

Abstract
Capillary zone electrophoresis-tandem mass spectrometry (CZE-MS/MS) has attracted attention recently for large-scale top-down proteomics that aims to characterize proteoforms in cells at a global scale and with high throughput. In this work, CZE-MS/MS with ultraviolet photodissociation (UVPD) was evaluated for large-scale top-down proteomics for the first time. Roughly, 600 proteoforms and 369 proteins were identified from a zebrafish brain sample via coupling size exclusion chromatography (SEC) fractionation to CZE-UVPD. The dataset represents one of the largest top-down proteomics datasets using UVPD. Single-shot CZE-UVPD identified 227 proteoforms of 139 proteins from one SEC fraction of the zebrafish brain sample. The SEC-CZE-UVPD system identified zebrafish brain proteoforms in a mass range of 3-21 kDa. The UVPD with 213-nm photons produced reasonably good gas-phase fragmentation of proteoforms. For instance, 75% backbone cleavages were observed for Parvalbumin-7 with about 12-kDa molecular weight. The system detected various post-translational modifications (PTMs) from the zebrafish brain sample, including N-terminal acetylation, trimethylation and myristoylation of N-terminal glycine. Two different proteoforms of calmodulin, with either only N-terminal acetylation or both N-terminal acetylation and K115 trimethylation, were identified in the zebrafish brain sample. To our best knowledge, there is no experimental evidence reported in the literature on the two proteoforms of calmodulin in the zebrafish brain.
Keywords: Capillary zone electrophoresis-tandem mass spectrometry, Ultraviolet photodissociation, Top-down proteomics, Zebrafish brain, Post-translational modifications
Graphical Abstract
Capillary electrophoresis-tandem mass spectrometry with ultraviolet photodissociation for top-down proteomics for the first time.

Introduction
Top-down proteomics is a powerful method for identifying and characterizing proteoforms in complex proteomes.1–3 Unlike bottom-up proteomics, top-down proteomics directly provides protein-level evidence and can accurately assign post-translational modifications (PTMs), combinations of PTMs, single nucleotide polymorphisms (SNPs), and splice variants to individual proteoforms.4–6 The ability to elucidate these forms of biological variation is vital for understanding the roles played by proteoforms in disease and development .7–8 Thousands of proteoforms and hundreds of proteins can now be identified and characterized using advanced liquid-phase separation methods, high-resolution mass spectrometers, and various gas-phase fragmentation techniques .9–14
Traditional reversed-phase liquid chromatography coupled to mass spectrometry (RPLC-MS) has been improved for top-down proteomics with the use of shorter bonded stationary phases (C2-C4) and longer columns.15 Capillary zone electrophoresis (CZE) offers an alternative and high-capacity separation of proteoforms based on their sizes and charges.16–19 The lack of stationary phase makes CZE especially useful for the separation of high-mass proteoforms (i.e., greater than 30 kDa).20,21 CZE coupled to MS creates a valuable platform for large-scale top-down proteomics with high sensitivity.22–23 Our recent studies using CZE-MS for proteomics have provided a good solution to the traditional limitations of CZE, including the low sample loading capacity and narrow separation window.11,24,25 The separation window here means a range of migration times over which proteoforms migrate out of the capillary. Microliter-scale loading capacity and hours of separation window were achieved using a dynamic pH junction-based online sample stacking technique 26 and a 1-meter-long LPA (linear polyacrylamide)-coated separation capillary.11,24, 25 Our very recent CZE-MS/MS-based top-down proteomics work has resulted in the identification (ID) of nearly 6,000 proteoforms and 850 proteins from an Escherichia coli (E. coli) whole-cell lysate via coupling size exclusion chromatography (SEC)-RPLC fractionation to the dynamic pH junction-based CZE-MS/MS.12
Extensive gas-phase fragmentation of proteins remains challenging. Many vibrational modes of intact proteins, proton sequestration by basic amino acids, and the loss of labile PTMs hinder the ability of collision-based dissociation methods, including higher energy collisional dissociation (HCD), to extensively fragment proteins.27,28 Direct dissociation methods, without or concurrent with internal energy redistribution prior to dissociation, include electron transfer dissociation (ETD), activated ion electron transfer dissociation (AI-ETD), and ultraviolet photodissociation (UVPD).27,29,30 AI-ETD disrupts non-covalent interactions, that can hold fragments together following fragmentation by ETD, by using infrared radiation to heat the ion.31 AI-ETD has been used for more efficient fragmentation of proteoforms in high-throughput top-down proteomics studies.30 UVPD utilizes high-energy photons to heat the proteins and directly dissociate along the backbone, generating a wide variety of fragments and superior sequence coverage.29 Close to complete sequence coverage of intact proteins using UVPD (193 nm) has been demonstrated, allowing for in-depth characterization of proteins.29 However, 213 nm UVPD is the only commercially available method currently. UVPD (193 nm) has been compared to HCD for high-throughput top-down proteomics in a recent study.13 UVPD (193 nm) was reported to yield better average proteoform sequence coverage compared to HCD.13 Coupling size-based protein fractionation to RPLC-UVPD led to 153 protein and 489 proteoform IDs from a HeLa cell lysate.13 RPLC-UVPD (193 nm) has also been applied for top-down characterization of histones and bottom-up proteomics.31,32
In this work, for the first time, we evaluated CZE-MS/MS using UVPD (213 nm)-based proteoform fragmentation for high-throughput top-down proteomics on an Orbitrap Fusion Lumos mass spectrometer. The commercialized UVPD source with a 213-nm laser was used in the experiment. SEC-based proteoform fractionation was coupled to CZE-UVPD for large-scale top-down proteomics of a zebrafish brain sample.
Experimental
Materials and Reagents
MS-grade water, acetonitrile (ACN), methanol (MeOH), formic acid (FA) and HPLC-grade acetic acid (AA) were purchased from Fisher Scientific (Pittsburgh, PA). Ammonium bicarbonate (NH4HCO3), urea, ammonium persulfate and 3-(trimethoxysilyl)propyl methacrylate, dithiothreitol (DTT), and iodoacetamide (IAA) were purchased from Sigma-Aldrich (St. Louis, MO). Hydrofluoric acid (HF, 48–51% solution in water) and acrylamide were purchased from Acros Organics (NJ, USA). Fused silica capillaries (50 μm i.d./360 μm o.d.) were purchased from Polymicro Technologies (Phoenix, AZ). Complete, mini protease inhibitor cocktail and PhosSTOP (EASYpacks) was from Roche (Indianapolis, IN).
Sample Preparation
Zebrafish brain samples were kindly provided by Professor Jose Cibelli’s group at the Department of Animal Science of Michigan State University. The whole protocol related to the zebrafish were performed in compliance with relevant laws or guidelines, and the protocol followed guidelines defined by the Institutional Animal Care and Use Committee of Michigan State University. Using zebrafish for scientific research has been approved by the Institutional Animal Care and Use Committee of Michigan State University. Male zebrafish brains were lysed in 8 M urea and 100 mM NH4HCO3 (pH 8.0) containing complete protease inhibitor cocktail and PhosSTOP (EASYpacks) from Roche (Indianapolis, IN). Homogenization with a Homogenizer 150 from Fisher Scientific (Pittsburgh, PA) and sonication with a Branson Sonifier 250 from VWR Scientific (Batavia, IL) were performed on ice for protein extraction. Samples were then centrifuged at 15,000 x g for 10 minutes to effectively separate lipids and cell debris from the proteins. The supernatant was collected for further preparation. The protein concentration was determined using a BCA assay. 0.7 mg of zebrafish brain proteins were reduced with DTT (1 M, 2 μL/mg of proteins) at 37 ºC for 30 minutes, alkylated with IAA (1 M, 5 μL/mg of proteins) at room temperature for 20 minutes in dark, and quenched with DTT (1M, 2 μL) before SEC-CZE-MS/MS analyses.
SEC Fractionation
An SEC column (4.6 × 300 mm, 3 μm particles, 300 Å pores) from Agilent was used for separation of the zebrafish brain sample with a mobile phase of 0.1% (v/v) FA, and a flow rate of 0.15 mL/min. The column temperature was kept at 40 ºC. All 0.7-mg zebrafish brain sample was loaded onto the SEC column for fraction collection. One fraction was collected from 8–11 minutes and then 1-minute/fraction from 11–20 minutes was performed. Each fraction was dried down with a vacuum concentrator and redissolved in 50 mM NH4HCO3 (pH 8.0) with a final concentration of ~2 mg/mL for CZE-MS/MS analysis.
CZE-ESI-MS and MS/MS
An ECE-001 CE autosampler and a commercialized electro-kinetically pumped sheath flow CE-MS interface from CMP Scientific (Brooklyn, NY) was coupled online to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, Waltham, MA) in the experiment.33,34 A fused silica capillary (1-meter-long, 50 μm i.d., 360 μm o.d.) was coated with linear polyacrylamide (LPA) according to references 24 and 35. The outer diameter of one end of the capillary was reduced to ~70–80 μm by etching with hydrofluoric acid based on reference 23. (Caution: use appropriate safety procedures while handling hydrofluoric acid solutions)
Samples (500 nL) were injected into the capillary by applying 5 psi for 95 s based on Poiseuille’s law. A separation voltage of 20 kV (120 min) was used for zebrafish brain samples. A pressure of 10 psi was used to flush the capillary between runs for 10 min with the background electrolyte (BGE). A glass capillary (1.0-mm o.d., 0.75-mm i.d., 10 cm long) was pulled with a Sutter P-1000 flaming/brown micropipette puller and used as the ESI emitter. The size of the emitter orifice was 20–40 μm. The voltage for ESI was about 2 kV. The BGE for CZE was 5% (v/v) acetic acid (pH 2.4) and the sheath buffer for ESI consisted of 0.2% (v/v) formic acid and 10% (v/v) methanol.
An Orbitrap Fusion Lumos was used in all CZE-UVPD (213 nm) experiments using data-dependent acquisition (DDA) methods with intact protein mode turned on. Advanced precursor determination was set to True, the default charge state was 10, and the ion transfer tube temperature was set to 275 ºC. For all MS experiments, orbitrap resolution was 120,000, the use of wide quad isolation was set to True, maximum injection time was 100 ms, the number of microscans was 4, and AGC target was 5e5. The scan range was 600–2000 m/z. A Top 5 DDA method was used for acquiring MS/MS spectra of proteoforms. The setting for performing dependent scan on single charge state per precursor only was set to False. An isolation window of 0.5 m/z was used for all experiments. The UVPD activation time was 50 ms. Orbitrap resolution was 120,000, scan range was 150–2000 m/z, maximum injection time was 246 ms, the AGC target was 5e5, and the number of microscans was 4. Charge states 7–24 were included and include charge states 25 and higher was set to True. Dynamic exclusion was set to 30 s in all experiments with low and high mass tolerance set to 1.5 m/z.
Data Analysis
All raw files were analyzed using Proteome Discoverer 2.2 (Thermo Fisher Scientific) with the ProSight PD Top Down High/High node.36 Proteoform MS/MS spectra were deconvoluted with the Xtract tool and filtered with a signal to noise ratio as 3. After that, the spectra were searched against a zebrafish protein database (downloaded from http://proteinaceous.net/database-warehouse-legacy/). There were three tiers involved in the database search: (1) Absolute mass search with 2.0 Da and 10 ppm precursor and fragment ion mass tolerances, (2) Biomarker search with 10 ppm precursor and fragment ion mass tolerances,36 and (3) Absolute mass search with 1000 Da and 10 ppm precursor and fragment ion mass tolerances. Fragment ions considered for UVPD included a and x, b and y, and c and z ions. False discovery rates (FDRs) were estimated using the target-decoy approach.37,38 A 1% proteoform spectrum matches (PrSMs)-level FDR and a 5% proteoform-level FDR were employed. Although the ProSight PD node integrated in Proteome Discoverer 2.2 can perform database search of individual raw files, it cannot combine raw files from different SEC fractions for database search. Therefore, the total numbers of protein and proteoform IDs from the SEC-CZE-MS/MS experiment were from our manual combinations of proteoform and protein IDs from each SEC fraction with the removal of redundant proteoforms and proteins.
Results & Discussion
As shown in Figure 1, proteins were extracted from zebrafish brains and fractionated by SEC into ten fractions based on their size. The SEC fractions were analyzed by CZE-MS/MS using UVPD (213 nm) for proteoform fragmentation. The ProSight PD software was used for database search for proteoform IDs. A 1% PrSM-level FDR and a 5% proteoform-level FDR were employed to filter the database search results. The workflow was able to identify proteoforms with high confidence. For example, one proteoform of calmodulin was identified with a good E-Value and a high proteoform characterization score (PCS), Figure 1. N-terminal acetylation and K115 trimethylation were also detected on this calmodulin proteoform.
Figure 1.
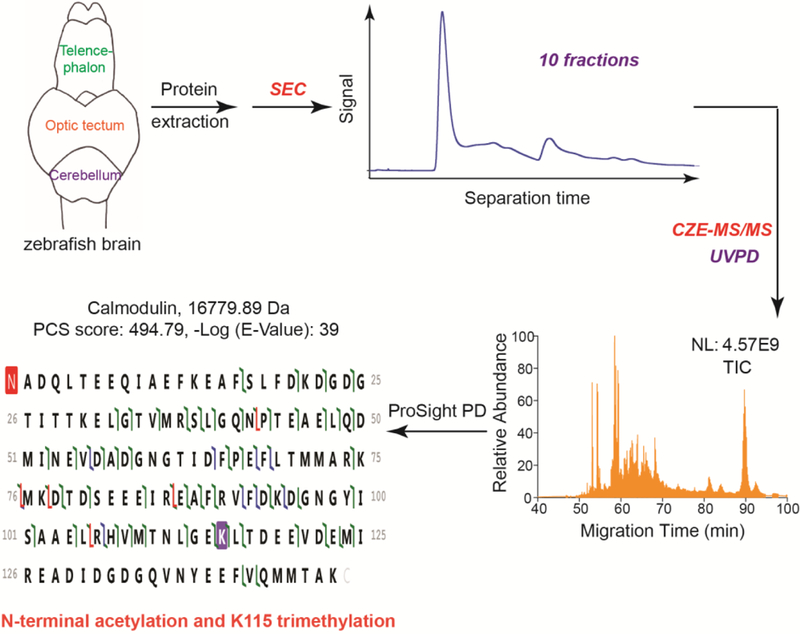
Diagram of the experimental design. NL: normalized level. TIC: total ion current.
In total, about 600 proteoforms and 369 proteins were identified from the zebrafish brain samples using the SEC-CZE-MS/MS in roughly 20 hours. The identified proteoforms from each SEC fraction are listed in Supporting Information I. The dataset represents one of the largest top-down proteomics datasets using UVPD. In the meantime, this work represents the first application of CZE-UVPD for top-down proteomics. The number of proteoform and protein IDs are not uniformly distributed across the ten SEC fractions, Figure 2A. Single-shot CZE-UVPD analysis of the SEC fraction 8 identified 227 proteoforms from 139 proteins. The number of proteoform and protein IDs in each SEC fraction are in ranges of 7–227 and 5–139. On average, ~95 proteoforms and ~62 proteins were identified per SEC fraction.
Figure 2.
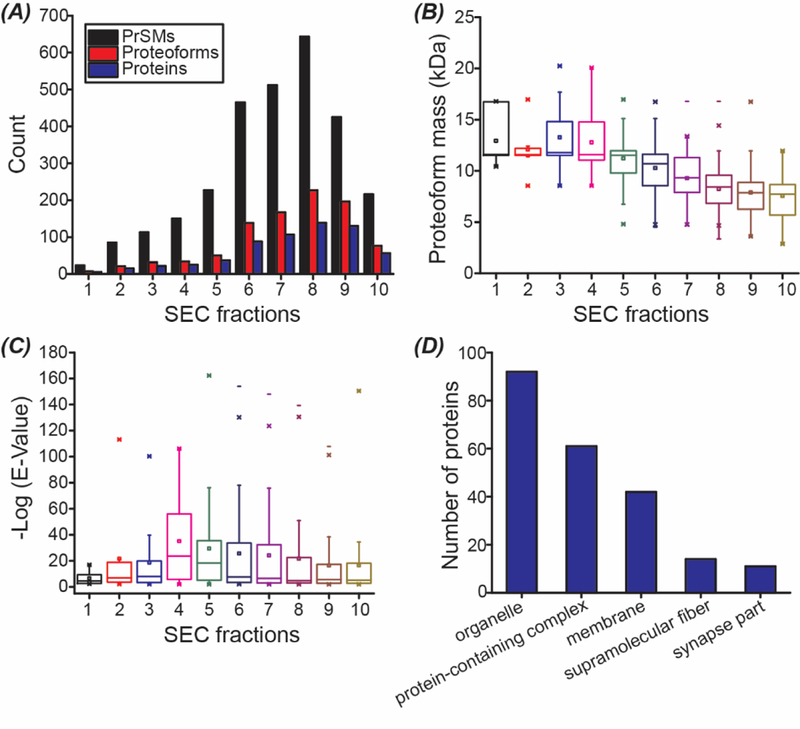
Summary of the SEC-CZE-UVPD data. (A) Numbers of proteoform spectra matches (PrSMs), proteoforms, and proteins vs. SEC fractions. (B) Box-plots of the masses of proteoforms identified from each SEC fraction. (C) Box-plots of the –log (E-Value) of identified proteoforms from each SEC fraction. (D) Cellular component distribution of identified proteins. The information was obtained from the UniProt website using the Retrieve/ID mapping tool.
SEC separates proteoforms based on their size. We plotted the mass distribution of the identified proteoforms in each SEC fraction, Figure 2B. On average, the identified proteoforms in early SEC fractions have larger masses than that in late SEC fractions, which agrees well with the separation principle of SEC. We noted that the adjacent SEC fractions have obvious overlaps regarding proteoform mass, suggesting the relatively low resolution of the SEC column used in the experiment for proteoform separation based on their masses. In our future study, we will increase the length of the SEC column for better separation resolution or try the serial SEC method developed by the Ge group recently.9 Our SEC-CZE-UVPD system identified proteoforms in a mass range of ~3–21 kDa. Top-down proteomics usually has difficulty in the identification of large proteins, partially due to the limited mass resolution of mass spectrometers and inefficient gas-phase fragmentation of large proteins.
In our work, a target-decoy approach was used to evaluate the FDR of proteoform IDs and a 5% proteoform-level FDR was used to filter the proteoform IDs. For each identified proteoform in Supporting Information I, a P-Score (Probability Score) and an E-Value (Expectation Value) were reported for the ID. Lower P-Scores and E-Values indicates better confidence in proteoform IDs; Higher –Log (P-Score) and –Log (E-Value) are better regarding the confidence of proteoform ID. For example, as shown in Figure 1, one proteoform of calmodulin was identified with –Log (E-Value) as 39, indicating a proteoform ID with very high confidence. The proteoform was well fragmented across the proteoform sequence, producing a good number of fragment ions. E-Value is a non-linear transformation of the number of matched fragment ions in a MS/MS spectrum. The –log (E-Value) ranges from 2 to over 160 for the identified proteoforms and the median value of –log (E-Value) is in a range of 4.6–24 for the ten SEC fractions, Figure 2C. Some of the proteoform IDs have –log (E-Values) well below 10, indicating small numbers of fragment ions produced during UVPD. Better UVPD fragmentation will be helpful for more confident identification and characterization of these proteoforms. We also analyzed the cellular component (CC) information of the identified proteins, Figure 2D. The Top 5 CCs are organelle, protein-containing complex, membrane, supramolecular fiber, and synapse part. Proteoforms of over 40 membrane proteins were identified in this work.
UVPD (213 nm) has produced reasonably good gas-phase fragmentation for some proteoforms. As shown in Figures 3A and 3B, 75% and 73% backbone cleavages for Parvalbumin-7 (11,932 Da) and Si:dkey-46i9.1 (7,184 Da) were observed using UVPD. For a relatively large protein, ATP synthase subunit d (18,158 Da), only 25% backbone cleavage was obtained, Figure 3C. The data suggest that the extensive fragmentation of large proteins is still challenging with the UVPD (213 nm). Interestingly, 87% backbone cleavage was reported by the Brodbelt’s group for carbonic anhydrase II (29 kDa) using UVPD (193 nm) under an optimal condition.29 The data suggest that the fragmentation performance of UVPD for large proteins can certainly be improved with further systematic optimizations. The dominant fragment ion types from UVPD (213 nm) for Parvalbumin-7 and Si:dkey-46i9.1 are a, x, y and z ions, Figure 3D. For the ATP synthase subunit d, a and y ions are the dominant ones. The data reasonably agrees with that reported in the literature.29
Figure 3.
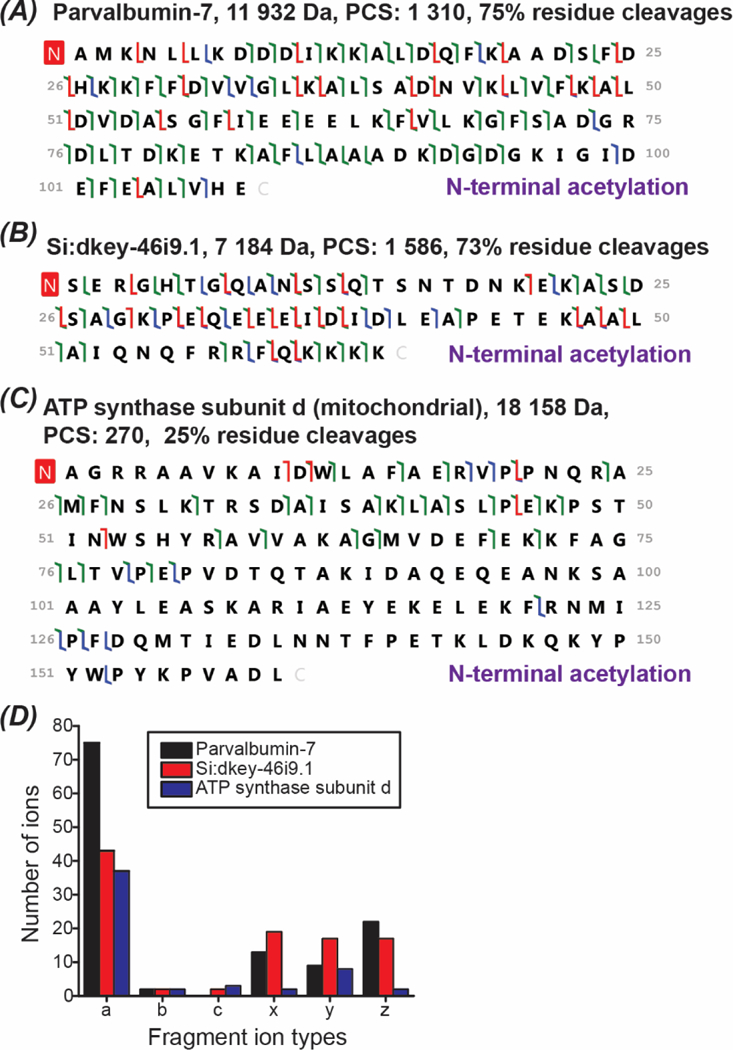
Proteoform fragmentation data. (A)-(C): sequences and fragmentation patterns of Parvalbumin-7, Si:dkey-46i9.1, and ATP synthase subunit d (mitochondrial). a/x ions, b/y ions, and c/z ions were marked in green, blue, and red. (D): Distribution of the fragment ion types for the three proteoforms shown in (A)-(C).
Our SEC-CZE-UVPD system detected various PTMs from the zebrafish brain sample, including N-terminal acetylation, trimethylation and myristoylation of N-terminal glycine. Acetylation was the most abundant PTM (most commonly N-terminal acetylation) with a total of 156 acetylated proteoforms. An example of an acetylated protein well-characterized by CZE-UVPD in the zebrafish brain sample is calmodulin from the calm1a gene. The calm1a gene has one ortholog in human. The calmodulin-calcium complex is known to control kinases, phosphatases, and other proteins. In this work, we detected two different proteoforms from the calm1a gene. One proteoform (Proteoform 1, theoretical mass: 16,739 Da) has only N-terminal acetylation and the other proteoform (Proteoform 2, theoretical mass: 16,781 Da) has both N-terminal acetylation and K115 trimethylation, Figures 4A and 4B. Both proteoforms were identified with good confidence with E-Values better than 10−20 (Proteoform 1) and 10−39 (Proteoform 2). The identification confidence of Proteoform 2 is much higher than that of Proteoform 1, indicated by the much lower E-Value. It has been reported that calmodulin in the human brain has both N-terminal acetylation and K115 trimethylation.39 However, there is no experimental evidence in the literature on the N-terminal acetylation and K115 trimethylation of calmodulin in zebrafish brain based on the information in the UniProt Protein knowledgebase (https://www.uniprot.org/uniprot/Q6PI52). Here we identified the two proteoforms of calmodulin from the zebrafish brain for the first time. We also compared the relative abundance of the two proteoforms of calmodulin in each SEC fraction based on their PrSMs.12 Proteoform 2 has much higher abundance than Proteoform 1 in all the SEC fractions, Figure 4C. The PrSM data agrees well with the proteoform intensity data, Figure 4D. We noted that the Proteoform 2 migrated slower than the Proteoform 1 during CZE separation, most likely because K115 trimethylation reduced the overall charge of the protein, Figure 4D.
Figure 4.

Data about proteoforms of the calmodulin. (A)-(B): sequences and fragmentation patterns of the Proteoform 1 and Proteoform 2 of calmodulin. a/x ions, b/y ions, and c/z ions were marked in green, blue, and red. (C): Distribution of the PrSMs of the Proteoform 1 and Proteoform 2 across different SEC fractions. (D): Extracted ion electropherogram of the Proteoform 1 and Proteoform 2 from the data of the SEC fraction 8. For peak extraction, m/z 1132.54 (+15) was used for the Proteoform 1 and m/z 1120.33 (+15) was used for the Proteoform 2. The mass tolerance was 20 ppm for peak extraction and Gaussian smoothing (5 points) was applied.
Conclusions
CZE-UVPD was applied for top-down proteomics for the first time. About 600 proteoforms and 369 proteins were identified from a zebrafish brain sample using the SEC-CZE-UVPD. The pilot data demonstrate the great potential of CZE-UVPD for large-scale top-down proteomics. We expect that further systematic optimizations of the UVPD fragmentation and further improvements in the SEC-CZE separations will boost the number of proteoform and protein IDs drastically.
We noted that UVPD (213 nm) did not reach extensive fragmentation for many identified proteoforms, which made the complete characterization of these proteoforms challenging. We expect that combinations of various fragmentation methods, e.g., HCD, ETD/AI-ETD, and UVPD, will be useful for improving the quality of proteoform characterization.
Supplementary Material
Acknowledgments
We thank Prof. Jose Cibelli’s group at Michigan State University (Department of Animal Science) for kindly providing the zebrafish brains for this project. We thank the support from the National Institute of General Medical Sciences, National Institutes of Health (NIH), through Grant R01GM125991 (L. Sun).
Footnotes
The authors declare no competing financial interest.
References
- [1].Toby TK, Fornelli L, Kelleher NL, Annu. Rev. Anal. Chem., 2016, 9, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL, Nature, 2011, 480, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhao Y, Sun L, Zhu G, Dovichi NJ, J. Proteome Res., 2016, 15, 3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Smith LM, Kelleher NL, Science, 2018, 359, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Evans VC, Barker G, Heesom KJ, Fan J, Bessant C, Mathews DA, Nat. Methods, 2012, 9, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang X, Slebos RJC, Wang D, Halvey PJ, Tabb DL, Lieber DC, Zhang B, J. Proteome Res., 2012, 11, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cravatt BF, Simon GM, Yates JR III, Nature, 2007, 450, 991. [DOI] [PubMed] [Google Scholar]
- [8].Lucitt BM, Price TS, Pizarro A, Wu W, Yocum AK, Seiler C, Pack MA, Blair IA, FitzGerald GA, Grosser T, Mol. Cell. Proteomics, 2008, 7, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cai W, Tucholski T, Chen B, Alpert AJ, McIlwain S, Kohmoto T, Jin S, Ge Y, Anal. Chem., 2017, 89, 5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liang Y, Jin Y, Wu Z, Tucholski T, Brown KA, Zhang L, Zhang Y, Ge Y, Anal. Chem., 2019, 91, 1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lubeckyj RA, McCool EN, Shen X, Kou Q, Liu X, Sun L, Anal. Chem., 2017, 89, 12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCool EN, Lubeckyj RA, Shen X, Chen D, Kou Q, Liu X, Sun L, Anal. Chem., 2018, 90, 5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cleland TP, DeHart CJ, Fellers RT, VanNispen AJ, Greer JB, LeDuc RD, Parker WR, Thomas PM, Kelleher NL, Brodbelt JS, J. Proteome Res., 2017, 16, 2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Riley NM, Sikora JW, Seckler HS, Greer JB, Fellers RT, LeDuc RD, Westphall MS, Thomas PM, Kelleher NL, Coon JJ, Anal. Chem., 2018, 90, 8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shen Y, Tolić N, Piehowski PD, Shukla AK, Kim S, Zhao R, Qu Y, Robinson E, Smith RD, Paša-Tolić L, J. Chromatogr. A, 2017, 1498, 99. [DOI] [PubMed] [Google Scholar]
- [16].Han X, Wang Y, Aslanian A, Fonslow B, Graczyk B, Davis TN, Yates III JR, J. Proteome Res., 2014, 13, 6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Haselberg R, de Jong GJ, Somsen GW, Anal. Chem., 2013, 85, 2289. [DOI] [PubMed] [Google Scholar]
- [18].Han X, Wang Y, Aslanian A, Bern M, Lavallée-Adam M, Yates JR 3rd, Anal. Chem., 2014, 86, 11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sun L, Knierman MD, Zhu G, Dovichi NJ, Anal. Chem., 2013, 85, 5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jorgenson JW, Lukacs KD, Science, 1983, 222, 266. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Compton PD, Tran JC, Ntai I, Kelleher NL, Proteomics, 2014, 14, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Valaskovic GA, Kelleher NL, McLafferty FW, Science, 1996, 273, 1199. [DOI] [PubMed] [Google Scholar]
- [23].Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ, Angew. Chem., Int. Ed. 2013, 52, 13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McCool EN, Lubeckyj R, Shen X, Kou Q, Liu X, Sun L, J. Vis. Exp., 2018, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen D, Shen X, Sun L, Analyst, 2017, 142, 2118. [DOI] [PubMed] [Google Scholar]
- [26].Britz-McKibbin P, Chen DD, Anal. Chem., 2000, 72, 1242. [DOI] [PubMed] [Google Scholar]
- [27].Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wysocki VH, Tsapralis G, Smith LL, Breci LA, J. Mass Spectrom., 2000, 35, 1399. [DOI] [PubMed] [Google Scholar]
- [29].Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS, J. Am. Chem. Soc., 2013, 135, 12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Riley NM, Sikora JW, Seckler HS, Greer JB, Fellers RT, LeDuc RD, Westphall MS, Thomas PM, Kelleher NL, Coon JJ, Anal. Chem., 2018, 90, 8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gargano AFG, Shaw JB, Zhou M, Wilkins CS, Fillmore TL, Moore RJ, Somsen GW, Paša-Tolić L, J. Proteome Res., 2018, 17, 3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Greer SM, Bern M, Becker C, Brodbelt JS, J. Proteome Res., 2018, 17, 1340. [DOI] [PubMed] [Google Scholar]
- [33].Wojcik R, Dada OO, Sadilek M, Dovichi NJ, Rapid Commun. Mass Spectrom., 2010, 24, 2554. [DOI] [PubMed] [Google Scholar]
- [34].Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ, J. Proteome Res., 2015, 14, 2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhu G, Sun L, Dovichi NJ, Talanta, 2016, 146, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zamdborg L, LeDuc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL, Nucleic Acids Res., 2007, 35, W701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Keller A, Nesvizhskii AI, Kolker E, Aebersold R, Anal. Chem., 2002, 74, 5383. [DOI] [PubMed] [Google Scholar]
- [38].Elias JE, Gygi SP, Nat. Methods, 2007, 4, 207. [DOI] [PubMed] [Google Scholar]
- [39].Sasagawa T, Ericsson LH, Walsh KA, Schreiber WE, Fischer EH, Titani K, Biochemistry, 1982, 21, 2565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.