

Abstract
Objectives
To investigate the efficacy and safety of peficitinib, an oral Janus kinase inhibitor, in patients with rheumatoid arthritis (RA).
Methods
In this double-blind phase III study, patients with RA and an inadequate response to prior disease-modifying anti-rheumatic drugs (DMARDs) were randomised to peficitinib 100 mg once daily, peficitinib 150 mg once daily, placebo or open-label etanercept for 52 weeks’ treatment; placebo-treated patients were switched at week 12 to peficitinib 100 or 150 mg once daily. The primary endpoint was American College of Rheumatology (ACR)20 response at week 12/early termination (ET). Secondary endpoints (assessed throughout) included ACR20, ACR50 and ACR70 response, changes from baseline in disease activity scores (DAS)28 and ACR core parameters, adverse events (AEs) and changes in clinical or laboratory measurements.
Results
In total, 507 patients received treatment. ACR20 response rates at week 12/ET were significantly higher in the peficitinib 100 mg (57.7%) and 150 mg (74.5%) groups versus placebo (30.7%) (p<0.001). ACR50/70 response rates were also higher for both peficitinib doses versus placebo. Improvements in ACR response were maintained until week 52. Changes from baseline in DAS28-C-reactive protein/erythrocyte sedimentation rate and the ACR core set were significantly greater for both peficitinib doses versus placebo at week 12/ET (p<0.001). AE incidence was similar across treatment arms. Incidence of serious infection and herpes zoster-related disease was higher with peficitinib versus placebo, but with no clear dose-dependent increase.
Conclusions
In patients with RA and inadequate response to DMARDs, peficitinib 100 mg once daily or 150 mg once daily was efficacious in reducing RA symptoms and was well tolerated compared with placebo.
Trial registration number
Keywords: rheumatoid arthritis, DAS28, disease activity, dmards (synthetic), treatment
Key messages.
Peficitinib, a pan-Janus kinase inhibitor, has been approved in Japan in 2019 as a once-daily rheumatoid arthritis (RA) therapy at doses of both 100 mg/day and 150 mg/day, with no dose adjustment in patients with renal injury.
This study was a randomised, double-blind, phase III trial conducted in patients who had an inadequate response to prior disease-modifying anti-rheumatic drug (DMARD) treatment. Patients were randomised to 52 weeks’ treatment with peficitinib 100 or 150 mg/day (alone or in combination with DMARDs), placebo or open-label etanercept.
The primary efficacy variable of American College of Rheumatology (ACR)20 response rate at week 12/early termination was significantly higher in both peficitinib groups compared with placebo. Doses of 150 mg/day provided numerically higher responses compared with 100 mg/day.
Both peficitinib doses were well tolerated up to 52 weeks and no clear dose dependency was observed in the incidence of herpes zoster-related disease (including varicella), serious infections or malignancies.
Based on these findings, peficitinib has the potential to be a valuable addition to the RA treatment armamentarium, particularly for the treatment of patients with RA who are unresponsive to conventional therapies.
Introduction
Rheumatoid arthritis (RA) affects 0.3% to 1% of the population worldwide.1 Biological agents such as tumour necrosis factor (TNF) inhibitors, used in combination with methotrexate (MTX), have been found to be effective in patients who are unresponsive to conventional disease-modifying anti-rheumatic drugs (DMARDs).2
The Janus kinase (JAK) family of non-receptor protein tyrosine kinases is more recently considered a promising alternative target for RA treatment.3–5 Two oral JAK inhibitors, tofacitinib and baricitinib, have so far been approved for use in the USA, the European Union and Japan.6–11 Peficitinib (ASP015K) is an oral JAK inhibitor which inhibits the activity of all JAK family members (JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2)) with similar potency (IC50 0.7 to 5.0 nM).12 A phase IIb study in Japanese patients with RA showed a dose-dependent, statistically significant improvement after 12 weeks in American College of Rheumatology (ACR)20 response rate (primary endpoint) for once-daily doses of peficitinib 50 mg, 100 mg and 150 mg and an acceptable safety profile.13 In addition, a dose-dependent increase in mean haemoglobin levels was observed, suggesting that JAK2 was not inhibited in the study even at the highest dose used.13 Peficitinib also demonstrated similar efficacy in one of two studies in non-Japanese populations14; in the other non-Japanese study, most of the improvements in ACR20/50/70 response rates were not significant due to a relatively high response rate in the placebo group.15
This study was undertaken to assess the efficacy and safety of peficitinib (100 or 150 mg/day), alone or in combination with DMARDs, in patients who had an inadequate response to prior DMARDs.
Methods
Study design
This was a randomised, placebo-controlled, double-blind, parallel-group, phase III confirmatory study conducted between August 2014 and November 2017 at 142 sites in Japan, 11 sites in Korea and 12 sites in Taiwan (online supplementary information). Following screening, patients were randomised in a 1:1:1:2 ratio to peficitinib 100 mg/day, peficitinib 150 mg/day, placebo or etanercept. The peficitinib doses used in this study were chosen from the upper two doses in former RA studies of peficitinib based on previous efficacy and safety findings (online supplementary methods). In accordance with guidance for this study from a regulatory authority, etanercept was designated as an open-label reference drug, mainly for a safety comparison versus treatment groups (online supplementary methods). Peficitinib and placebo were taken orally once daily; etanercept (50 mg) was injected subcutaneously once weekly. Patients in the peficitinib 100 mg, peficitinib 150 mg and etanercept arms received their allocated treatment for 52 weeks. Patients in the placebo arm were switched at week 12 under blinded conditions, based on the randomisation to either peficitinib 100 mg or peficitinib 150 mg performed at baseline; this dose was maintained until the end of treatment (EOT) (online supplementary figure 1).
annrheumdis-2019-215163supp001.pdf (4.3MB, pdf)
Patients
Patients aged ≥20 years with RA (according to the 1987 American College of Rheumatology criteria (ACR) or the 2010 ACR/European League Against Rheumatism criteria), were enrolled. Eligibility criteria at screening included active RA, defined as ≥6/68 tender/painful joints and ≥6/66 swollen joints, C-reactive protein (CRP) >0.50 mg/dL at screening and an inadequate response to, or intolerance of, at least one DMARD administered for ≥90 days prior to screening (DMARD-IR). Exclusion criteria included an inadequate response to ≥3 biological DMARDs as determined by the investigator, a diagnosis of inflammatory arthritis other than RA and laboratory abnormalities. Further details of inclusion and exclusion criteria can be found in the online supplementary methods.
Outcomes
Efficacy assessments
The primary efficacy endpoint was the response rate according to ACR20 improvement criteria16 at week 12/early termination (ET). Key secondary endpoints assessed throughout included response rates according to ACR20/50/70 improvement criteria17, changes from baseline in 28-joint disease activity score (DAS) based on CRP (DAS28-CRP) and erythrocyte sedimentation rate (ESR) (DAS28-ESR), rates of remission defined as DAS28-CRP<2.6 and DAS28-ESR<2.6, rates of DAS28-CRP≤3.2, changes from baseline in CRP and ESR values, changes from baseline in tender joint count at 68 joints and swollen joint count at 66 joints and changes from baseline in Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI). Patient and physician-reported outcomes included Subject’s Global Assessment of disease activity (SGA), Subject’s Global Assessment of Pain (SGAP), and Physician’s Global Assessment of disease activity (PGA) using a 100 mm visual analogue scale (VAS) for each, plus Health Assessment Questionnaire – Disability Index (HAQ-DI) scores.
Safety
Treatment-emergent adverse events (TEAEs) were defined as any adverse event (AE), as reported by the investigator, that started or worsened in severity after the initial dose of study or reference drug through week 52 or follow-up period, including the incidence of venous thromboembolism (VTE) (online supplementary figure 1). Serious infections, malignancies and herpes zoster-related disease (including varicella) were assessed per 100 patient-years. Mean (SD) changes from baseline in haematological, biochemical and select laboratory parameters were recorded throughout.
Statistical analyses
Based on the calculation that 62 patients per arm would provide 90% power to detect a significant difference with a two-sided significance level of 0.05, and following guidance from the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use18 and the Japan Ministry of Health, Labour and Welfare19 on methodology for adequate assessment of AEs (online supplementary methods), the planned sample size was 100 patients for each of the peficitinib 100 mg, peficitinib 150 mg and placebo arms, and 200 patients in the etanercept group, taking into consideration incidence rates for serious infections from post-marketing studies for approved biological drugs and feasibility. As a result of discussions with a regulatory authority, etanercept was set as an open-label anti-TNF reference arm and was not included in statistical comparisons with placebo.
The primary analysis was conducted on the full analysis set (FAS), which consisted of all patients who were randomised and received at least one dose of study or reference treatment. For the ACR20/50/70 responses at week 12/ET, pairwise comparisons with placebo (using a logistic regression model with treatment group as the factor and prior biological DMARD-IR, concomitant DMARD use at baseline and study region as the covariates), multiplicity adjustment and sensitivity analyses were conducted as detailed in the online supplementary methods. The null hypotheses were tested at a two-sided significance level of 0.05. Safety analyses were conducted on the safety analysis set (SAF), which included all patients who received at least one dose of study treatment.
Results
Patient demographics, baseline characteristics and treatment compliance
Of 724 patients screened, 509 were randomised: 102, 104, 102 and 201 to the placebo, peficitinib 100 mg, peficitinib 150 mg and etanercept groups, respectively. Of the randomised patients, one patient in each of the placebo and etanercept arms did not receive treatment; the SAF/FAS therefore included a total of 507 patients (figure 1). The majority of patients were female (366/507, 72.2%) and the mean age was 55.3 years. Most patients were from Japan (415/507, 81.9%), 54 (10.7%) were from Korea and 38 (7.5%) were from Taiwan (table 1).
Figure 1.

Patient disposition. *The number of patients who were allocated at randomisation to either peficitinib 100 mg or peficitinib 150 mg starting from week 12. †Discontinuation up to week 12: discontinued at any time from date of randomisation through day 85. ‡Discontinuation for overall period: discontinued at any time from first dose of study drug through the last dose day for overall period.
Table 1.
Patient demographics and baseline characteristics (SAF)
| Placebo (N=101) | Peficitinib 100 mg (N=104) | Peficitinib 150 mg (N=102) | Peficitinib 100 mg +150 mg (N=206) | Etanercept (open-label arm) (N=200) | Total (N=507) | |
| Female, n (%) | 73 (72.3) | 77 (74.0) | 78 (76.5) | 155 (75.2) | 138 (69.0) | 366 (72.2) |
| Age in years, mean (SD) | 56.3 (11.7) | 54.1 (12.2) | 55.0 (12.8) | 54.5 (12.5) | 55.5 (11.6) | 55.3 (12.0) |
| <65 years, n (%) | 71 (70.3) | 83 (79.8) | 75 (73.5) | 158 (76.7) | 154 (77.0) | 383 (75.5) |
| Study region, n (%) | ||||||
| Japan | 83 (82.2) | 85 (81.7) | 83 (81.4) | 168 (81.6) | 164 (82.0) | 415 (81.9) |
| Korea | 10 (9.9) | 11 (10.6) | 11 (10.8) | 22 (10.7) | 22 (11.0) | 54 (10.7) |
| Taiwan | 8 (7.9) | 8 (7.7) | 8 (7.8) | 16 (7.8) | 14 (7.0) | 38 (7.5) |
| Body weight in kg, mean (SD) | 58.52 (13.00) | 59.90 (12.35) | 57.69 (11.35) | 58.81 (11.89) | 58.29 (12.34) | 58.54 (12.27) |
| RA duration in years, mean (SD) | 6.98 (6.57) | 8.75 (7.12) | 10.39 (8.23) | 9.56 (7.71) | 9.17 (8.00) | 8.89 (7.66) |
| Tender joint count (68-joint), mean (SD) | 16.2 (10.7) | 15.0 (9.4) | 15.4 (9.5) | 15.2 (9.4) | 14.9 (9.3) | 15.3 (9.6) |
| Swollen joint count (66-joint), mean (SD) | 12.9 (7.2) | 12.4 (6.3) | 12.8 (7.1) | 12.6 (6.7) | 11.9 (6.8) | 12.4 (6.8) |
| Physician's Global Assessment of disease activity (100 mm VAS), mean (SD) | 61.93 (19.35) | 60.21 (20.11) | 58.46 (19.35) | 59.34 (19.71) | 58.17 (19.87) | 59.39 (19.71) |
| Subject’s Global Assessment of disease activity (100 mm VAS), mean (SD) | 58.99 (25.70) | 57.54 (24.78) | 59.52 (25.73) | 58.52 (25.21) | 57.52 (26.92) | 58.22 (25.95) |
| Subject’s Global Assessment of Pain (100 mm VAS), mean (SD) | 57.56 (25.07) | 57.31 (26.71) | 58.02 (25.66) | 57.67 (26.13) | 55.79 (26.54) | 56.90 (26.05) |
| DAS28-CRP, mean (SD) | 5.43 (1.03) | 5.29 (0.98) | 5.41 (0.96) | 5.35 (0.97) | 5.27 (0.94) | 5.33 (0.97) |
| DAS28-ESR, mean (SD) | 6.03 (1.13) | 5.94 (1.07) | 6.01 (1.03) | 5.98 (1.05) | 5.87 (1.09) | 5.94 (1.08) |
| HAQ-DI score, mean (SD) | 1.00 (0.66) | 0.92 (0.69) | 1.03 (0.67) | 0.97 (0.68) | 1.03 (0.75) | 1.00 (0.70) |
| CRP (mg/dL), mean (SD) | 2.258 (2.224) | 2.296 (2.566) | 2.561 (2.597) | 2.428 (2.579) | 2.055 (2.144) | 2.247 (2.346) |
| ESR (mm/hr), mean (SD) | 47.9 (27.6) | 49.6 (27.4) | 50.6 (29.7) | 50.1 (28.5) | 47.4 (29.8) | 48.6 (28.8) |
| SDAI score, mean (SD) | 35.48 (12.95) | 33.57 (12.71) | 34.15 (12.78) | 33.86 (12.72) | 32.91 (12.09) | 33.81 (12.53) |
| CDAI score, mean (SD) | 33.21 (12.14) | 31.27 (11.40) | 31.59 (11.97) | 31.43 (11.66) | 30.86 (11.40) | 31.56 (11.67) |
| Prior non-biological DMARD use only, n (%) | 90 (89.1) | 90 (86.5) | 89 (87.3) | 179 (86.9) | 162 (81.0) | 431 (85.0) |
| Prior biological DMARD use only, n (%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Prior non-biological and biological DMARD use, n (%) | 11 (10.9) | 14 (13.5) | 13 (12.7) | 27 (13.1) | 38 (19.0) | 76 (15.0) |
| Prior MTX use, n (%) | 89 (88.1) | 94 (90.4) | 92 (90.2) | 186 (90.3) | 179 (89.5) | 454 (89.5) |
| Number of prior DMARDs (including biologicals), n (%) | ||||||
| 1 | 14 (13.9) | 19 (18.3) | 16 (15.7) | 35 (17.0) | 24 (12.0) | 73 (14.4) |
| 2 | 56 (55.4) | 57 (54.8) | 57 (55.9) | 114 (55.3) | 109 (54.5) | 279 (55.0) |
| ≥3 | 31 (30.7) | 28 (26.9) | 29 (28.4) | 57 (27.7) | 67 (33.5) | 155 (30.6) |
| Concomitant DMARD at baseline, n (%) | 87 (86.1) | 91 (87.5) | 89 (87.3) | 180 (87.4) | 176 (88.0) | 443 (87.4) |
| MTX | 57 (56.4) | 63 (60.6) | 62 (60.8) | 125 (60.7) | 117 (58.5) | 299 (59.0) |
| DMARD except for MTX only | 30 (29.7) | 28 (26.9) | 27 (26.5) | 55 (26.7) | 59 (29.5) | 144 (28.4) |
| None | 14 (13.9) | 13 (12.5) | 13 (12.7) | 26 (12.6) | 24 (12.0) | 64 (12.6) |
| MTX dose at baseline, mg/week | 11.35 (3.82) | 11.28 (2.98) | 10.77 (3.16) | 11.03 (3.07) | 11.12 (3.78) | 11.13 (3.50) |
| Prior biological DMARD-IR | 5 (5.0) | 9 (8.7) | 7 (6.9) | 16 (7.8) | 15 (7.5) | 36 (7.1) |
CDAI, Clinical Disease Activity Index; CRP, C-reactive protein; DAS, disease activity score; DMARD, disease-modifying antirheumatic drug; ESR, erythrocyte sedimentation rate; HAQ-DI, Health Assessment Questionnaire - Disability Index; IR, inadequate response; MTX, methotrexate; RA, rheumatoid arthritis; SAF, safety analysis set; SDAI, Simplified Disease Activity Index; VAS, visual analogue scale.
For the overall period, the proportion of patients who discontinued the study ranged from 16.9% to 29.8% across the treatment groups: the primary reasons for discontinuation across treatment arms were lack of efficacy (32/509, 6.3%) and AEs (29/509, 5.7%). Reasons for discontinuation in each arm are described in figure 1.
Baseline disease activity and RA history were mostly balanced between the treatment arms. At baseline, a total of 443/507 (87.4%) patients received concomitant DMARDs, of whom 299 (59.0%) received MTX and 144 (28.4%) received DMARDs other than MTX only. An inadequate response to previous biological DMARDs was reported in 36 (7.1%) patients. Mean treatment compliance for the overall study period was 96.44% to 97.70% for all treatment groups.
Efficacy
ACR20/50/70 response rates
The primary efficacy variable, ACR20 response rate at Week 12/ET (last observation carried forward (LOCF)), was 57.7%, 74.5%, 83.5% and 30.7%, in the peficitinib 100 mg, peficitinib 150 mg, etanercept and placebo groups, respectively (figure 2A). Significant differences versus placebo of 27.0% (OR: 3.13, 95% CI 1.76 to 5.58) with peficitinib 100 mg and 43.8% (OR: 6.59, 95% CI 3.56 to 12.20) with peficitinib 150 mg were observed (p<0.001 for both comparisons) (figure 2A). The primary analysis of the ACR20 response rate was also demonstrated to be robust using sensitivity analyses (online supplementary table 1). Subgroup analyses indicated that both peficitinib doses produced numerically higher ACR20 response rates than placebo regardless of number of prior biological DMARDs, prior biological DMARD-IR, concomitant DMARD use or MTX dose at baseline; however, these subgroup analyses were not powered for statistical comparisons (online supplementary table 1).
Figure 2.


(A) ACR20, ACR50 and ACR70 response rates at week 12/ET (FAS). (B) Response rates for ACR20 from baseline until week 52 and EOT (FAS). (C) Response rates for ACR50 from baseline until week 52 and EOT (FAS). (D) Response rates for ACR70 from baseline until week 52 and EOT (FAS). For all timepoints except for week 12/ET and EOT, observed data are plotted. For week 12/ET and EOT, in the case of early termination, ACR components were analysed using the LOCF method first, and then ACR20/50/70 responses were calculated. A pairwise comparison with the placebo group was performed using a logistic regression model with treatment group as the factor and inadequate response to prior biological DMARD use, concomitant DMARD use during the study period and region as covariates. P values were calculated using Wald’s Chi-square test with a closed testing procedure for multiplicity adjustment for ACR20 and no multiplicity adjustment for ACR50/70. 95% CI were based on a normal approximation to the binomial distribution (continuity corrected). Etanercept was an open-label reference arm and was not included in statistical comparisons with placebo. *The odds ratio for the treatment difference in ACR70 between peficitinib 100 mg and placebo was not estimable with the planned logistic regression model (p=0.009 with an ad-hoc analysis using a logistic regression model with treatment group as the only explanatory variable). †Includes LOCF. ACR, American College of Rheumatology; DMARD, disease-modifying anti-rheumatic drugs; EOT, end of treatment; ET, early termination; FAS, full analysis set; LOCF, last observation carried forward; N/E, not estimable.
ACR50 and ACR70 response rates at week 12/ET were also significantly higher for peficitinib 100 mg and 150 mg compared with placebo (figure 2A; online supplementary figure 3). The increase in ACR20/50/70 response rates versus placebo was maintained throughout the study period in the peficitinib 100 mg group, the peficitinib 150 mg group and the etanercept group (figure 2B–D; online supplementary table 3). For the patients who switched from placebo to peficitinib 100/150 mg at week 12, the response rates were initially lower compared with those in the other treatment arms, but were improved at week 16 and then maintained or increased through week 52 (figure 2B–D).
Key secondary efficacy endpoints
The proportions of patients achieving DAS28-CRP<2.6, DAS28-ESR<2.6 and DAS28-CRP≤3.2 scores at week 12/ET were significantly increased in the peficitinib 100 mg and 150 mg groups compared with placebo, except for the DAS28-ESR<2.6 scores in the peficitinib 100 mg group due to difficulty in estimating the odds ratio. The proportion of patients achieving these criteria was higher at week 52/ET than at week 12/ET (online supplementary figure 2).
The changes from baseline in DAS28-CRP and the ACR core set at week 12/ET (LOCF) were significantly greater in the peficitinib 100 mg and 150 mg groups compared with placebo (p<0.001). In addition, the change from baseline was greater for all outcomes in the peficitinib 150 mg group than in the peficitinib 100 mg group (figure 3).
Figure 3.


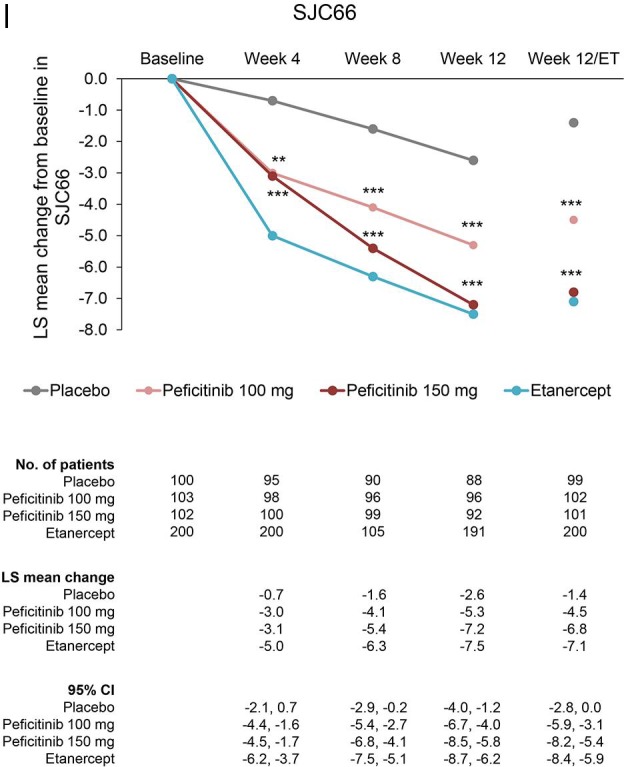
Changes from baseline to week 12/ET in DAS28-CRP scores and ACR core parameters (CRP, ESR, HAQ-DI, SGA, SGAP, PGA, TJC68 and SJC66) (FAS). For all timepoints except for week 12/ET, observed data are plotted. For week 12/ET, in the case of early termination, the LOCF method was used. Data are plotted as least-squares means with 95% CI based on ANCOVA model: change from baseline = treatment + baseline value + prior biological DMARD-IR + concomitant DMARD use + study region (Japan, Korea or Taiwan). Statistical comparisons with placebo were performed using analysis of covariance with no multiplicity adjustment. Etanercept was an open-label reference arm and was not included in statistical comparisons with placebo. *p<0.05; **p<0.01; ***p<0.001. ACR, American College of Rheumatology; ANCOVA, analysis of covariance; CRP, C-reactive protein; DAS28-CRP, Disease Activity Scores in 28 joints using CRP; DMARD, disease-modifying anti-rheumatic drugs; ESR, erythrocyte sedimentation rate; ET, early termination; FAS, full analysis set; HAQ-DI, Health Assessment Questionnaire – Disability Index; IR, inadequate response; LOCF, last observation carried forward; PGA, Physician’s Global Assessment of disease activity; SGA, Subject’s Global Assessment of disease activity; SGAP, Subject’s Global Assessment of Pain; SJC66, swollen joint count at 66 joints; TJC68, tender joint count at 68 joints.
The proportions of patients with CDAI score ≤2.8, CDAI score ≤10 (low disease activity), SDAI score ≤3.3 and SDAI score ≤11 (low disease activity) were higher with peficitinib 100 mg and 150 mg compared with placebo, although p values could not be estimated in all instances (online supplementary file 1). Changes from baseline in CDAI and SDAI scores were significantly higher with both peficitinib doses than with placebo from week four through to week 12/ET (online supplementary file 1).
Safety
Treatment-emergent adverse events
The incidence of investigator-reported TEAEs from week 0 to week 12 was similar across treatment arms at 53.5% to 59.5% (SAF). For the overall study period, TEAEs were reported in 87.3% to 89.0% patients across all treatment arms, and no major differences in incidence were observed compared with the week 0 to 12 period (table 2A). The majority of investigator-reported TEAEs were mild or moderate in severity (National Cancer Institute Common Terminology Criteria for Adverse Events grades 1 to 2). No deaths were reported during the study. After the end of study, one patient in the etanercept group died from thyroid cancer reported during the study; this event was considered possibly related to etanercept. From baseline to week 12/ET and to the end of the study period, investigator-reported TEAEs leading to permanent discontinuation of study drug occurred in ≥3 patients in each group. An overview of TEAEs from week 12 to week 52 is presented in the online supplementary table 4.
Table 2A.
TEAEs for weeks 0 to 12 and overall period (SAF)
| Weeks 0 to 12 | Overall period | ||||||||
| Placebo (N=101) | Peficitinib 100 mg (N=104) | Peficitinib 150 mg (N=102) | Peficitinib 100 mg + 150 mg (N=206) | Etanercept (open-label arm) (N=200) | Peficitinib 100 mg (N=104) | Peficitinib 150 mg (N=102) | Peficitinib 100 mg + 150 mg (N=206) | Etanercept (open-label arm) (N=200) | |
| All TEAEs | 54 (53.5) | 59 (56.7) | 55 (53.9) | 114 (55.3) | 119 (59.5) | 92 (88.5) | 89 (87.3) | 181 (87.9) | 178 (89.0) |
| Drug-related TEAEs* | 29 (28.7) | 33 (31.7) | 38 (37.3) | 71 (34.5) | 75 (37.5) | 63 (60.6) | 63 (61.8) | 126 (61.2) | 122 (61.0) |
| SAEs | 4 (4.0) | 3 (2.9) | 2 (2.0) | 5 (2.4) | 4 (2.0) | 7 (6.7) | 8 (7.8) | 15 (7.3) | 18 (9.0) |
| Drug-related SAEs* | 3 (3.0) | 2 (1.9) | 1 (1.0) | 3 (1.5) | 4 (2.0) | 3 (2.9) | 3 (2.9) | 6 (2.9) | 9 (4.5) |
| ≥Grade 3 TEAE† | 8 (7.9) | 6 (5.8) | 3 (2.9) | 9 (4.4) | 6 (3.0) | 14 (13.5) | 19 (18.6) | 33 (16.0) | 29 (14.5) |
| TEAEs leading to permanent discontinuation of study drug or reference drug | |||||||||
| All | 4 (4.0) | 6 (5.8) | 3 (2.9) | 9 (4.4) | 5 (2.5) | 13 (12.5) | 6 (5.9) | 19 (9.2) | 13 (6.5) |
| Drug-related* | 1 (1.0) | 4 (3.8) | 2 (2.0) | 6 (2.9) | 5 (2.5) | 7 (6.7) | 4 (3.9) | 11 (5.3) | 11 (5.5) |
| SAEs | 2 (2.0) | 2 (1.9) | 2 (2.0) | 4 (1.9) | 2 (1.0) | 6 (5.8) | 2 (2.0) | 8 (3.9) | 5 (2.5) |
| Drug-related SAEs* | 1 (1.0) | 1 (1.0) | 1 (1.0) | 2 (1.0) | 2 (1.0) | 3 (2.9) | 1 (1.0) | 4 (1.9) | 4 (2.0) |
Treatment-emergent adverse events are defined as any adverse event that started or worsened in severity after initial dose of study drug or reference drug through the follow-up period. All values are n (%).
*Possible or probable, as assessed by the investigator or records where relationship is missing.
†Based on National Cancer Institute Common Terminology Criteria for Adverse Events grading: grade 3 = severe or medically significant, grade 4 = life threatening, grade 5 = death related to AE.
AE, adverse event; SAE, serious adverse event; SAF, safety analysis set; TEAE, treatment-emergent adverse event.
Table 2B.
Incidence of serious infections, herpes zoster-related disease and malignancy per 100 patient-years, from week 0 to week 52 (SAF)
| Placebo (N=101) | Peficitinib 100 mg (N=104) | Peficitinib 150 mg (N=102) | Peficitinib 100 mg + 150 mg (N=206) |
Peficitinib total* (N=296) | Etanercept (open-label arm) (N=200) | |
| Serious infections | ||||||
| Patient-years | 22.6 | 88.2 | 92.1 | 180.3 | 245.7 | 195.5 |
| Number of patients who had at least one incidence | 0 | 1 | 2 | 3 | 5 | 4 |
| Incidence rate/100 patient-years (95% CI) | 0.0 | 1.1 (0.2 to 8.1) | 2.2 (0.5 to 8.7) | 1.7 (0.5 to 5.2) | 2.0 (0.8 to 4.9) | 2.0 (0.8 to 5.5) |
| Herpes zoster-related disease (including varicella) | ||||||
| Patient-years | 22.6 | 86.8 | 90.9 | 177.7 | 241.2 | 194.0 |
| Number of patients who had at least one incidence | 0 | 5 | 4 | 9 | 14 | 5 |
| Incidence rate/100 patient-years (95% CI) | 0.0 | 5.8 (2.4 to 13.8) | 4.4 (1.7 to 11.7) | 5.1 (2.6 to 9.7) | 5.8 (3.4 to 9.8) | 2.6 (1.1 to 6.2) |
| Malignancies | ||||||
| Patient-years | 22.6 | 88.1 | 92.8 | 180.9 | 246.4 | 197.3 |
| Number of patients who had at least one incidence | 0 | 2 | 0 | 2 | 3 | 1 |
| Incidence rate/100 patient-years (95% CI) | 0.0 | 2.3 (0.6 to 9.1) | 0.0 (- to -) | 1.1 (0.3 to 4.4) | 1.2 (0.4 to 3.8) | 0.5 (0.1 to 3.6) |
Patient-years was calculated from initial dose up to first incidence of the event for patients who had at least one event, and from initial dose through follow-up for patients who had no events; incidence rate is calculated as (100 × number of patients who had at least one incidence/total patient-years).
*Included adverse events occurring during treatment with peficitinib in patients who were initially treated with placebo and switched to peficitinib at week 12.
SAF, safety analysis set.
The incidence per 100 patient-years of serious infection, herpes zoster-related disease (including varicella) and malignancies was higher in the groups treated with peficitinib compared with placebo, although there was no clear dose-dependent increase (table 2B). No occurrence of VTE was reported during the study.
Clinical laboratory evaluations
At week 12/ET, reductions in platelet counts and dose-dependent increases in haemoglobin level were observed in the peficitinib 100 mg and 150 mg groups (table 3). Reductions in absolute neutrophil counts were also observed; this parameter also tended to decrease in the placebo group. Dose-dependent increases in creatine kinase, creatinine, low-density lipoprotein cholesterol and high-density lipoprotein cholesterol were observed in the peficitinib 100 mg and 150 mg groups until week 12/ET. There were no major changes from week 12/ET to week 52/ET in any of these parameters.
Table 3.
Change from baseline in haematological and biochemical parameters to week 12/ET and week 52/ET (SAF)
| Baseline result | Change to week 12/ET | Change to Week 52/ET | |||||||||
| Placebo (N=100) | Peficitinib 100 mg (N=102) | Peficitinib 150 mg (N=101) | Etanercept (open-label arm) (N=200) | Placebo (N=100) | Peficitinib 100 mg (N=102) | Peficitinib 150 mg (N=101) | Etanercept (open-label arm) (N=200) | Peficitinib 100 mg (N=102) | Peficitinib 150 mg (N=101) | Etanercept (open-label arm) (N=200) | |
| Absolute neutrophil count (106/L) | 5610.9 (2282.7) | 5326.0 (2024.4) | 5955.9 (2410.4) | 5477.5 (1828.7) | −465.0 (1812.9) | −102.9 (1741.9) | −937.6 (1836.0) | −1359.5 (1690.6) | −289.2 (2142.6) | −1160.4 (1832.9) | −1459.5 (1704.1) |
| Haemoglobin (g/L) | 121.3 (14.6) | 121.5 (13.3) | 122.3 (14.0) | 123.7 (14.6) | −0.4 (8.5) | 1.4 (7.5) | 4.1 (8.4) | 4.5 (8.0) | 2.3 (10.8) | 5.5 (11.6) | 5.2 (10.1) |
| Platelets (109/L) | 303.1 (78.4) | 318.9 (89.2) | 316.3 (83.6) | 304.1 (97.1) | 1.8 (46.8) | −19.3 (49.0) | −47.2 (54.3) | −55.4 (53.5) | −24.3 (65.1) | −45.9 (62.8) | −56.3 (67.4) |
| LDL cholesterol (mmol/L) | 2.770 (0.870) | 2.737 (0.790) | 2.851 (0.782) | 2.704 (0.767) | 0.020 (0.487) | 0.130 (0.422) | 0.281 (0.713) | 0.124 (0.503) | 0.209 (0.547) | 0.385 (0.723) | 0.105 (0.561) |
| HDL cholesterol (mmol/L) | 1.510 (0.395) | 1.595 (0.417) | 1.562 (0.424) | 1.572 (0.470) | 0.023 (0.240) | 0.261 (0.297) | 0.363 (0.313) | 0.110 (0.246) | 0.251 (0.265) | 0.439 (0.405) | 0.061 (0.303) |
| Serum creatinine (μmol/L) | 55.22 (14.80) | 54.62 (14.96) | 52.83 (15.84) | 53.52 (12.52) | −0.25 (5.32) | 3.90 (6.26) | 5.40 (6.60) | 3.54 (7.18) | 4.47 (7.57) | 8.39 (6.54) | 4.24 (7.46) |
| Creatine kinase (U/L)* | 63.1 (54.6) | 67.8 (45.8) | 70.1 (45.8) | 80.0 (155.8) | 0.6 (22.3) | 55.5 (118.6) | 67.7 (61.1) | 79.5 (1117.7) | 91.9 (213.9) | 102.2 (105.4) | 86.4 (1117.8) |
| ALT (U/L) | 18.3 (13.4) | 17.9 (10.0) | 21.1 (17.5) | 19.0 (13.9) | 2.0 (12.4) | 2.3 (8.9) | 6.9 (17.4) | 4.6 (15.9) | 3.8 (12.2) | 5.9 (22.9) | 3.2 (15.0) |
| AST (U/L) | 23.1 (12.8) | 22.2 (9.5) | 25.0 (16.9) | 23.2 (10.4) | 0.8 (7.9) | 2.7 (7.1) | 4.9 (14.6) | 4.3 (18.6) | 4.3 (10.3) | 5.1 (16.4) | 3.8 (19.0) |
Data are presented as mean (SD).
*Median changes in creatine kinase from baseline to Wweek 12/ET were −1.0, 36.5, 52.0 and 8.5 U/L in the placebo, peficitinib 100 mg, peficitinib 150 mg and etanercept groups, respectively; changes to week 52 ET were 38.5, 75.0 and 10.0 U/L in the peficitinib 100 mg, peficitinib 150 mg and etanercept groups, respectively.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ET, early termination; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SAF, safety analysis set.
Discussion
This was the first phase III, randomised, double-blind, placebo-controlled trial of peficitinib, a novel oral JAK inhibitor, in patients with RA who had an inadequate response to DMARDs. Results showed statistically significant increases by week 12 in ACR20 response rates compared with placebo for peficitinib doses of 100 mg/day and 150 mg/day. The results for the secondary efficacy variables, including ACR50, ACR70, DAS28-CRP and patient-reported outcomes such as HAQ-DI, supported the results for the primary efficacy variable. ACR20/50/70 showed rapid responses through the first 4 to 8 weeks of treatment, which were either maintained or improved during long-term treatment up to week 52 (EOT). Even in the placebo group, improvements were observed after switching to peficitinib 100 mg or 150 mg and then maintained during the study. Treatment with etanercept appeared to provide numerically greater response rates than either peficitinib 100 mg or 150 mg, across all outcomes measured.
Of note, ACR20 response rates at week 12 following peficitinib 100 mg or 150 mg treatment were similar to those observed in previous phase III studies of tofacitinib or baricitinib.20 21 Across five phase III clinical trials of tofacitinib 5 mg twice daily in patients with active RA and an inadequate response to prior DMARDs, MTX or TNF inhibitors, ACR20 response rates at week 12 ranged from 41.7% to 60.7%.20 Similarly, the RA-BUILD study investigated a once-daily dose of baricitinib 4 mg in patients with active RA and an inadequate response or intolerance to prior conventional synthetic DMARDs; the ACR20 response rate at week 12 was 62%.21 In the present study, the ACR20 response rates were 57.7% and 74.5% in the peficitinib 100 mg and 150 mg groups, respectively. However, the inclusion criteria for specific DMARD-IR varied across previous studies evaluating the efficacy of other JAK inhibitors, including tofacitinib and baricitinib20 21; outcomes may therefore not be directly comparable with the current study.
Safety analysis indicated that peficitinib was well tolerated for up to 52 weeks. The incidence of TEAEs was similar for both peficitinib arms compared with placebo, and no new safety signals were observed in clinical laboratory results. Of note, a mean decrease in haemoglobin levels, which might be attributable to JAK2 inhibition, was not observed even with the highest dose of 150 mg/day. These safety results are consistent with those from short-term (12 week) treatment with 25 to 150 mg/day peficitinib monotherapy.13
With regard to TEAEs of special interests, no apparent dose dependency was observed in the incidence of herpes zoster-related disease (including varicella), serious infections or malignancies. Previous safety data for the use of tofacitinib suggest that Asian populations may be specifically at risk of developing herpes zoster infection when receiving JAK inhibitors.22 In line with this, the incidence of herpes zoster-related disease was approximately doubled with peficitinib compared with etanercept in our study, although rates remained within the range previously observed with tofacitinib and baricitinib in Japanese/Korean/Taiwanese populations.22–25 As historical data of vaccination for herpes zoster or varicella in each patient were not captured in this study, the relationship between the observed incidence and vaccination rate is unknown and further investigation would be required to elucidate this. Lastly, although previous studies have shown an association between JAK inhibitors and VTE,26 no incidence of VTE was observed in our study.
Greater clinical improvements were observed with peficitinib 150 mg versus peficitinib 100 mg. In this study, no marked increase in AEs was observed with peficitinib 150 mg compared with 100 mg and both doses demonstrated efficacy and tolerability, thus broadening the potential treatment options for those with refractory disease. However, further evaluation is needed to establish whether peficitinib >150 mg/day may provide even greater clinical benefits, without additional safety concerns.
The strengths of this study include the 52-week treatment period, which allowed for assessment of long-term efficacy and safety. The study population comprised patients who previously had an inadequate response or intolerance to either conventional synthetic or biological DMARDs, thus representing a patient population with more restricted treatment options. One limitation of this study is that placebo treatment was of shorter duration (12 weeks) compared with the peficitinib and etanercept arms, for ethical reasons; comparisons between placebo and peficitinib arms are therefore limited. In addition, the study design did not allow for a statistical comparison of treatment differences in the open-label etanercept arm relative to the other arms. No radiographical assessments were conducted for this study, and it is therefore uncertain whether peficitinib inhibits radiographical progression in this population. Moreover, the patient population was drawn from Japan, Korea and Taiwan and although the results reflect populations of more than one country, they lack global diversity. The baseline HAQ-DI score of 1.0 for the study population was low, especially considering the mean DAS-CRP/ESR of 5.3 to 6.0 and mean RA duration of almost 9 years, but these parameters are in line with findings from previous studies of tofacitinib27 28 and baricitinib25 in Japanese populations.
As previously described, kinase assays showed peficitinib inhibits the activity of all JAK family members with similar potency (IC50 0.7 to 5.0 nM).12 In contrast, currently marketed JAK inhibitors have greater selectivity for certain JAK isoforms: tofacitinib inhibits JAK1, JAK2, JAK3 and to a lesser extent TYK2,12 29 while baricitinib has greater potency against JAK1 and JAK2 than against JAK3 and TYK2.30 In an animal study, TYK2-deficient mice were resistant to collagen antibody-induced arthritis, suggesting a crucial role of TYK2 in the development of inflammatory arthritis.31 We therefore consider that TYK2 inhibition may contribute to the clinical efficacy of peficitinib in RA patients. However, the clinical significance of the mouse model findings is unclear and additional investigation is necessary to elucidate the involvement of TYK2 in the pathogenesis of RA.
In conclusion, peficitinib at doses of 100 mg/day and 150 mg/day was superior to placebo in reducing RA symptoms, and these improvements were maintained throughout the study period. Peficitinib was generally well tolerated in long-term treatment up to 52 weeks. Peficitinib may therefore be a viable treatment option for patients who fail to respond to previous lines of therapy with other biological and non-biological DMARDs.
Footnotes
Handling editor: Josef S Smolen
YT and TT contributed equally.
Contributors: All authors met the following criteria for authorship: substantial contributions to the acquisition, analysis and interpretation of data for the work; contribution to drafting the work and revising it critically; giving the final approval of the version submitted and agreeing to be accountable for all aspects of the work.
Funding: This study was supported by Astellas Pharma, Inc.
Competing interests: YT reports speaking fees and/or honoraria from Daiichi Sankyo, Astellas, Eli Lilly, Chugai, Sanofi, AbbVie, Pfizer, YL Biologics, Bristol-Myers Squibb, GlaxoSmithKline, UCB, Mitsubishi-Tanabe, Novartis, Eisai, Takeda, Janssen and Asahi Kasei, and research grants from Mitsubishi Tanabe, Bristol-Myers Squibb, Eisai, Chugai, Takeda, AbbVie, Astellas, Daiichi Sankyo, Ono, MSD and Taisho Toyama. TT has received grants from Astellas Pharma, Inc., Chugai Pharmaceutical Co. Ltd, Daiichi Sankyo Co. Ltd, Takeda Pharmaceutical Co. Ltd, AbbVie GK, Asahi Kasei Pharma Corp., Mitsubishi Tanabe Pharma Co., Pfizer Japan, Inc., Eisai Co. Ltd, AYUMI Pharmaceutical Corp., Nippon Kayaku Co. Ltd, and Novartis Pharma K.K.; speaking fees from AbbVie GK, Bristol-Myers Squibb K.K., Chugai Pharmaceutical Co. Ltd, Mitsubishi Tanabe Pharma Co., Pfizer Japan, Inc., Astellas Pharma, Inc., Daiichi Sankyo Co. Ltd, Eisai Co. Ltd, Sanofi K.K., Teijin Pharma Ltd, Takeda Pharmaceutical Co. Ltd, and Novartis Pharma K.K.; consultancy fees from Astra Zeneca K.K., Eli Lilly Japan K.K., Novartis Pharma K.K., Mitsubishi Tanabe Pharma Co., AbbVie GK, Nippon Kayaku Co. Ltd, Janssen Pharmaceutical K.K., Astellas Pharma, Inc., Taiho Pharmaceutical Co. Ltd, Chugai Pharmaceutical Co. Ltd, Taisho Toyama Pharmaceutical Co. Ltd, GlaxoSmithKline K.K., and UCB Japan Co. Ltd. ST reports personal fees from Amgen, Inc., Asahi Kasei Pharma Corporation, Amgen Astellas BioPharma K.K., Ono Pharmaceutical Co. Ltd, KYOCERA Medical Corporation, Daiichi Sankyo Co. Ltd, Teijin Pharma Ltd, Eli Lilly Japan K.K., and Pfizer Japan, Inc.; endowments from Astellas Pharma, Inc., AYUMI Pharmaceutical Corporation, Pfizer Japan, Inc., Bristol-Myers Squibb, Daiichi Sankyo Co. Ltd, and Chugai Pharmaceutical Co. Ltd; and grants from Japan Agency for Medical Research and Development (AMED), Japan Society for the Promotion of Science (JSPS)/Grant-in-Aid for Scientific Research (A), and Japan Society for the Promotion of Science (JSPS)/Grant-in-Aid for Exploratory Research outside the submitted work. AK reports grants from AbbVie, Eisai, Mitsubishi-Tanabe, Pfizer Japan, Takeda Pharmaceutical, Astellas Pharma, MSD, Ono Pharmaceutical, Teijin Pharma, Kyowa Hakko-Kirin, Sumitomo-Dainippon Pharma, Kissei Pharmaceutical, Boehringer Ingelheim, AstraZeneca, Otsuka Pharmaceutical, Chugai Pharmaceutical, Santen Pharmaceutical and Daiichi Sankyo; participation in speakers’ bureaux for AbbVie, Eisai, Takeda Pharmaceutical, Ono Pharmaceutical, Astellas Pharma, Mitsubishi-Tanabe, Pfizer Japan, Chugai Pharmaceutical, MSD, and Bristol-Myers Squibb K.K.; and consultant fees from Astellas Pharma and Janssen Pharmaceutical K.K. YWS reports a grant from Astellas Pharma, Inc. YHC reports grants for research and clinical trials from Taiwan Ministry of Science and Technology, Taiwan Department of Health, Taichung Veterans General Hospital, National Yang-Ming University, GlaxoSmithKline, Pfizer, Bristol-Myers Squibb, Novartis, AbbVie, Johnson & Johnson, Roche, Sanofi, Chugai Pharma Taiwan Ltd, Boehringer Ingelheim, UCB, MSD, AstraZeneca and Astellas; and honoraria and consultant fees from Pfizer, Novartis, AbbVie, Johnson & Johnson, Bristol-Myers Squibb, Roche, Lilly, GlaxoSmithKline, AstraZeneca, Sanofi, MSD, Chugai Pharma Taiwan Ltd, Astellas, Inova Diagnostics, UCB, Agnitio Science Technology, United Biopharma and Thermo Fisher. MR, HI, SU, YK, RA, TS and EY are employees of Astellas Pharma, Inc.
Patient consent for publication: Not required.
Ethics approval: This study was conducted in accordance with Good Clinical Practice, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines, and local laws and regulations. The protocol and amendments were approved by an Institutional Review Board at each study site, and safety data were reviewed by the independent Data and Safety Monitoring Board. Each patient provided written informed consent prior to treatment initiation.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Data are available upon reasonable request.
References
- 1. World Health Organization Chronic rheumatic conditions, 2019. Available: https://www.who.int/chp/topics/rheumatic/en/
- 2. O'Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med Overseas Ed 2004;350:2591–602. 10.1056/NEJMra040226 [DOI] [PubMed] [Google Scholar]
- 3. O'Shea JJ, Kontzias A, Yamaoka K, et al. . Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis 2013;72 Suppl 2:ii111–5. 10.1136/annrheumdis-2012-202576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tanaka Y. Recent progress and perspective in JAK inhibitors for rheumatoid arthritis: from bench to bedside. J Biochem 2015;158:173–9. 10.1093/jb/mvv069 [DOI] [PubMed] [Google Scholar]
- 5. Tanaka Y, Maeshima K, Maeshima Y, et al. . In vitro and in vivo analysis of a JAK inhibitor in rheumatoid arthritis. Ann Rheum Dis 2012;71 Suppl 2:i70–4. 10.1136/annrheumdis-2011-200595 [DOI] [PubMed] [Google Scholar]
- 6. Japan Ministry of Health Labour and Welfare Xeljanz tablets 5 mg report, 2013. Available: https://www.pmda.go.jp/files/000153609.pdf
- 7. Japan Ministry of Health Labour and Welfare Olumiant tablets 2 mg, 4mg report, 2017. Available: http://www.pmda.go.jp/drugs/2017/P20170724002/530471000_22900AMX00582_A100_1.pdf
- 8. Committee for Medicinal Products for Human Use (CHMP) Olumiant - Assessment report, 2016. Available: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004085/WC500223725.pdf
- 9. Committee for Medicinal Products for Human Use (CHMP) Xeljanz - Assessment report, 2017. Available: https://www.ema.europa.eu/documents/assessment-report/xeljanz-epar-public-assessment-report_en.pdf
- 10. US Food and Drug Administration NDA approval for Xeljanz (tofacitinib) 5 mg tablets, 2012. Available: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/203214Orig1s000ltr.pdf
- 11. US Food and Drug Administration NDA approval for Olumiant (baricitinib) 2 mg tablets, 2018. Available: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2018/207924Orig1s000ltr.pdf
- 12. Ito M, Yamazaki S, Yamagami K, et al. . A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J Pharmacol Sci 2017;133:25–33. 10.1016/j.jphs.2016.12.001 [DOI] [PubMed] [Google Scholar]
- 13. Takeuchi T, Tanaka Y, Iwasaki M, et al. . Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann Rheum Dis 2016;75:1057–64. 10.1136/annrheumdis-2015-208279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Genovese MC, Greenwald M, Codding C, et al. . Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol 2017;69:932–42. 10.1002/art.40054 [DOI] [PubMed] [Google Scholar]
- 15. Kivitz AJ, Gutierrez‐Ureña SR, Poiley J, et al. . Peficitinib, a JAK inhibitor, in the treatment of moderate-to-severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol 2017;69:709–19. 10.1002/art.39955 [DOI] [PubMed] [Google Scholar]
- 16. Felson DT, Anderson JJ, Boers M, et al. . American College of Rheumatology Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. 10.1002/art.1780380602 [DOI] [PubMed] [Google Scholar]
- 17. Felson D, American College of Rheumatology Committee to Reevaluate Improvement Criteria . A proposed revision to the ACR20: the hybrid measure of American College of Rheumatology response. Arthritis Rheum 2007;57:193–202. 10.1002/art.22552 [DOI] [PubMed] [Google Scholar]
- 18. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use The extent of population exposure to assess clinical safety for drugs intended for long-term treatment of non-life threatening conditions, 1994. Available: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E1/Step4/E1_Guideline.pdf
- 19. Japan Ministry of Health Labour and Welfare Guidelines on methodology for clinical assessment of antirheumatic drugs, 2006. Available: https://www.pmda.go.jp/files/000206217.pdf
- 20. Bird P, Bensen W, El-Zorkany B, et al. . Tofacitinib 5 mg twice daily in patients with rheumatoid arthritis and inadequate response to disease-modifying antirheumatic drugs: a comprehensive review of phase 3 efficacy and safety. J Clin Rheumatol 2019;25:115–26. 10.1097/RHU.0000000000000786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dougados M, van der Heijde D, Chen Y-C, et al. . Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis 2017;76:88–95. 10.1136/annrheumdis-2016-210094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winthrop KL, Yamanaka H, Valdez H, et al. . Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol 2014;66:2675–84. 10.1002/art.38745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen SB, Tanaka Y, Mariette X, et al. . Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 2017;76:1253–62. 10.1136/annrheumdis-2016-210457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka Y, Atsumi T, Amano K, et al. . Efficacy and safety of baricitinib in Japanese patients with rheumatoid arthritis: subgroup analyses of four multinational phase 3 randomized trials. Mod Rheumatol 2018;28:583–91. 10.1080/14397595.2017.1392057 [DOI] [PubMed] [Google Scholar]
- 25. Tanaka Y, Ishii T, Cai Z, et al. . Efficacy and safety of baricitinib in Japanese patients with active rheumatoid arthritis: a 52-week, randomized, single-blind, extension study. Mod Rheumatol 2018;28:20–9. 10.1080/14397595.2017.1307899 [DOI] [PubMed] [Google Scholar]
- 26. Scott IC, Hider SL, Scott DL. Thromboembolism with Janus kinase (JAK) inhibitors for rheumatoid arthritis: how real is the risk? Drug Saf 2018;41:645–53. 10.1007/s40264-018-0651-5 [DOI] [PubMed] [Google Scholar]
- 27. Tanaka Y, Suzuki M, Nakamura H, et al. . Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res 2011;63:1150–8. 10.1002/acr.20494 [DOI] [PubMed] [Google Scholar]
- 28. Tanaka Y, Takeuchi T, Yamanaka H, et al. . Efficacy and safety of tofacitinib as monotherapy in Japanese patients with active rheumatoid arthritis: a 12-week, randomized, phase 2 study. Mod Rheumatol 2015;25:514–21. 10.3109/14397595.2014.995875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Changelian PS, Flanagan ME, Ball DJ, et al. . Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003;302:875–8. 10.1126/science.1087061 [DOI] [PubMed] [Google Scholar]
- 30. Fridman JS, Scherle PA, Collins R, et al. . Selective inhibition of JAK1 and Jak2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 2010;184:5298–307. 10.4049/jimmunol.0902819 [DOI] [PubMed] [Google Scholar]
- 31. Ishizaki M, Muromoto R, Akimoto T, et al. . Tyk2 deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Int Immunol 2011;23:575–82. 10.1093/intimm/dxr057 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2019-215163supp001.pdf (4.3MB, pdf)