao A, Coustan-Smith E, & Campana D (2003). Minimal residual disease monitoring by flow cytometry. Best Pract Res Clin Haematol, 16(4), 599–612. [DOI] [PubMed] [Google Scholar]
- Wang L, & Hoffman RA (2017). Standardization, Calibration, and Control in Flow Cytometry. Current Protocols in Cytometry, 79(1),1.3.1–1.3.27. doi: 10.1002/cpcy.14 [DOI] [PubMed] [Google Scholar]
- Wood B, Jevremovic D, Bene MC, Yan M, Jacobs P, & Litwin V (2013). Validation of cell-based fluorescence assays: practice guidelines from the ICSH and ICCS - part V - assay performance criteria. Cytometry B Clin Cytom, 84(5), 315–323. doi: 10.1002/cyto.b.21108 [DOI] [PubMed] [Google Scholar]