

C─H alkylation of heteroarenes using ketones and aldehydes as alkyl radical equivalents is achieved.
Abstract
The polar nature of the C═O bond commonly allows it to undergo direct attack by nucleophiles at the electrophilic carbon atom in which ketones and aldehydes act as alkyl carbocation equivalents. In contrast, transformations in which ketones and aldehydes act as alkyl radical equivalents (generated in carbonyl carbon) are unknown. Here, we describe a new catalytic activation mode that combines proton-coupled electron transfer (PCET) with spin-center shift (SCS) and enables C─H alkylation of heteroarenes using ketones and aldehydes as alkyl radical equivalents. This transformation proceeded via reductive PCET activation of the ketones and aldehydes to form α-oxy radicals, addition of the radicals to the N-heteroarenes to form C─C bonds, and SCS to cleave the C─O bonds of the resulting alcohols. This mild protocol represents a general use of abundant, commercially available, ketones and aldehydes as latent alkyl radical equivalents.
INTRODUCTION
The carbonyl group is central to many widely used synthetic methods in organic chemistry, including Grignard reactions, Wittig olefinations, and reductive aminations (1). The polar nature of the C═O bond allows it to undergo direct attack by nucleophiles at the electrophilic carbon atom in which ketones and aldehydes act as alkyl carbocation equivalents. In contrast, Li’s group recently reported that carbonyls could be used as latent alkyl carbanion equivalents via polarity reversal (2). In addition, aldehydes have been widely used as alkyl radical precursors through oxidative decarbonylation (3–6). However, transformations in which ketones and aldehydes act as alkyl radical equivalents (generated in carbonyl carbon) are unknown (Fig. 1A). Here, we describe a new catalytic activation mode that combines proton-coupled electron transfer (PCET) with a ribonucleotide reductase (RNR) class I reaction and results in C─H alkylation of heteroarenes, with ketones and aldehydes acting as alkyl radical equivalents.
Fig. 1. The combination of PCET and an RNR class I reaction enables heteroarene C─H alkylation using ketones and aldehydes as alkyl radical equivalents.
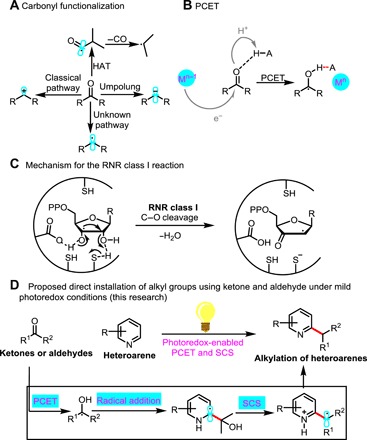
(A) The polarity of the C═O bond. HAT, hydrogen atom transfer. (B) A new catalyst system for reductive PCET. (C) The RNR class I reaction. (D) The method reported herein.
PCET is known to occur in numerous important biological redox processes (7), and in recent years, PCET catalysis has been used as a general mode of substrate activation in organic synthesis (Fig. 1B). For example, Knowles’ group developed a catalyst system for reductive PCET activation of ketones to directly furnish reactive ketyl radicals (8–11). In addition, it is known that deoxyribonucleoside diphosphates, the monomeric precursors of DNA, are formed by radical deoxygenation of ribonucleoside diphosphates catalyzed by RNRs in all organisms (12, 13). This enzymatic process involves a crucial (3,2)-spin-center shift (SCS) that leads to both β–C─O bond cleavage and elimination of water (Fig. 1C) (14–16). Jin and MacMillan (17) recently reported a heteroaromatic C─H alkylation reaction that mimics this process. Considering the efficiency of reductive PCET activation of ketones and aldehydes to form α-oxy radicals and enzymatic cleavage of C─O bonds, we wondered whether PCET and an RNR class I reaction could be combined to achieve otherwise challenging bond constructions involving ketones and aldehydes.
Our group is interested in visible light–mediated Minisci reactions (18–24), and we recently reported photoredox-mediated C─H alkylation reactions of heteroarenes with alkyl halides and amines (25, 26). Because ketones and aldehydes are readily available and inexpensive, we wondered whether it would be possible to generate alkyl radicals from ketones and aldehydes and then use the radicals as alkylating agents in heteroaromatic C─H functionalization reactions under mild, visible light–mediated photoredox conditions. However, we must overcome two challenges: First, as stated above, ketones and aldehydes generally act as electrophilic alkyl groups and are therefore difficult to couple to N-heteroarenes owing to polarity mismatch (1). Second, it is difficult to get alkylation products that forge new bonds at the carbonyl carbon for aldehydes due to decarbonylation (3–6). Here, we report that a protocol for this transformation has been successfully realized by means of reductive PCET activation of ketones and aldehydes to afford α-oxy radicals, addition of the radicals to N-heteroarenes to form C─C bonds, and subsequent SCS to cleave the C─O bonds of the resulting alcohols (Fig. 1D). This alkylation strategy represents a general use of abundant commercially available ketones and aldehydes as latent alkyl radical equivalents.
RESULTS
Design plan
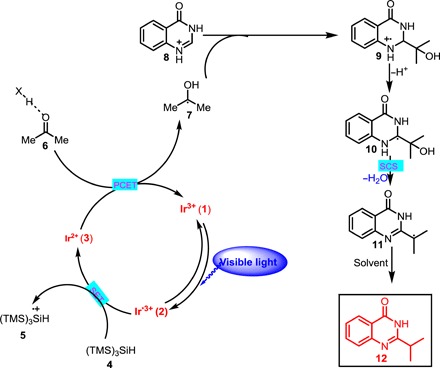
We hypothesized that light-mediated C─H alkylation reactions of heteroarenes might proceed via the mechanism outlined in Fig. 2. Upon exposure to visible light, the photocatalyst, Ir[dF(CF3)ppy]2(dtbbpy)PF6 [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, dtbbpy = 4,4-di-tert-butyl-2,2-bipyridine] (1), would produce long-lived photoexcited *IrIII complex 2 (excited-state lifetime, 2.3 μs) (27). This complex [E1/2red (*IrIII/IrII) = +1.21 V versus saturated calomel electrode (SCE) in CH3CN] would be reduced by tris(trimethylsilyl)silane (4) [TTMS; Ep (4+•/4) = +0.73 V versus SCE in CH3CN] to generate more strongly reducing IrII complex 3 and a TTMS+• radical cation (5) (28, 29). Concurrently, a Brønsted acid would reversibly form a hydrogen-bonded complex with the ketone substrate. Subsequent PCET, with electron transfer from the redox catalyst occurring concomitantly with proton transfer to the ketone oxygen along the hydrogen-bond coordinate, would produce α-oxy radical 7 and regenerate 1 to complete the catalytic photoredox cycle. α-Oxy radical 7 would then add to protonated electron-deficient heteroarene 8 via a Minisci-type pathway to afford aminyl radical cation 9 (30). The α–C─H bond of 9 would be sufficiently acidic to undergo deprotonation to form α-amino radical 10, which would be primed to undergo SCS to eliminate H2O and generate open-shell benzylic radical 11 (17, 31). Radical 11 would then abstract a hydrogen atom from the solvent to give the desired alkylation product 12.
Fig. 2. Proposed mechanism for direct alkylation of heteroaromatic using ketones and aldehydes as alkyl radical sources.
Optimization of the reaction conditions
To test this mechanistic hypothesis, we began by examining the alkylation reaction between 4-hydroxyquinazoline and acetone (as the solvent) using a variety of photocatalysts and reductants (Table 1). With Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst, TTMS as the reductant, and trifluoroacetic acid (TFA) as the proton source, we obtained the desired product 12 in 96% yield upon irradiation with blue light (entry 1). Using the above-described conditions, we then varied the photocatalyst (entries 2 to 4). However, [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 proved to be the most effective catalyst. Next, a number of reductants were screened and gave lower yields (entries 5 to 8). The use of TTMS as the reductant was crucial; the amine reductants that are commonly used in photoredox reactions are incompatible with Minisci reaction conditions (which are acidic), and Hantzsch dihydropyridine, another commonly used reductant, gave only a 40% yield of 12, because it reduced the electron-deficient heteroarene in this reaction system. Control experiments confirmed that the photocatalyst, the reductant, the acid, and light were necessary: None of the desired alkylation product was observed if any of these components was omitted (entries 9 to 12). Last, using cyclohexanone (30 eq) as the alkylating agent instead of acetone, we screened a variety of solvents and found acetonitrile to be optimal, giving alkylated product in 92% yield (table S1).
Table 1. Optimization of conditions for alkylation of 4-hydroxyquinazoline with acetone.
Reaction conditions: 4-hydroxyquinazoline (0.3 mmol), photocatalyst (0.003 mmol), reductant (0.6 mmol), TFA (0.6 mmol), and acetone (3.0 ml) under Ar atmosphere. The yield was determined by 1H NMR spectroscopy using dibromomethane as the internal standard. Reaction was performed in the absence of light for entry 9. Reaction was performed in the absence of photocatalyst for entry 10. Reaction was performed in the absence of TTMS for entry 11. Reaction was performed in the absence of TFA for entry 12. rt, room temperature; NR, no reaction; DIPEA, N,N-diisopropylethylamine; HEH, diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate.

| Entry | Photocatalyst | Reductant | Yield (%) |
| 1 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | TTMS | 96 |
| 2 | Ir(ppy)3 | TTMS | NR |
| 3 | [Ru(bpy)3](PF6)2 | TTMS | NR |
| 4 | Eosin-Y | TTMS | NR |
| 5 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Et3SiH | 82 |
| 6 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | Ph3SiH | NR |
| 7 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | DIPEA | NR |
| 8 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | HEH | 40 |
| 9 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | TTMS | NR |
| 10 | — | TTMS | NR |
| 11 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | NR | |
| 12 | Ir[dF(CF3)ppy]2(dtbbpy)PF6 | TTMS | NR |
Substrate scope
With the optimized reaction conditions in hand, we investigated the scope of the reaction with respect to the ketone and aldehyde substrates by using 4-hydroxyquinazoline as the heteroarene (Table 2). Unlike the isopropylation reactions, in which acetone was used as the solvent, the alkylation reactions with other ketones and aldehydes (30 eq) were typically carried out in acetonitrile. The protocol was amenable to a wide variety of ketones and aldehydes, giving the desired products in fair to excellent yields. Specifically, cyclic ketones with various ring sizes afforded products 13 to 20 in 49 to 93% yields. The alkylation reaction was not restricted to cyclic ketones. Reactions of acyclic ketones afforded products 21 to 23, although the yields (36 to 47%) were lower than those obtained with the cyclic ketones, and the yields decreased gradually as the length of the alkyl chain on the ketone was increased. We speculate that as the chain length was increased, steric bulk increased concomitantly and hindered the attack of the ketyl radicals (tertiary alkyl radicals) on the protonated heteroarene. The steric bulk proximal to the ketone functionality was not acceptable in this reaction. In addition, the reaction was amenable to scale up; when it was carried out on a 6-mmol scale, 12 was isolated in 72% yield.
Table 2. Exploration of substrate scope.
Reactions were performed on a 0.3-mmol scale, unless otherwise noted. Isolated yields are given. We used Hantzsch dihydropyridine as reductant for 48. See the Supplementary Materials for experimental details.
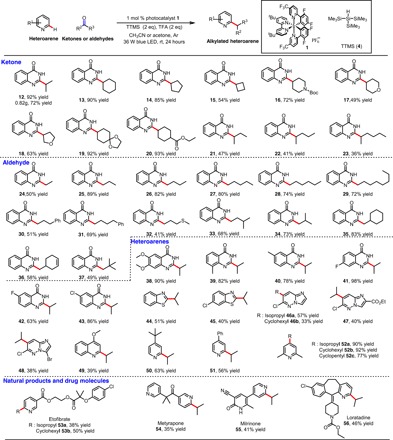
Gratifyingly, we found that various aldehydes also underwent the desired reaction to give the corresponding products in 41 to 89% yields (24 to 37); these yields were higher than those of the acyclic ketones, owing to the lower steric bulk of the secondary alkyl radicals generated from the aldehydes. We found that steric bulk proximal to the aldehyde functionality was acceptable, as exemplified by the successful reactions of isopropyl, cyclohexyl, and tert-butyl aldehydes 33 to 37.
Next, we explored the scope of the alkylation reaction with respect to the N-heteroarene substrates (Table 2). Various electron-deficient heteroarenes were readily alkylated at the most electrophilic position to afford the desired products in fair to excellent yields. Specifically, reactions of 4-hydroxyquinazoline substrates with electron-withdrawing or electron-donating substituents (e.g., methyl, methoxyl, and halogen) proceeded smoothly with excellent selectivity for the C2 position to afford 38 to 43 in 63 to 98% isolated yields. Benzothiazoles and imidazo[1,2-b]pyridazines were also acceptable substrates, giving corresponding products 44 and 45 (51 and 40%) and 46 to 48 (33 to 57%). Unsubstituted quinoline and quinolines with varying substituents (e.g., methyl, ester group, amide group, and halogen) were not acceptable substrates; only 4-methoxyquinoline could give the desired product 49 in 39% yield. In addition, the use of 4-tert-butyl-pyridine, 4-phenylpyridine, or 2,6-dimethylpyridine resulted in selective monoalkylation at C2, C2, and C4, respectively (50 to 52, 56 to 92% yields). The selectivity for monoalkylated products 50 and 51 may have been due to the reduced electrophilicity of the monoalkylated heteroarenes, which slowed down the second alkylation. Notably, halide, ether, amine, amide, ester, acetal, alkene, and tert-butyl carbamate functional groups were tolerated under these mild reaction conditions.
Last, we used our alkylation protocol for late-stage functionalization of several complex natural products and drug molecules (Table 2) (32). For instance, etofibrate, which contains clofibrate and niacin moieties, was selectively alkylated on the pyridine ring to give 53 in moderate yields. Metyrapone, an inhibitor of cortisol biosynthesis, was selectively monoalkylated to afford 54 in 35% yield. Milrinone, a phosphodiesterase 3 inhibitor and vasodilator, afforded a moderate yield of alkylated product 55. The antihistamine drug loratadine could be alkylated selectively at the C2 position of the pyridine ring to give 56.
DISCUSSION
We conducted mechanistic studies to support the proposed pathway outlined in Fig. 3. The oxidation potential of TTMS in CH3CN was determined to be +0.71 V versus SCE, indicating that direct oxidation of TTMS (E0 = +0.71 versus SCE) by Ir*3+ [E1/2red (*IrIII/IrII) = +1.21 V versus SCE] was thermodynamically favored (29); Stern-Volmer fluorescence quenching experiments demonstrated that the excited state of the photocatalyst could be quenched by TTMS (Fig. 3A). When pivaldehyde (57) was used as the alkylating agent, the product of tert-radical addition (58) was obtained in 45% yield. This result indicates that the ketyl radical underwent α-scission to form a tert-radical (Fig. 3B). With 2-pentanone (59) as the alkylating agent, the product of ethyl radical addition (39) was obtained in 16% yield, indicating that the corresponding ketyl radical underwent β-scission to form an ethyl radical (Fig. 3B). A reaction with dearomatized compound 60 under the developed reaction conditions was conducted, showing trace conversion to 40, thus demonstrating that 60 was not the reaction intermediate (Fig. 3C). When we conducted the reaction in the presence of cyclohexanol instead of cyclohexanone, the desired product 13 was not detected. Thus, demonstrating cyclohexanol was not the reaction intermediate (Fig. 3C). When we conducted the reaction in the absence of ketones and aldehydes, the ring-opened product 65 was not detected, indicating that the bis-alpha-amino radical has not been formed (Fig. 3C).
Fig. 3. Mechanistic studies in support of the proposed pathway.
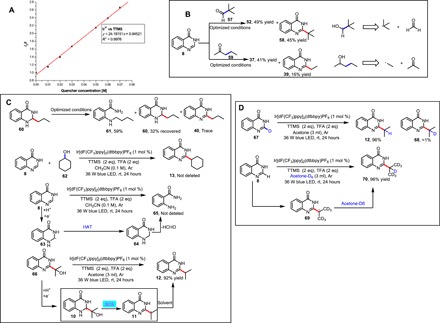
(A) Ir[dF(CF3)ppy]2(dtbbpy)PF6 emission quenching with TTMS. (B) Proof of the corresponding α-oxy radicals. (C) Mechanistic studies support the spin-center shift elimination pathway. (D) Confirmation of the source of hydrogen atoms at the benzylic position of product. rt, room temperature.
Furthermore, a series of experiments were conducted to investigate the proposed SCS elimination (Fig. 3C). When hydroxylated intermediate 66 was exposed to the optimized reaction conditions, the desired product was obtained in 92% yield, indicating that the corresponding α-amino radical (10) was generated and that the deoxygenated product was subsequently formed via an SCS pathway (Fig. 3C). None of the desired alkylation product was generated from 66 in the absence of photocatalyst, reductant, acid, or light (table S4).
To confirm the source of hydrogen atoms at the benzylic position of product, we performed two different deuterium-labeling experiments (Fig. 3D). When C2-deuterated 4-hydroxyquinazoline was treated under reaction conditions, we obtained a 96% yield of the product with no deuterium incorporation at the benzylic position. When the reaction was carried out in acetone-d6, we obtained a 96% yield of the product with deuterium incorporation at the benzylic position. This result indicates that open-shell species 11 abstracted a hydrogen atom from acetone to give the desired alkylation product 12 (Fig. 3D).
In summary, we have developed a new catalytic activation mode that involves a combination of PCET and an RNR class I reaction to enable C─H alkylation of heteroarenes using ketones and aldehydes as alkyl radical sources under mild conditions without the need for oxidants or high temperatures. This transformation proceeded via reductive PCET activation of the ketones and aldehydes to form α-oxyl radicals, addition of the radicals to the N-heteroarenes to form C─C bonds, and SCS to cleave the C─O bonds of the resulting alcohols. This alkylation strategy represents a general use of abundant commercially available ketones and aldehydes as latent alkyl radical equivalents. The robustness of this protocol was demonstrated by late-stage functionalization of several complex nitrogen-containing natural products and drugs.
MATERIALS AND METHODS
Experimental design
Reagents were purchased from commercial sources and were used as received. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker AVANCE 400 Ultrashield NMR spectrometers. Chemical shifts (δ) were given in parts per million and were measured downfield from internal tetramethylsilane. High-resolution mass spectrometry data were obtained on a Fourier-transform ion cyclotron resonance mass spectrometry instrument (IonSpec 7.0T). The melting points were determined on an X-4 microscope melting point apparatus and are uncorrected. Conversion was monitored by thin-layer chromatography. Flash column chromatography was performed over silica gel (100 to 200 mesh). Blue light-emitting diode (LED) (36 W, λmax = 470 nm) purchased from JIADENG (LS) was used for blue light irradiation. A fan attached to the apparatus was used to maintain the reaction temperature at room temperature (rt).
General procedure for the alkylation of N-heteroarenes
Ir[dF(CF3)ppy)]2(dtbbpy)PF6 (3.36 mg, 0.003 mmol, 1 mol %), heteroarene (0.3 mmol, 1.0 eq), ketones or aldehydes (9.0 mmol, 30 eq), TTMS (185 μl, 0.6 mmol, 2.0 eq), TFA (45 μl, 0.6 mmol, 2.0 eq), and CH3CN (2.0 ml) were added to a 10-ml glass vial. The reaction mixture was degassed by bubbling with Ar for 15 s with an outlet needle, and the vial was sealed with a polytetrafluoroethylene (PTFE) cap. The mixture was then stirred rapidly and irradiated with a 36-W blue LED (approximately 2 cm away from the light source) at room temperature for 24 hours. The reaction mixture was concentrated in vacuum to remove the CH3CN. The mixture was diluted with 10 ml of aqueous 1 M NaHCO3 solution and extracted with 3 × 20 ml dichloromethane (DCM). The combined organic extracts were washed with 40 ml of brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude product by flash chromatography on silica gel using the indicated solvent system afforded the desired product.
General procedure for the isopropylation of N-heteroarenes
Ir[dF(CF3)ppy)]2(dtbbpy)PF6 (3.36 mg, 0.003 mmol, 1 mol %), heteroarene (0.3 mmol, 1.0 eq), HEH (155 mg, 0.6 mmol, 2.0 eq) or TTMS (185 μl, 0.6 mmol, 2.0 eq), TFA (45 μl, 0.6 mmol, 2.0 eq), and acetone (3.0 ml) were added to a 10-ml glass vial. The reaction mixture was degassed by bubbling with Ar for 15 s with an outlet needle, and the vial was sealed with a PTFE cap. The mixture was then stirred rapidly and irradiated with a 36-W blue LED (approximately 2 cm away from the light source) at room temperature for 24 hours. The reaction mixture was concentrated in vacuum to remove the acetone. The mixture was diluted with 10 ml of aqueous 1 M NaHCO3 solution and extracted with DCM (3 × 20 ml). The combined organic extracts were washed with 40 ml of brine, dried over Na2SO4, and concentrated in vacuo. Purification of the crude product by flash chromatography on silica gel using the indicated solvent system afforded the desired product.
Supplementary Material
Acknowledgments
Funding: We are grateful to the National Natural Science Foundation of China (21732002 and 21672117) for financial support for our programs. Author contributions: J.D., Z.W., and X.W. performed and analyzed experiments. J.D., H.S., Y.L., and Q.W. designed experiments to develop this reaction and probe its utility. J.D. prepared this manuscript. H.S., Y.L., and Q.W. helped to revise the manuscript. Competing interests: All authors are inventors on a patent application related to this work filed by the China National Patent Office (no. 201910582763.4, filed 1 July 2019). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax9955/DC1
Section S1. General information
Section S2. Reaction optimization
Section S3. Investigation of the mechanism
Section S4. Experimental procedures and product characterization
Section S5. Copies of 1H NMR and 13C NMR spectra for new compounds
Table S1. Screening of different solvents.
Table S2. Screening of the amount of cyclohexanone.
Table S3. Light on and off experiments.
Table S4. Control experiments of intermediate 66.
Fig. S1. Control experiments.
Fig. S2. Emission quenching experiments (Stern-Volmer studies).
REFERENCES AND NOTES
- 1.F. A. Carey, R. J. Sundberg, Advanced Organic Chemistry: Part B: Reactions and Synthesis (Springer, 2001). [Google Scholar]
- 2.Wang H., Dai X.-J., Li C.-J., Aldehydes as alkyl carbanion equivalents for additions to carbonyl compounds. Nat. Chem. 9, 374–378 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Chatgilialoglu C., Crich D., Komatsu M., Ryu I., Chemistry of acyl radicals. Chem. Rev. 99, 1991–2070 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Paul S., Guin J., Dioxygen-mediated decarbonylative C-H alkylation of heteroaromatic bases with aldehydes. Chem. Eur. J. 21, 17618–17622 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Tang R.-J., Kang L., Yang L., Metal-free oxidative decarbonylative coupling of aliphatic aldehydes with azaarenes: Successful minisci-type alkylation of various heterocycles. Adv. Synth. Catal. 357, 2055–2060 (2015). [Google Scholar]
- 6.Bohman B., Berntsson B., Dixon R. C. M., Stewart C. D., Barrow R. A., Alkylations and hydroxymethylations of pyrazines via green minisci-type reactions. Org. Lett. 16, 2787–2789 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Weinberg D. R., Gagliardi C. J., Hull J. F., Murphy C. F., Kent C. A., Westlake B. C., Paul A., Ess D. H., McCafferty D. G., Meyer T. J., Proton-coupled electron transfer. Chem. Rev. 112, 4016–4093 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Tarantino K. T., Liu P., Knowles R. R., Catalytic ketyl-olefin cyclizations enabled by proton-coupled electron transfer. J. Am. Chem. Soc. 135, 10022–10025 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Rono L. J., Yayla H. G., Wang D. Y., Armstrong M. F., Knowles R. R., Enantioselective photoredox catalysis enabled by proton-coupled electron transfer: Development of an asymmetric aza-pinacol cyclization. J. Am. Chem. Soc. 135, 17735–17738 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Gentry E. C., Knowles R. R., Synthetic applications of proton-coupled electron transfer. Acc. Chem. Res. 49, 1546–1556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu G., Knowles R. R., Rate–driving force relationships in the multisite proton-coupled electron transfer activation of ketones. J. Am. Chem. Soc. 141, 2721–2730 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.S. Licht, J. Stubbe, Mechanistic investigations of ribonucleotide reductases, in Comprehensive Natural Products Chemistry, C. D. Poulter, Ed. (Elsevier, 1999). [Google Scholar]
- 13.Eklund H., Uhlin U., Farnegardh M., Logan D. T., Nordlund P., Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 77, 177–268 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Lenz R., Giese B., Studies on the mechanism of ribonucleotide reductases. J. Am. Chem. Soc. 119, 2784–2794 (1997). [Google Scholar]
- 15.Himo F., Siegbahn P. E. M., Quantum chemical studies of radical-containing enzymes. Chem. Rev. 103, 2421–2456 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Wessig P., Muehling O., Spin-centershift(SCS)–aversatileconceptinbiological and synthetic chemistry. Eur. J. Org. Chem. 14, 2219–2232 (2007). [Google Scholar]
- 17.Jin J., MacMillan D. W. C., Alcohols as alkylating agents in heteroarene C–H functionalization. Nature 525, 87–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tauber J., Imbr D., Opatz T., Radical addition to iminium ions and cationic heterocycles. Molecules 19, 16190–16222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minisci F., Vismara E., Fontana F., Recent developments of free-radical substitutions of heteroaromatic bases. Heterocycles 28, 489–519 (1989). [Google Scholar]
- 20.Minisci F., Fontana F., Vismara E., Substitutions by nucleophilic free radicals: A new general reaction of heteroaromatic bases. J. Heterocyclic Chem. 27, 79–96 (1990). [Google Scholar]
- 21.Duncton M. A. J., Minisci reactions: Versatile C-H functionalizations for medicinal chemists. Med. Chem. Commun. 2, 1135–1161 (2011). [Google Scholar]
- 22.Proctor R. S. J., Phipps R. J., Recent advances in minisci-type reactions. Angew. Chem. Int. Ed. 58, 13666–13699 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Ji Y., Brueckl T., Baxter R. D., Fujiwara Y., Seiple I. B., Su S., Blackmond D. G., Baran P. S., Innate C–H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. U.S.A. 108, 14411–14415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Proctor R. S. J., Davis H. J., Phipps R. J., Catalytic enantioselective Minisci-type addition to heteroarenes. Science 360, 419–422 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Dong J., Lyu X., Wang Z., Wang X., Song H., Liu Y., Wang Q., Visible-light-mediated Minisci C–H alkylation of heteroarenes with unactivated alkyl halides using O2 as an oxidant. Chem. Sci. 10, 976–982 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong J., Xia Q., Lyu X., Yan C., Song H., Liu Y., Wang Q., Photoredox-mediated direct cross-dehydrogenative coupling of heteroarenes and amines. Org. Lett. 20, 5661–5665 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Lowry M. S., Goldsmith J. I., Slinker J. D., Rohl R., Pascal R. A., Malliaras G. G., Bernhard S., Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium(III) complex. Chem. Mater. 17, 5712–5719 (2005). [Google Scholar]
- 28.Zhu J., Cui W.-C., Wang S., Yao Z.-J., Radical hydrosilylation of alkynes catalyzed by eosin Y and thiol under visible light irradiation. Org. Lett. 20, 3174–3178 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Le C., Chen T. Q., Liang T., Zhang P., MacMillan D. W. C., A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 360, 1010–1014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qi L., Chen Y., Polarity-reversed allylations of aldehydes, ketones, and imines enabled by Hantzsch ester in photoredox catalysis. Angew. Chem. Int. Ed. 55, 13312–13315 (2016). [DOI] [PubMed] [Google Scholar]
- 31.McNally A., Prier C. K., MacMillan D. W. C., Discovery of an a-amino C–H arylation reaction using the strategy of accelerated serendipity. Science 334, 1114–1117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brückl T., Baxter R. D., Ishihara R. Y., Baran P. S., Innate and guided C–H functionalization logic. Acc. Chem. Res. 45, 826–839 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutierrez-Bonet A., Remeur C., Matsui J. K., Molander G. A., Late-stage C−H alkylation of heterocycles and 1,4-quinones via oxidative homolysis of 1,4-dihydropyridines. J. Am. Chem. Soc. 139, 12251–12258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garza-Sanchez R. A., Tlahuext-Aca A., Tavakoli G., Glorius F., Visible light-mediated direct decarboxylative C–H functionalization of heteroarenes. ACS Catal. 7, 4057–4061 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax9955/DC1
Section S1. General information
Section S2. Reaction optimization
Section S3. Investigation of the mechanism
Section S4. Experimental procedures and product characterization
Section S5. Copies of 1H NMR and 13C NMR spectra for new compounds
Table S1. Screening of different solvents.
Table S2. Screening of the amount of cyclohexanone.
Table S3. Light on and off experiments.
Table S4. Control experiments of intermediate 66.
Fig. S1. Control experiments.
Fig. S2. Emission quenching experiments (Stern-Volmer studies).