

Abstract
Objective
To evaluate the efficacy and safety of the oral Janus kinase (JAK) inhibitor peficitinib versus placebo in Japanese patients with rheumatoid arthritis (RA).
Methods
In this multicentre, double-blind, parallel-group, placebo-controlled phase III study, patients with RA and inadequate response to methotrexate (MTX) were randomised 1:1:1 to placebo, peficitinib 100 mg once daily or peficitinib 150 mg once daily with MTX for 52 weeks. Based on baseline randomisation, at week 12, non-responders receiving placebo were switched to peficitinib until the end of treatment; the remaining patients were switched to peficitinib at week 28. Primary efficacy variables were American College of Rheumatology (ACR)20 response rate at week 12/early termination (ET) and change from baseline in van der Heijde-modified total Sharp score (mTSS) at week 28/ET.
Results
519 patients were randomised and treated. Significantly more (p<0.001) peficitinib (58.6%, 100 mg; 64.4%, 150 mg) than placebo (21.8%) recipients achieved ACR20 response at week 12/ET. Significantly lower (p<0.001) mean changes from baseline in mTSS at week 28/ET occurred in peficitinib (1.62, 100 mg; 1.03, 150 mg) than placebo (3.37) recipients. Peficitinib was associated with haematological and biochemical parameter changes, and increased incidence of serious infections and herpes zoster-related disease. One death from suicide occurred in a patient in the placebo group after switching to peficitinib 100 mg.
Conclusions
In Japanese patients with RA and inadequate response to MTX, peficitinib demonstrated significant superiority versus placebo in reducing RA symptoms and suppressing joint destruction. Peficitinib had an acceptable safety and tolerability profile, with no new safety signals compared with other JAK inhibitors.
Trial registration number
Keywords: rheumatoid arthritis, DAS28, disease activity, methotrexate, treatment
Key messages.
What is already known about this subject?
Peficitinib inhibits the kinase activities of all Janus kinase (JAK) family members (pan-JAK inhibition) and was approved in Japan in 2019 as a once-daily rheumatoid arthritis (RA) therapy in both 100 and 150 mg/day regimens, with no dose adjustment for renal injury.
What does this study add?
This study was a randomised, double-blind, phase III study conducted in patients who had an inadequate response to methotrexate (MTX). Patients from Japan were randomised to 52 weeks’ treatment with peficitinib 100 or 150 mg/day, or placebo, in combination with MTX.
Peficitinib demonstrated superiority over placebo at both doses in reducing RA symptoms and suppressing joint destruction, according to the primary efficacy variables of ACR response and van der Heijde-modified total Sharp score.
The efficacy of peficitinib 150 mg/day was numerically superior to the 100 mg/day dose, with no apparent dose dependency from a safety perspective and similar safety signals, such as serious infections, herpes zoster and malignancies, to those of other JAK inhibitors.
How might this impact on clinical practice or future developments?
Based on these results, peficitinib may be a valuable addition to the treatment options for RA, particularly for patients with RA who are unresponsive to conventional treatments.
Introduction
Despite the advances in the management of rheumatoid arthritis (RA),1 a vast unmet need remains in relation to progressive disability, reduced quality of life, systemic comorbidities, premature death and high socioeconomic costs.2–5 RA therapy includes conventional synthetic disease-modifying antirheumatic drugs (csDMARDs)6 7 and biological disease-modifying antirheumatic drugs (DMARDs), mostly in combination with methotrexate (MTX), with the objective of achieving remission or low disease activity.7 8 However, current treatment modalities have restricted efficacy: 30%–40% of patients fail to respond to biological treatment, and only 20%–25% of patients achieve complete remission.9 Accordingly, new treatment options with mechanisms of action distinct from those of csDMARDs or biological agents are needed.
A key event in the pathogenesis and progression of RA is the activation of the Janus kinase (JAK)/signal transducers and activators of transcription signal transduction pathway.10–13 The role of JAKs in RA has led to the development of targeted small-molecule JAK inhibitors.14 Peficitinib (ASP015K) is an oral JAK inhibitor that inhibits kinase activities of all JAK family members: JAK1, JAK2, JAK3 and tyrosine kinase 2.15 16 An increase in haemoglobin levels corroborates the observation that peficitinib is associated with relatively mild inhibition of JAK2.17 18 In Japan, two other JAK inhibitors, tofacitinib and baricitinib, are currently available for patients with RA and an inadequate response to conventional therapies.16
A phase IIb study in Japanese patients with moderate-to-severe RA treated with peficitinib monotherapy for 12 weeks demonstrated that peficitinib was efficacious and had an acceptable safety profile.17 Two other phase IIb studies of peficitinib have also been conducted in non-Japanese populations.19 20 A separate study of peficitinib (RAJ3) has recently completed in patients with an inadequate response to DMARDs. Here we report the results of the RAJ4 study that assessed the efficacy and safety of two dosage regimens of peficitinib in combination with MTX, compared with placebo, in Japanese patients with RA and an inadequate response to MTX. The upper two doses (100 and 150 mg/day) were selected for this study based on previous efficacy and safety findings.17
Methods
Study design
This was a randomised, phase III, placebo-controlled, double-blind, parallel-group confirmatory study conducted in 161 centres in Japan between 25 July 2014 and 28 November 2017. Patients were randomly assigned 1:1:1 to receive peficitinib 100 mg, peficitinib 150 mg or placebo once daily, orally, in combination with a stable dose of MTX (≤16 mg/week) for 52 weeks (online supplementary methods). The peficitinib dose (100 or 150 mg) to which the patients in the placebo group were switched was determined randomly at baseline (online supplementary figure 1). Inadequate responders (<20% improvement from baseline in tender joint count at 68 joints (TJC68) and swollen joint count at 66 joints (SJC66)) in the placebo group were switched in a double-blind manner at week 12 to either peficitinib 100 or 150 mg; dosage was maintained until end of treatment (EOT). Patients still receiving placebo at week 28 were switched to either peficitinib 100 or 150 mg. The study was registered at ClinicalTrials.gov.
annrheumdis-2019-215164supp001.pdf (4.3MB, pdf)
Patients
Eligible patients were ≥20 years old, had RA for <10 years (fulfilling the 1987 American College of Rheumatology (ACR)21 or the 2010 ACR/European League Against Rheumatism (EULAR) criteria22), had evidence of active disease (≥6/68 TJC and ≥6/66 SJC), had C reactive protein (CRP) of ≥1.00 mg/dL and bone erosion in ≥1 joint (confirmed at the local site using van der Heijde-modified total Sharp score (mTSS)) and met the ACR 1991 Revised Criteria for the Classification of Global Functional Status in RA class I, II or III at screening.23 Eligible patients must also have had an inadequate response to MTX ≥8 mg/week for ≥28 days prior to baseline when administered continuously for ≥90 days prior to screening. Patients with an inadequate response to MTX at a dose of <8 mg/week were eligible if they were unable to tolerate a higher dose. Patients had to continue a stable dose of MTX (≤16 mg/week) ≥28 days prior to screening until EOT. Exclusion criteria included treatment with biological DMARDs within specified periods prior to baseline or other JAK inhibitors, infections or laboratory abnormalities, or a history of or concurrent malignant tumour (online supplementary methods).
Outcomes
Efficacy assessments
The primary efficacy endpoints were ACR20 response rate24 at week 12/early termination (ET) and change from baseline in mTSS25 at week 28/ET. Hand and foot radiographs were scored by two central readers independently, and if necessary by an adjudicator. All were blinded to the (time) order of the films and clinical information (online supplementary methods). The mean scores assigned by the two primary readers were used for the analyses.
Key secondary efficacy endpoints included ACR20/50/70 response, Disease Activity Score (DAS) 28-CRP26 and DAS28-erythrocyte sedimentation rate (ESR), CRP, ESR, Physician’s Global Assessment of Disease Activity (PGA), TJC68, SJC66, Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI). The proportion of patients achieving ACR/EULAR remission (≤1/68 TJC, ≤1/66 SJC, CRP≤1 mg/dL and Subject's Global Assessment of Disease Activity (SGA)≤10 mm on a visual analogue scale (VAS)) was also assessed. Structural progression assessments included changes from baseline in mTSS (at week 52/ET), erosion and joint space narrowing (JSN) scores (at weeks 28/ET and 52/ET), and rates of nonprogression (≤0.5 unit change from baseline in mTSS). Patient-reported outcomes included Health Assessment Questionnaire-Disability Index (HAQ-DI),27 SGA and Subject’s Global Assessment of Pain (SGAP,28 both measured on a 0–100 mm VAS).
Safety
Key safety variables included treatment-emergent adverse events (TEAEs), including the incidence of venous thromboembolism (VTE), from the initial dose of study drug through week 52 or follow-up period (online supplementary figure 1) and mean (SD) change from baseline in haematological and biochemical parameters after initial dose of study drug through week 52 or withdrawal. TEAEs for serious infections, herpes zoster-related disease (including varicella) and malignancies were assessed per 100 patient-years.
Statistical analyses
Primary analysis was performed on the full analysis set (FAS), comprising all randomised patients who received at least one dose of study drug; this was the primary data set for efficacy analyses. The assessment of joint destruction was performed on patients with at least one film at baseline and at week 12 or later (including week 12 assessment; see online supplementary methods). Safety analyses were performed on the safety analysis set (SAF), comprising all patients who received at least one dose of study drug. While the definitions of the FAS and SAF were different, ultimately these comprised the same patients.
For ACR20 response at week 12/ET, pairwise comparisons with placebo were performed for each peficitinib dose using Fisher’s exact test in the primary analysis. For mTSS change from baseline at week 28/ET, pairwise comparisons with placebo were performed at each peficitinib dose using rank analysis of covariance with treatment group as factor and baseline rank mTSS as covariate in the primary analysis. A closed testing procedure was used for multiplicity adjustment in the primary analysis (online supplementary methods).
To detect a difference between peficitinib and placebo, a sample size of 510 patients (170 per treatment group) was estimated to provide 90% power at a two-sided 0.05 significance level, allowing for a dropout rate of approximately 10%.
For missing data, the last observation carried forward (LOCF) was used for ACR components, DAS28 and safety variables at weeks 12/ET, 28/ET and 52/ET. For mTSS, erosion score and JSN score, linear extrapolation was used at weeks 28/ET and 52/ET (see online supplementary methods). Two-sided tests at the 0.05 significance level were used for statistical comparisons of peficitinib treatment groups versus placebo. Statistical analyses were performed using SAS V.9.4.
To assess the robustness of findings from the primary efficacy analysis, sensitivity analyses were performed (online supplementary tables 1 and 2).
Results
Patient demographics, baseline characteristics and treatment compliance
A total of 780 patients were screened; 519 patients were randomised and treated with the study drug, and 518 (99.8%) patients were included in the FAS and SAF. One patient was excluded from all analyses due to a major protocol violation (prescription of peficitinib 100 mg outside of study parameters). The completion rate of the study was numerically higher in patients randomised to peficitinib 100 or 150 mg at baseline (83.9%–84.6%) than placebo (77.6%–78.8%) (figure 1). There were 364 (70.3%) female patients. The mean age ranged from 55.3 to 58.5 years. The mean duration of RA ranged from 4.30 to 4.41 years. The mean prior biological DMARD use ranged from 15.5% to 22.4%, and the dose of concomitant MTX at baseline ranged from 9.78 to 10.09 mg/week. Patient characteristics were similar between treatment groups, except for age. Baseline disease activity and RA history were also similar between treatment groups, except for baseline SGA (table 1).
Figure 1.

Patient disposition. *The number of patients who were allocated at randomisation to either peficitinib 100 mg or peficitinib 150 mg starting from week 12 (in the case of inadequate response) or week 28 (for placebo responders). †Discontinuation up to week 12: discontinued at any time from date of randomisation before day 85. ‡Discontinuation for overall period: discontinued at any time from start of initial dosing of study drug through follow-up.
Table 1.
Patient demographics and baseline characteristics*
| Placebo (n=170) | Peficitinib 100 mg (n=174) | Peficitinib 150 mg (n=174) | Peficitinib 100 mg +150 mg (n=348) | Total (n=518) | |
| Female, n (%) | 121 (71.2) | 118 (67.8) | 125 (71.8) | 243 (69.8) | 364 (70.3) |
| Age in years, mean (SD) | 55.3 (12.1) | 58.5 (10.8) | 56.2 (11.6) | 57.4 (11.2) | 56.7 (11.6) |
| <65 years, n (%) | 125 (73.5) | 116 (66.7) | 131 (75.3) | 247 (71.0) | 372 (71.8) |
| Body weight in kg, mean (SD) | 58.92 (13.30) | 57.39 (12.32) | 58.20 (12.49) | 57.79 (12.39) | 58.16 (12.70) |
| RA duration in years,† mean (SD) | 4.30 (2.93) | 4.41 (2.96) | 4.37 (3.09) | 4.39 (3.02) | 4.36 (2.99) |
| Tender joint count (68 joints),‡ mean (SD) | 15.4 (9.4) | 14.0 (8.6) | 14.5 (7.8) | 14.2 (8.2) | 14.6 (8.6) |
| Swollen joint count (66 joints),‡ mean (SD) | 13.6 (7.0) | 12.8 (6.8) | 13.1 (6.9) | 13.0 (6.8) | 13.2 (6.9) |
| Physician’s Global Assessment of Disease Activity (100 mm VAS),§ mean (SD) |
60.98 (19.59) | 58.87 (19.67) | 60.86 (19.09) | 59.87 (19.38) | 60.23 (19.43) |
| Subject’s Global Assessment of Disease Activity (100 mm VAS),§ mean (SD) |
58.18 (23.90) | 51.70 (25.25) | 55.44 (24.49) | 53.57 (24.91) | 55.07 (24.65) |
| Subject’s Global Assessment of Pain (100 mm VAS),§ mean (SD) |
56.75 (25.29) | 51.12 (26.14) | 55.09 (24.89) | 53.10 (25.56) | 54.30 (25.51) |
| mTSS,‡ mean (SD) | 28.40 (36.28) | 25.23 (35.50) | 25.00 (32.38) | 25.11 (33.92) | 26.19 (34.71) |
| Erosion score,‡ mean (SD) | 11.03 (17.96) | 10.34 (17.47) | 9.76 (15.93) | 10.05 (16.69) | 10.37 (17.11) |
| Joint space narrowing score,‡ mean (SD) | 17.37 (20.13) | 14.89 (19.47) | 15.23 (18.33) | 15.06 (18.88) | 15.82 (19.31) |
| DAS28-CRP,‡ mean (SD) | 5.41 (0.85) | 5.21 (0.94) | 5.36 (0.92) | 5.29 (0.93) | 5.33 (0.91) |
| DAS28-CRP ≤3.2, n (%) | 0 | 3 (1.7) | 3 (1.7) | 6 (1.7) | 6 (1.2) |
| DAS28-CRP >3.2–≤5.1, n (%) | 63 (37.3) | 76 (43.7) | 59 (33.9) | 135 (38.8) | 198 (38.3) |
| DAS28-CRP >5.1, n (%) | 106 (62.7) | 95 (54.6) | 112 (64.4) | 207 (59.5) | 313 (60.5) |
| Missing, n | 1 | 0 | 0 | 0 | 1 |
| DAS28-ESR,¶ mean (SD) | 6.05 (0.88) | 5.83 (0.99) | 5.98 (1.00) | 5.91 (1.00) | 5.95 (0.96) |
| DAS28-ESR ≤3.2, n (%) | 0 | 1 (0.6) | 1 (0.6) | 2 (0.6) | 2 (0.4) |
| DAS28-ESR >3.2–≤5.1, n (%) | 24 (14.2) | 30 (17.2) | 29 (16.8) | 59 (17.0) | 83 (16.1) |
| DAS28-ESR>5.1, n (%) | 145 (85.8) | 143 (82.2) | 143 (82.7) | 286 (82.4) | 431 (83.5) |
| Missing, n | 1 | 0 | 1 | 1 | 2 |
| HAQ-DI score,† mean (SD) | 1.05 (0.66) | 0.91 (0.65) | 1.02 (0.62) | 0.96 (0.64) | 0.99 (0.65) |
| CRP (mg/dL), mean (SD) | 2.622 (2.146) | 2.432 (2.076) | 2.524 (2.183) | 2.478 (2.127) | 2.525 (2.132) |
| ESR (mm/hr), mean (SD) | 53.8 (26.9) | 50.4 (26.2) | 51.5 (26.8) | 51.0 (26.5) | 51.9 (26.6) |
| CDAI score,‡ mean (SD) | 31.56 (10.60) | 29.88 (11.73) | 31.51 (11.39) | 30.69 (11.57) | 30.98 (11.26) |
| SDAI score,‡ mean (SD) | 34.18 (11.14) | 32.31 (12.26) | 34.03 (11.99) | 33.17 (12.14) | 33.50 (11.82) |
| Prior biological DMARD use, n (%) | 38 (22.4) | 33 (19.0) | 27 (15.5) | 60 (17.2) | 98 (18.9) |
| MTX dose at baseline mg/week, mean (SD) | 9.78 (3.08) | 10.09 (2.75) | 9.88 (2.81) | 9.99 (2.78) | 9.92 (2.88) |
| >0–≤8, n (%) | 84 (49.7) | 63 (36.6) | 76 (44.2) | 139 (40.4) | 223 (43.5) |
| >8–≤12, n (%) | 60 (35.5) | 88 (51.2) | 74 (43.0) | 162 (47.1) | 222 (43.3) |
| >12, n (%) | 25 (14.8) | 21 (12.2) | 22 (12.8) | 43 (12.5) | 68 (13.3) |
| Missing, n | 1 | 2 | 2 | 4 | 5 |
| Positive for anti-CCP antibodies,** % | 88.8 | 89.7 | 92.5 | 91.1 | 90.3 |
| Positive for rheumatoid factor,** % | 67.6 | 66.1 | 67.8 | 67.0 | 67.2 |
| Number of prior DMARDs (including biologicals) | |||||
| 1 | 56 (32.9) | 57 (32.8) | 66 (37.9) | 123 (35.3) | 179 (34.6) |
| 2 | 83 (48.8) | 90 (51.7) | 83 (47.7) | 173 (49.7) | 256 (49.4) |
| ≥3 | 31 (18.2) | 27 (15.5) | 25 (14.4) | 52 (14.9) | 83 (16.0) |
| Prior non-biological DMARD use, except for MTX, n (%) | 97 (57.1) | 105 (60.3) | 95 (54.6) | 200 (57.5) | 297 (57.3) |
*All values are n (mean) unless otherwise indicated.
†Duration of RA (years) was calculated as (date of baseline mTSS taken − onset date of RA + 1)/365.25.
‡Higher values indicate greater levels of disease activity.
§Possible VAS scores range from 0 to 100, with higher scores indicating greater disease activity.
¶Possible HAQ-DI scores range from 0 to 3, with higher scores indicating greater disability.
**Patients with ‘high positive’ readings, defined as 3 × upper limit of normal range.
CCP, cyclic citrullinated peptide; CDAI, Clinical Disease Activity Index; CRP, C-reactive protein; DAS28-CRP, disease activity score for 28 joints based on CRP; DAS28-ESR, disease activity score for 28 joints based on erythrocyte sedimentation rate; DMARD, disease-modifying antirheumatic drug; ESR, erythrocyte sedimentation rate; HAQ-DI, Health Assessment Questionnaire-Disability Index; MTX, methotrexate; RA, rheumatoid arthritis; SDAI, Simplified Disease Activity Index; mTSS, van der Heijde-modified total Sharp score.
Efficacy
ACR20/50/70 response rates
ACR20 response rates at week 12/ET (LOCF) were 37/170 (21.8%, placebo), 102/174 (58.6%, peficitinib 100 mg) and 112/174 (64.4%, peficitinib 150 mg). Significant differences versus placebo of 36.9% (95% CI 26.7% to 47.0%) for peficitinib 100 mg and 42.6% (95% CI 32.6% to 52.6%) for peficitinib 150 mg were observed (p<0.001 for both comparisons). ACR50 and ACR70 responses were significantly greater for both peficitinib groups than placebo (figure 2A). Mean changes from baseline in mTSS at week 28/ET (linear extrapolation) were 3.37 (placebo), 1.62 (peficitinib 100 mg) and 1.03 (peficitinib 150 mg). The rank analysis of covariance model demonstrated that differences from placebo were significant for both peficitinib doses (p<0.001) (figure 2B). Sensitivity analyses showed the robustness of both primary efficacy analyses (online supplementary tables 1 and 2).
Figure 2A.

ACR20/50/70 responses at week 12/ET (FAS). Data shown are the LOCF. A closed testing procedure was used for multiplicity adjustment for ACR20 and no multiplicity adjustment for ACR50 and ACR70. *Fisher’s exact test.
Figure 2B.

Change from baseline in mTSS, JSN and erosion scores at week 28/ET (linear extrapolation) (FAS). A closed testing procedure was used for multiplicity adjustment for mTSS. †Based on rank analysis of covariance model: rank of mTSS change = treatment + baseline rank of mTSS; rank of JSN score change = treatment + baseline rank of JSN score; rank of erosion score change = treatment + baseline rank of erosion score.
Key secondary efficacy endpoints
ACR/EULAR remission at week 12/ET was achieved by 1/169 (0.6%), 10/172 (5.8%) and 17/171 (9.9%) patients in the placebo, peficitinib 100 and 150 mg groups, respectively (LOCF and FAS). The treatment difference versus placebo was 5.2% (p=0.011) for peficitinib 100 mg and 9.3% (p<0.001) for peficitinib 150 mg.
Significantly reduced mean changes from baseline in both JSN and erosion score were observed in both peficitinib groups compared with placebo (figure 2B). Cumulative probability plot for the change in mTSS score up to weeks 28/ET and 52/ET illustrates that peficitinib 100 and 150 mg were more efficacious than placebo for prevention of joint destruction (figure 2C). Significantly greater proportions of peficitinib-treated patients showed a change from baseline in mTSS ≤0.5 (figure 2D) and mTSS ≤0 (online supplementary figure 2A) when compared with placebo. Significantly greater proportions of placebo-treated than peficitinib-treated patients showed a change from baseline in mTSS ≥3 (online supplementary figure 2B). A significantly greater proportion of peficitinib-treated patients achieved CDAI ≤2.8 and SDAI ≤3.3 (remission), as well as CDAI 10 and SDAI≤11 (low disease activity) at weeks 12/ET and 28/ET (online supplementary figure 3A–D) when compared with placebo.
Figure 2C.

Cumulative probability plots for change from baseline in mTSS score at weeks 28/ET and 52/ET (FAS).
Figure 2D.

Proportions of patients showing a change from baseline in mTSS of ≤0.5 at weeks 28/ET and 52/ET (linear extrapolation (FAS)). Comparisons performed using Fisher’s exact test with no multiplicity adjustment.
From weeks 12 to 52, ACR20 response rates were maintained in the peficitinib groups, while respective ACR50 and ACR70 response rates further improved during this period. ACR20/50/70 response rates improved at weeks 16 or 32 after switching from placebo to peficitinib; the respective responses were maintained through week 52 (figure 2E–G).
Figure 2E.

ACR20 response over time to week 52 and EOT (FAS).
Figure 2F.

ACR50 response over time to week 52 and EOT (FAS).
Figure 2G.

ACR70 response over time to week 52 and EOT (FAS). Patients were switched from placebo to peficitinib 100 or 150 mg at week 12 for inadequate responders in the placebo group (as determined by <20% improvement from baseline in TJC68 and SJC66). Patients remaining in the placebo group at week 12 were switched from placebo to peficitinib 100 or 150 mg at week 28. Patients responding to placebo were maintained on placebo until week 28; ACR20 response rates at week 28 were therefore higher than those at week 12 for the placebo/peficitinib 100 mg (week 28) and placebo/peficitinib 150 mg (week 28) groups. ACR components were LOCF first and then the ACR response was calculated. LOCF data were included at EOT. ACR, American College of Rheumatology; EOT, end of treatment; ET, early termination; FAS, full analysis set; JSN, joint space narrowing; LOCF, last observation carried forward; mTSS, van der Heijde-modified total Sharp score; TJC68, tender joint count at 68 joints; SJC66, swollen joint count at 66 joints.
Compared with placebo, significant improvements in mean changes over time from baseline to week 28/ET were observed in both peficitinib groups for DAS28-CRP or other ACR core set (figure 3A–I and online supplementary table 3), ACR20/50/70 excluding ACR70 at Week 4 (online supplementary figure 4A–C and online supplementary table 3) and CDAI and SDAI scores (online supplementary figure 5A–B). Proportions of patients achieving DAS28-CRP<2.6, DAS28-CRP≤3.2 and DAS28-ESR<2.6 at weeks 12/ET and 28/ET were significantly greater for both peficitinib groups than placebo (p<0.001 for all comparisons, online supplementary figure 6A–C).
Figure 3.


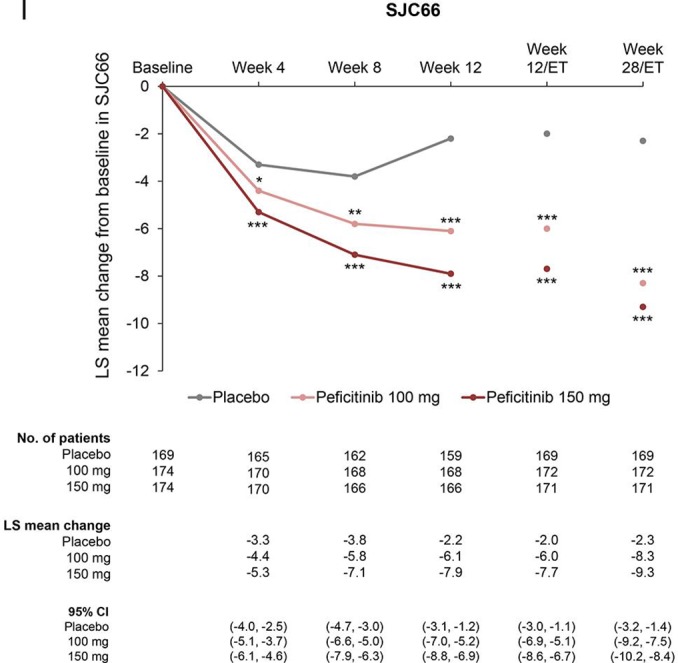
Change from baseline over time in (A) DAS28-CRP, (B) HAQ-DI, (C) CRP (mg/dL), (D) ESR (mm/hour), (E) SGAP (100 mm VAS), (F) SGA (100 mm VAS), (G) PGA (100 mm VAS) (H) TJC68 and (I) SJC66 (FAS). Figure shows observed data for weeks 0–12, and last observation carried forward for weeks 12/ET and 28/ET. No multiplicity adjustment. Statistical comparisons were conducted according to analysis of covariance model. *P<0.05, **P<0.01, ***P<0.001. CRP, C-reactive protein; DAS28-CRP, disease activity score in 28 joints based on CRP; ESR, erythrocyte sedimentation rate; FAS, full analysis set; HAQ-DI, Health Assessment Questionnaire – Disability Index; PGA, Physician’s Global Assessment of disease activity; SGA, Subject’s Global Assessment of disease activity; SGAP, Subject’s Global Assessment of Pain; SJC66, swollen joint count at 66 joints; TJC68, tender joint count at 68 joints; VAS, visual analogue scale.
Safety
Treatment-emergent adverse events
In the first 12 weeks of the study, investigator-reported TEAE incidence was greater in the peficitinib 100 mg (89 patients, 51.1%) and 150 mg (104 patients, 59.8%) groups than in the placebo group (84 patients, 49.4%). Investigator-reported drug-related TEAEs were also more common in the peficitinib 100 mg (57 patients, 32.8%) and 150 mg (80 patients, 46.0%) groups versus the placebo group (47 patients, 27.6%). Most TEAEs were grade 1 or 2 in severity and investigator-reported drug-related serious adverse events (SAEs) were reported at a similar incidence in all three treatment groups. Investigator-reported drug-related adverse events (AEs) leading to permanent discontinuation of study drug were reported in 3 (1.7%), 5 (2.9%) and 6 (3.5%) patients in the peficitinib 100 mg group, 150 mg group and placebo group, respectively (table 2A).
Table 2.
Safety summary. (A) Treatment-emergent adverse events (SAF); (B) TEAEs for serious infections, herpes zoster-related disease and malignancies for overall study period (SAF)
| (A) | Weeks 0–12 | Overall period | |||||
| Placebo (N=170) | Peficitinib 100 mg (N=174) | Peficitinib 150 mg (N=174) | Peficitinib 100 mg +150 mg (N=348) | Peficitinib 100 mg (N=174) | Peficitinib 150 mg (N=174) | Peficitinib 100 mg +150 mg (N=348) | |
| AEs | 84 (49.4) | 89 (51.1) | 104 (59.8) | 193 (55.5) | 154 (88.5) | 153 (87.9) | 307 (88.2) |
| Drug-related AEs* | 47 (27.6) | 57 (32.8) | 80 (46.0) | 137 (39.4) | 118 (67.8) | 123 (70.7) | 241 (69.3) |
| Deaths | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| SAEs | 4 (2.4) | 5 (2.9) | 3 (1.7) | 8 (2.3) | 19 (10.9) | 13 (7.5) | 32 (9.2) |
| Drug-related SAEs* | 2 (1.2) | 3 (1.7) | 3 (1.7) | 6 (1.7) | 10 (5.7) | 8 (4.6) | 18 (5.2) |
| ≥Grade 3 AEs† | 8 (4.7) | 9 (5.2) | 16 (9.2) | 25 (7.2) | 29 (16.7) | 32 (18.4) | 61 (17.5) |
| TEAEs leading to permanent discontinuation of study drug | |||||||
| All | 7 (4.1) | 5 (2.9) | 5 (2.9) | 10 (2.9) | 13 (7.5) | 12 (6.9) | 25 (7.2) |
| Drug-related AEs* | 6 (3.5) | 3 (1.7) | 5 (2.9) | 8 (2.3) | 7 (4.0) | 11 (6.3) | 18 (5.2) |
| SAEs | 3 (1.8) | 1 (0.6) | 0 | 1 (0.3) | 6 (3.4) | 4 (2.3) | 10 (2.9) |
| Drug-related SAEs* | 2 (1.2) | 1 (0.6) | 0 | 1 (0.3) | 4 (2.3) | 3 (1.7) | 7 (2.0) |
All values are n (%).
*Possible or probable, as assessed by the investigator or records where relationship is missing.
†Based on National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) grade: grade 3 = severe or medically significant, grade 4 = life threatening, grade 5 = death related to AE.
AE, adverse event; SAE, severe adverse event; SAF, safety analysis set; TEAE, treatment-emergent adverse event.
| (B) | Placebo (N=170) |
Peficitinib 100 mg (N=174) | Peficitinib 150 mg (N=174) | Peficitinib 100 mg +150 mg (N=348) | Peficitinib total* (N=496) |
| Serious infections | |||||
| Patient-years | 62.9 | 159.5 | 160.8 | 320.3 | 407.8 |
| Number of patients who had at least one incidence | 0 | 6 | 6 | 12 | 14 |
| Incidence rate/100 patient-years (95% CI) | 0.0 | 3.8 (1.7, 8.4) | 3.7 (1.7, 8.3) | 3.7 (2.1, 6.6) | 3.4 (2.0, 5.8) |
| Herpes zoster-related disease (including varicella) | |||||
| Patient-years | 62.6 | 156.2 | 159.8 | 316.0 | 402.9 |
| Number of patients who had at least one incidence | 2 | 13 | 6 | 19 | 23 |
| Incidence rate/100 patient-years (95% CI) | 3.2 (0.8, 12.8) | 8.3 (4.8, 14.3) | 3.8 (1.7, 8.4) | 6.0 (3.8, 9.4) | 5.7 (3.8, 8.6) |
| Malignancies | |||||
| Patient-years | 62.9 | 162.2 | 163.0 | 325.2 | 413.2 |
| Number of patients who had at least one incidence | 1 | 1 | 0 | 1 | 1 |
| Incidence rate/100 patient-years (95% CI) | 1.6 (0.2, 11.3) | 0.6 (0.1, 4.4) | 0.0 | 0.3 (0.0, 2.2) | 0.2 (0.0, 1.7) |
Patient-years was calculated from initial dose up to first incidence of the event for patients who had at least one event, and from initial dose through follow-up for patients who had no events; incidence rate is calculated as (100 × number of patients who had at least one incidence/total patient-years).
*Including adverse events during treatment with peficitinib in patients who were initially treated with placebo and switched to peficitinib at week 12 or 28.
CI, confidence interval.
During the overall study period, TEAEs were reported in 154 (88.5%) and 153 (87.9%) patients in the peficitinib 100 and 150 mg groups, respectively. Investigator-reported drug-related TEAEs were reported in 118 (67.8%) and 123 (70.7%) patients in the peficitinib 100 and 150 mg groups, respectively. Most TEAEs were grade 1 or 2 in severity. Investigator-reported drug-related SAEs were reported in 10 (5.7%) and 8 (4.6%) patients in the peficitinib 100 and 150 mg groups, respectively. Investigator-reported drug-related AEs leading to permanent discontinuation of study drug were reported in 7 (4.0%) and 11 (6.3%) patients in the peficitinib 100 and 150 mg groups, respectively (table 2A).
No deaths were reported up to week 28 (table 2A, online supplementary table 4). One death due to suicide was reported in a 69-year-old man in the placebo group after switching to peficitinib 100 mg at week 28 (online supplementary table 5). The investigator considered the suicide was not related to the study drug because he had concurrent depression (online supplementary narrative).
In the overall period, the incidence of serious infections and herpes zoster-related diseases (including varicella) was higher in the peficitinib groups than in the placebo group; no dose-dependent association was observed. There was no report of multiple dermatomes and generalised herpes zoster in this study. No apparent dose dependency was observed in the incidence of malignancy in the treatment groups (table 2B). VTE was not observed in the study.
Clinical laboratory evaluations
At week 12/ET, a decrease in absolute neutrophil count and an increase in haemoglobin were observed in the peficitinib groups compared with the placebo group. No major differences were observed in mean changes from baseline in haematological parameters at EOT compared with week 12/ET (table 3).
Table 3.
Haematological and biochemical parameters
| Baseline result | Change to week 12/ET | Change to week 28/ET | Change to Week 52/ET | ||||||||
| Placebo (N=170) | Peficitinib 100 mg (N=174) | Peficitinib 150 mg (N=174) | Placebo (N=170) | Peficitinib 100 mg (N=174) | Peficitinib 150 mg (N=174) | Placebo (N=170) | Peficitinib 100 mg (N=174) | Peficitinib 150 mg (N=174) | Peficitinib 100 mg (N=170) |
Peficitinib 150 mg (N=170) |
|
| Absolute neutrophil count (106/L) | 5803.5 (2171.9) | 5746.0 (1901.3) | 5679.9 (2058.8) | −41.4 (1496.4) | −579.7 (1717.0) | −720.5 (1823.8) | −89.9 (1611.6) | −640.7 (1782.2) | −802.9 (1701.5) | −694.8 (1725.7) | −740.4 (1937.3) |
| Haemoglobin (g/L) | 119.0 (13.0) | 121.4 (14.6) | 122.4 (13.2) | 0.2 (7.4) | 4.5 (8.4) | 5.0 (7.7) | 0.2 (8.6) | 5.1 (9.8) | 5.3 (10.6) | 4.7 (10.5) | 4.4 (10.2) |
| Platelets (109/L) | 327.4 (73.7) | 320.9 (84.6) | 320.9 (82.2) | 1.6 (47.1) | −40.7 (49.4) | −48.8 (48.9) | −1.4 (59.0) | −47.9 (55.9) | −49.1 (56.6) | −50.2 (57.0) | −46.9 (61.4) |
| LDL cholesterol (mmol/L) | 2.799 (0.665) | 2.843 (0.706) | 2.842 (0.742) | 0.016 (0.387) | 0.281 (0.533) | 0.246 (0.468) | 0.033 (0.441) | 0.306 (0.569) | 0.310 (0.617) | 0.261 (0.618) | 0.353 (0.599) |
| HDL cholesterol (mmol/L) | 1.620 (0.424) | 1.628 (0.427) | 1.567 (0.424) | 0.011 (0.221) | 0.235 (0.322) | 0.330 (0.294) | 0.018 (0.247) | 0.320 (0.364) | 0.396 (0.373) | 0.340 (0.353) | 0.403 (0.353) |
| Serum creatinine (μmol/L) | 51.22 (11.12) | 51.21 (13.53) | 51.01 (12.38) | −0.13 (5.19) | 4.73 (4.61) | 5.24 (6.61) | 0.64 (5.99) | 6.50 (5.61) | 7.26 (6.70) | 7.31 (5.46) | 9.13 (7.93) |
| Creatine kinase (U/L) | 60.3 (35.3) | 63.3 (36.6) | 61.9 (49.4) | −1.7 (26.6) | 49.0 (46.1) | 61.9 (93.3) | 4.6 (45.1) | 62.8 (61.4) | 72.6 (75.8) | 74.0 (72.3) | 80.1 (79.2) |
| ALT (U/L) | 20.5 (16.0) | 20.1 (16.3) | 20.2 (13.6) | −0.2 (12.8) | 3.8 (16.7) | 6.9 (19.6) | −0.7 (15.0) | 4.2 (17.3) | 7.9 (19.8) | 3.2 (16.5) | 7.5 (19.9) |
| AST (U/L) | 23.4 (11.9) | 23.6 (12.5) | 23.6 (11.6) | −0.1 (10.8) | 3.6 (11.9) | 5.6 (14.4) | 0.2 (12.9) | 4.3 (12.0) | 6.5 (14.8) | 4.2 (12.4) | 7.0 (16.0) |
Results shown are mean (SD).
ALT, alanine aminotransferase; AST, aspartate aminotransferase; EOT, end of treatment; ET, early termination; HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Compared with the placebo group at week 12/ET, increases in creatine kinase, creatinine, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol were observed in peficitinib groups. No major differences were observed in the mean changes from baseline in biochemical parameters at EOT compared with week 12/ET (table 3).
Discussion
This phase III study in Japanese patients with RA and an inadequate response to MTX demonstrated that peficitinib is associated with statistically significant improvements in ACR20 response rates and change from baseline mTSS at both 100 and 150 mg/day doses, compared with placebo. The secondary efficacy variables supported the results of the primary efficacy analysis, with generally significant improvements at weeks 12/ET and 28/ET for both peficitinib doses versus placebo. The efficacy variables observed with both peficitinib doses were maintained throughout the study period. Improvements in the placebo group after switching to either peficitinib dose were also maintained throughout the remaining study period. Generally, the efficacy of peficitinib 150 mg was numerically greater than that of peficitinib 100 mg, particularly with respect to depth of response (ACR70 and radiographic inhibition). The results from this study expand on those from a previous phase IIb study.17 Peficitinib also demonstrated similar results to those in a study of non-Japanese populations.19 However, they differed from those in another non-Japanese study due to a higher-than-expected response rate in the placebo group.20 There appears to be a lack of consistency in the proportional placebo response rate across geographical regions, and the reasons for this are not yet understood.
Peficitinib was generally well tolerated over 52 weeks of treatment. Most TEAEs were grade 1 or 2 in severity. The rate of discontinuation due to drug-related AEs was similar between the peficitinib 100 and 150 mg groups. The safety signals were comparable with those of other currently available JAK inhibitors.29–32 Decreased neutrophils and changes in haematological and biochemical parameters have been observed in both tofacitinib-31 and baricitinib-treated RA patients,29 and an increased incidence of herpes zoster has also been reported in patients with RA treated with tofacitinib30 or baricitinib.33 While there are concerns of an increased risk of thromboembolic events with JAK inhibitors,34 no such events, such as VTE, were reported in the patients in our study.
This study recruited patients with predominantly severe RA with inadequate response to prior therapy with MTX, thus representing a group of patients with potentially refractory disease. It should be borne in mind that the recommended MTX dose in Japan (up to 16 mg/week), which was used in the present study, is generally lower than that recommended in non-Japanese populations.7 35 The improvements in clinical signs and symptoms achieved with once-daily peficitinib in combination with MTX were robust and durable. Evidence of structural efficacy was also observed with peficitinib: radiographic changes were significantly reduced with peficitinib compared with placebo. However, changes from baseline in mTSS at week 28/ET remained at 1.62 and 1.03 for peficitinib 100 and 150 mg dose groups, respectively. Indeed, mTSS tends to be higher in Japanese patients than in other populations.36–39 This may also be explained by the baseline CRP levels and RA duration in patients included in the study, as high CRP levels40 and early RA36 37 could contribute to the progression of joint destruction.
Limitations of the study include the shorter duration of placebo compared with active treatment, which was necessary for ethical reasons but makes comparisons difficult. Another limitation is that a peficitinib dose higher than 150 mg/day was not evaluated in the present study; further evaluation might be needed to confirm whether 150 mg/day is the maximal dose from an efficacy perspective. The efficacy and safety of peficitinib in combination with RA treatments other than MTX were not assessed in this study, but have been assessed in another phase III trial, which showed results comparable with those in the present study.41 An extension study is also ongoing to further evaluate safety and efficacy of peficitinib (NCT01638013), specifically to confirm the persistence of treatment response and to ascertain that peficitinib’s safety profile does not differ greatly from the safety profile of other JAK inhibitors over long-term treatment. In addition, the patient population was drawn only from Japan, which may lack global diversity.
In conclusion, this study provides evidence that once-daily peficitinib 100 and 150 mg demonstrates robust clinical and structural efficacy in patients with RA who have an inadequate response to MTX.
Acknowledgments
The authors thank the patients who were involved in this study, as well as the investigators and the study team. A list of study centres is provided in the online supplementary information. The study was sponsored by Astellas Pharma, Inc. Writing and editorial support were provided by Derek Ho, PhD, and Rhian Harper Owen, PhD, for Cello Health MedErgy, and were funded by Astellas Pharma, Inc.
Footnotes
Handling editor: Josef S Smolen
TT and YT contributed equally.
Contributors: All authors met the following criteria for authorship: substantial contributions to the acquisition, analysis and interpretation of data for the work; contribution to drafting the work and revising it critically; giving the final approval of the version submitted; and agreeing to be accountable for all aspects of the work.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: TT has received grants from Astellas Pharma, Inc, Chugai Pharmaceutical Co, Ltd, Daiichi Sankyo Co, Ltd, Takeda Pharmaceutical Co, Ltd, AbbVie GK, Asahi Kasei Pharma Corp, Mitsubishi Tanabe Pharma Co, Pfizer Japan, Inc, Eisai Co, Ltd, AYUMI Pharmaceutical Corp, Nippon Kayaku Co, Ltd, and Novartis Pharma KK; speaking fees from AbbVie GK, Bristol-Myers KK, Chugai Pharmaceutical Co, Ltd, Mitsubishi Tanabe Pharma Co, Pfizer Japan, Inc, Astellas Pharma, Inc, Daiichi Sankyo Co, Ltd, Eisai Co, Ltd, Sanofi KK, Teijin Pharma Ltd, Takeda Pharmaceutical Co, Ltd, and Novartis Pharma KK; consultancy fees from AstraZeneca KK, Eli Lilly Japan KK, Novartis Pharma KK, Mitsubishi Tanabe Pharma Co, AbbVie GK, Nippon Kayaku Co, Ltd, Janssen Pharmaceutical KK, Astellas Pharma, Inc, Taiho Pharmaceutical Co, Ltd, Chugai Pharmaceutical Co, Ltd, Taisho Toyama Pharmaceutical Co, Ltd, GlaxoSmithKline KK, and UCB Japan Co, Ltd. YT reports speaking fees and/or honoraria from Daiichi Sankyo, Astellas, Eli Lilly, Chugai, Sanofi, AbbVie, Pfizer, YL Biologics, Bristol-Myers, GlaxoSmithKline, UCB, Mitsubishi-Tanabe, Novartis, Eisai, Takeda, Janssen and Asahi Kasei, and research grants from Mitsubishi-Tanabe, Bristol-Myers, Eisai, Chugai, Takeda, AbbVie, Astellas, Daiichi Sankyo, Ono, MSD and Taisho Toyama. ST reports personal fees from Amgen, Inc, Asahi Kasei Pharma Corporation, Amgen Astellas BioPharma KK, Ono Pharmaceutical Co, Ltd, KYOCERA Medical Corporation, Daiichi Sankyo Co, Ltd, Teijin Pharma Ltd, Eli Lilly Japan KK and Pfizer Japan, Inc; endowments from Astellas Pharma, Inc, AYUMI Pharmaceutical Corporation, Pfizer Japan, Inc, Bristol-Myers Squibb, Daiichi Sankyo Co, Ltd, and Chugai Pharmaceutical Co, Ltd; and grants from Japan Agency for Medical Research and Development, Japan Society for the Promotion of Science (JSPS)/Grant-in-Aid for Scientific Research (A), and JSPS/Grant-in-Aid for Exploratory Research outside the submitted work. AK has received grants from AbbVie, Eisai, Mitsubishi-Tanabe, Pfizer Japan, Takeda Pharmaceutical, Astellas Pharma, MSD, Ono Pharmaceutical, Teijin Pharma, Kyowa Hakko-Kirin, Sumitomo-Dainippon Pharma, Kissei Pharmaceutical, Boehringer Ingelheim, AstraZeneca, Otsuka Pharmaceutical, Chugai Pharmaceutical, Santen Pharmaceutical and Daiichi Sankyo; participated in speaker’s bureaux for AbbVie, Eisai, Takeda Pharmaceutical, Ono Pharmaceutical, Astellas Pharma, Mitsubishi Tanabe, Pfizer Japan, Chugai Pharmaceutical, MSD, and Bristol-Myers KK; and received consultancy fees from Astellas Pharma and Janssen Pharmaceutical KK. KK reports consultancy fees from Eisai Co, Ltd, and Toyama Chemical Co, Ltd and speaker fees from Daiichi Sankyo Co, Ltd, Astellas Pharma, Eli Lilly, Chugai, AbbVie, Pfizer, Bristol-Myers Squibb, UCB, Mitsubishi Tanabe, Takeda, Janssen, Asahi Kasei and AYUMI Pharmaceutical Corporation. MR, HI, SU, YK, TS and EY are employees of Astellas Pharma, Inc. MI has nothing to declare. DvdH has received consultancy honoraria from AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi, Eli Lilly, Galapagos, Gilead, GlaxoSmithKline, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda and UCB, and is the owner of Imaging Rheumatology.
Patient consent for publication: Not required.
Ethics approval: An institutional review board at each study site reviewed and approved the protocol and amendments. The study was conducted in accordance with Good Clinical Practice, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines, and applicable laws and regulations. Written informed consent was obtained from each participant.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Data are available upon reasonable request.
References
- 1. Kiadaliri AA, Felson DT, Neogi T, et al. Brief report: rheumatoid arthritis as the underlying cause of death in Thirty-One countries, 1987-2011: trend analysis of World Health Organization mortality database. Arthritis Rheumatol 2017;69:1560–5. 10.1002/art.40091 [DOI] [PubMed] [Google Scholar]
- 2. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature 2003;423:356–61. 10.1038/nature01661 [DOI] [PubMed] [Google Scholar]
- 3. Strand V, Singh JA. Improved health-related quality of life with effective disease-modifying antirheumatic drugs: evidence from randomized controlled trials. Am J Manag Care 2007;13 Suppl 9:S237–51. [PubMed] [Google Scholar]
- 4. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011;365:2205–19. 10.1056/NEJMra1004965 [DOI] [PubMed] [Google Scholar]
- 5. Taylor PC, Moore A, Vasilescu R, et al. A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatol Int 2016;36:685–95. 10.1007/s00296-015-3415-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O’Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med 2004;350:2591–602. 10.1056/NEJMra040226 [DOI] [PubMed] [Google Scholar]
- 7. Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 2017;76:960–77. 10.1136/annrheumdis-2016-210715 [DOI] [PubMed] [Google Scholar]
- 8. Smolen JS, Aletaha D, Bijlsma JWJ, et al. Treating rheumatoid arthritis to target: recommendations of an international Task Force. Ann Rheum Dis 2010;69:631–7. 10.1136/ard.2009.123919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zampeli E, Vlachoyiannopoulos PG, Tzioufas AG. Treatment of rheumatoid arthritis: unraveling the conundrum. J Autoimmun 2015;65:1–18. 10.1016/j.jaut.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 10. Malemud CJ. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther Adv Musculoskelet Dis 2018;10:117–27. 10.1177/1759720X18776224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka Y, Iwata S, Yamaoka K. JAK and SYK: emerging their relevance to the treatment of inflammatory diseases. Inflamm Regen 2011;31:237–44. 10.2492/inflammregen.31.237 [DOI] [Google Scholar]
- 12. Tanaka Y, Maeshima Y, Yamaoka K. In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis 2012;71(Suppl 2):i70–4. 10.1136/annrheumdis-2011-200595 [DOI] [PubMed] [Google Scholar]
- 13. Tanaka Y, Yamaoka K. JAK inhibitor tofacitinib for treating rheumatoid arthritis: from basic to clinical. Mod Rheumatol 2013;23:415–24. 10.3109/s10165-012-0799-2 [DOI] [PubMed] [Google Scholar]
- 14. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Exp Opin Investig Drugs 2014;23:1067–77. 10.1517/13543784.2014.918604 [DOI] [PubMed] [Google Scholar]
- 15. Ito M, Yamazaki S, Yamagami K, et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J Pharmacol Sci 2017;133:25–33. 10.1016/j.jphs.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 16. Yamaoka K. Janus kinase inhibitors for rheumatoid arthritis. Curr Opin Chem Biol 2016;32:29–33. 10.1016/j.cbpa.2016.03.006 [DOI] [PubMed] [Google Scholar]
- 17. Takeuchi T, Tanaka Y, Iwasaki M, et al. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis 2016;75:1057–64. 10.1136/annrheumdis-2015-208279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998;93:385–95. 10.1016/S0092-8674(00)81167-8 [DOI] [PubMed] [Google Scholar]
- 19. Genovese MC, Greenwald M, Codding C, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol 2017;69:932–42. 10.1002/art.40054 [DOI] [PubMed] [Google Scholar]
- 20. Kivitz AJ, Gutierrez-Ureña SR, Poiley J, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate-to-severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol 2017;69:709–19. 10.1002/art.39955 [DOI] [PubMed] [Google Scholar]
- 21. Arnett FC, Edworthy SM, Bloch DA, et al. The American rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. 10.1002/art.1780310302 [DOI] [PubMed] [Google Scholar]
- 22. Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. 10.1002/art.27584 [DOI] [PubMed] [Google Scholar]
- 23. Hochberg MC, Chang RW, Dwosh I, et al. The American College of rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum 1992;35:498–502. 10.1002/art.1780350502 [DOI] [PubMed] [Google Scholar]
- 24. Felson DT, Anderson JJ, Boers M, et al. American College of rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. 10.1002/art.1780380602 [DOI] [PubMed] [Google Scholar]
- 25. van der Heijde D. How to read radiographs according to the Sharp/van Der Heijde method. J Rheumatol 2000;27:261–3. [PubMed] [Google Scholar]
- 26. Fransen J, van Riel PLCM. The disease activity score and the EULAR response criteria. Clin Exp Rheumatol 2005;23(5 Suppl 39):S93–9. [PubMed] [Google Scholar]
- 27. Bruce B, Fries JF. The Stanford health assessment questionnaire: dimensions and practical applications. Health Qual Life Outcomes 2003;1:20 10.1186/1477-7525-1-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fries JF, Spitz P, Kraines RG, et al. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. 10.1002/art.1780230202 [DOI] [PubMed] [Google Scholar]
- 29. Dougados M, van der Heijde D, Chen Y-C, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis 2017;76:88–95. 10.1136/annrheumdis-2016-210094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winthrop KL, Yamanaka H, Valdez H, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol 2014;66:2675–84. 10.1002/art.38745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamanaka H, Tanaka Y, Takeuchi T, et al. Tofacitinib, an oral Janus kinase inhibitor, as monotherapy or with background methotrexate, in Japanese patients with rheumatoid arthritis: an open-label, long-term extension study. Arthritis Res Ther 2016;18:1–12. 10.1186/s13075-016-0932-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tanaka Y, Takeuchi T, Yamanaka H, et al. Efficacy and safety of tofacitinib as monotherapy in Japanese patients with active rheumatoid arthritis: a 12-week, randomized, phase 2 study. Mod Rheumatol 2015;25:514–21. 10.3109/14397595.2014.995875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smolen J, Genovese M, Takeuchi T, et al. THU0166 Safety Profile of Baricitinib in Patients with Active RA: An Integrated Analysis: Table 1. Ann Rheum Dis 2016;75(Suppl 2):243.2–4. 10.1136/annrheumdis-2016-eular.1612 [DOI] [Google Scholar]
- 34. Scott IC, Hider SL, Scott DL. Thromboembolism with Janus kinase (JAK) inhibitors for rheumatoid arthritis: how real is the risk? Drug Saf 2018;41:645–53. 10.1007/s40264-018-0651-5 [DOI] [PubMed] [Google Scholar]
- 35. Takahashi C, Kaneko Y, Okano Y, et al. Association of erythrocyte methotrexate-polyglutamate levels with the efficacy and hepatotoxicity of methotrexate in patients with rheumatoid arthritis: a 76-week prospective study. RMD Open 2017;3:e000363 10.1136/rmdopen-2016-000363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an X ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007;66:1162–7. 10.1136/ard.2006.068064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takeuchi T, Miyasaka N, Zang C, et al. A phase 3 randomized, double-blind, multicenter comparative study evaluating the effect of etanercept versus methotrexate on radiographic outcomes, disease activity, and safety in Japanese subjects with active rheumatoid arthritis. Mod Rheumatol 2013;23:623–33. 10.3109/s10165-012-0742-6 [DOI] [PubMed] [Google Scholar]
- 38. Yamamoto K, Takeuchi T, Yamanaka H, et al. Efficacy and safety of certolizumab pegol without methotrexate co-administration in Japanese patients with active rheumatoid arthritis: the HIKARI randomized, placebo-controlled trial. Mod Rheumatol 2014;24:552–60. 10.3109/14397595.2013.843764 [DOI] [PubMed] [Google Scholar]
- 39. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The premier study: a multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26–37. 10.1002/art.21519 [DOI] [PubMed] [Google Scholar]
- 40. Atsumi T, Yamamoto K, Takeuchi T, et al. The first double-blind, randomised, parallel-group certolizumab pegol study in methotrexate-naive early rheumatoid arthritis patients with poor prognostic factors, C-OPERA, shows inhibition of radiographic progression. Ann Rheum Dis 2016;75:75–83. 10.1136/annrheumdis-2015-207511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abstract. RAJ3 Efficacy and safety of the novel oral Janus kinase inhibitor, Peficitinib (ASP015K), in a phase 3, double-blind, placebo-controlled, randomized study of patients with RA who had an inadequate response to DMARDs. ACR/ARHP 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2019-215164supp001.pdf (4.3MB, pdf)