

Abstract
Background.
Src signaling is markedly upregulated at the invasive glioblastoma (GBM) front following bevacizumab administration. The Src family kinase inhibitor, dasatinib, can effectively block bevacizumab-induced glioma invasion in preclinical models. We hypothesized that combining bevacizumab with dasatinib could increase bevacizumab efficacy in recurrent GBM.
Methods.
Following completion of the phase I component, in the phase II trial (NCT00892177), recurrent GBM patients were randomized 2:1 to receive 100 mg of oral dasatinib twice daily (arm A) or placebo (arm B) on days 1–14 of each 14-day cycle combined with 10 mg/kg intravenous bevacizumab on day 1 of each 14-day cycle. The primary endpoint was 6-month progression-free survival (PFS6).
Results.
In the 121 evaluable patients, the PFS6 rate was numerically, but not statistically, higher in arm A versus arm B (28.9% [95% CI, 19.5–40.0%] versus 18.4% [95% CI, 7.7–34.4%]; p=0.22). Similarly, there was no significant difference in median OS between arms (7.3 months and 7.7 months, respectively; p=0.93). Objective response rate was 15.7% in arm A and 26.3% in arm B (p=0.52), but with significantly longer duration in arm A patients (16.3 vs 2 mo). Incidence of grade ≥3 toxicity was comparable between arms, with hematologic toxicities occurring more frequently in arm A versus arm B (15.7% versus 7.9%). Correlative tissue analysis showed an association between pSRC/LYN signaling in patient tumors and outcome.
Conclusion.
Despite upregulation of Src signaling in GBM, the combination of bevacizumab with dasatinib did not significantly improve outcomes of patients with recurrent GBM, compared with bevacizumab alone.
Keywords: bevacizumab, dasatinib, Src family kinase inhibitors, recurrent glioblastoma, phase II trial
Precis
In a randomized comparative phase II trial in recurrent GBM the combination the Src inhibitor dasatinib with bevacizumab led to PFS6 rate that was numerically but not statistically higher in the combination arm (28.9 vs 18.4%, p=0.22) as compared to single agent bevacizumab. Addition of dasatinib resulted in significantly longer response duration (16.3 vs 2 mo, p=0.0012).
Introduction
Glioblastoma (GBM) is the most frequent and aggressive glioma histology, accounting for approximately 15% of all primary brain tumors and 46% of primary malignant brain tumors in adults.1 Patients diagnosed with GBM have an extremely poor prognosis despite multimodality treatment. The standard care for newly diagnosed patients involves maximal surgical resection followed by radiotherapy with concomitant and adjuvant temozolomide; however, the median survival with this approach ranges from 16 to 18 months.2, 3 A high rate of recurrence is seen with current therapies for GBM and various agents have shown limited clinical benefit in this setting.4 Due to the highly heterogeneous nature of GBM, with several different signaling pathways involved in its pathogenesis, a combination of therapies targeting different pathways may prove to be beneficial.
GBM is characterized by intense angiogenesis that inevitably promotes tumor growth and rapid progression.5, 6 The clinical development of anti-angiogenic agents led to major therapeutic advancements in the treatment of cancer and, in 2009, the vascular endothelial growth factor (VEGF) targeting antibody bevacizumab received accelerated approval by the FDA for the treatment of patients with recurrent GBM, based on durable objective response rates observed in two prospective phase II studies;7–9 this was converted to regular approval in December 2017. Nevertheless, bevacizumab treatment regimens have only proven active in up to 30% of patients with very modest or conclusive impact overall survival, indicating a need for novel therapeutic combinations to be explored.10, 11 Furthermore, disease progression on bevacizumab treatment can be associated with increased tumor invasiveness, with patients progressing with a diffuse or multi-focal tumor recurrence.12 In vitro and in vivo xenograft model studies have confirmed that there is an increase in tumor invading cells following bevacizumab treatment,13–15 which represents a possible mechanism of resistance that undermines bevacizumab efficacy.
An increase in Src family kinase (SFK) activation is commonly found in GBM and it is associated with increased tumor cell invasion and motility.16 SFK signaling was found to be markedly upregulated at the invasive GBM front following bevacizumab administration.14 Dasatinib (BMS-354825) is a potent, oral ATP competitive, multi-targeted kinase inhibitor that inhibits all members of the Src family of kinases. Administration of this broad spectrum SFK inhibitor effectively inhibited bevacizumab-induced glioma invasion in an orthotopic xenograft model.14 Therefore, the combination of dasatinib with bevacizumab could have the potential to increase bevacizumab efficacy in GBM by blocking resistance mechanisms.
Here, we report the results of a placebo-controlled phase I/randomized phase II trial designed to evaluate the efficacy and safety of bevacizumab in combination with the broad spectrum Src kinase inhibitor dasatinib, compared with single-agent bevacizumab in recurrent GBM.
Methods
Study design and treatment
In the phase I portion of the study, patients were enrolled in cohorts of 3 in a standard 3+3 phase I design. The primary objective of the phase I study was to determine the maximum tolerated dose (MTD). There were 4 pre-specified dose combination levels from 5 mg/kg intravenous (i.v.) bevacizumab every 2 weeks (q2wks) plus 50 mg dasatanib orally (p.o.) twice daily (starting dose level 0) up to 10 mg/kg i.v. bevacizumab q2wks plus 100 mg dasatinib p.o. twice daily (dose level 3). Patients were assessed for dose limiting toxicities (DLTs) during the first four weeks of treatment (i.e., at the end of cycle 2). DLTs were defined as the following toxicities (considered at least possibly related to the study treatment): 1) Grade 3/4 thrombocytopenia, grade 4 anemia, or grade 4 neutropenia lasting >7 days; 2) any non-hematologic grade 3+ toxicity, excluding alopecia and venous thromboembolism; 3) Grade 3/4 nausea, vomiting, diarrhea, and electrolyte abnormalities, such as hypocalcemia and hypophosphatemia if they cannot be controlled by medical therapy; 4) failure to recover from toxicities to be eligible for re-treatment with bevacizumab and dasatinib ≤28 days of the last dose of the two drugs.
Following completion of the phase I dose escalation, in the randomized, double-blind, placebo-controlled, phase II portion of the study, patients were randomized 2:1 to receive either 100 mg twice-daily dasatinib p.o. or placebo from day 1 to 14 of each 14-day cycle and 10 mg/kg i.v. bevacizumab (over 90 minutes) on day 1 of each 14-day cycle. Patients received study treatment until disease progression, unacceptable toxicity, decision to withdraw from study, or changes in the patient condition that in the investigator’s judgement would prevent or render the patient unacceptable for further treatment.
The primary objective of the study was to estimate the efficacy of bevacizumab in combination with dasatinib (arm A) in recurrent GBM, as measured by PFS6, compared with the efficacy of bevacizumab with placebo (arm B). The secondary objectives were to determine objective response, time to disease progression (TTP), and overall survival (OS), as well as to assess the safety and toxicity of bevacizumab combined with dasatinib. Patient-reported quality of life (QOL) outcomes were also evaluated. Exploratory endpoints included associations between molecular (Src pathway activation in baseline tissue) and outcomes.
The study received institutional review board approval and all patients provided written informed consent in accordance with federal and institutional guidelines.
Patient eligibility
Main inclusion criteria were patients ≥18 years of age; histologically confirmed GBM, including variant gliosarcoma, as determined by pre-registration central pathology review; evidence of tumor progression and measurable and evaluable disease by magnetic resonance imaging (MRI) or computed tomography (CT) scan; Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2; acceptable hematologic, liver and renal function; ≥12 weeks from completion of prior radiotherapy; ≥4 weeks from prior chemotherapy (6 weeks for nitrosourea-based regimens); up to 2 prior chemotherapy regimens with ≤1 regimen for recurrent disease; no prior treatment with angiogenesis inhibitors or dasatinib. Main exclusion criteria were inadequately controlled hypertension; co-morbid systemic illnesses, or other severe concurrent disease; other active malignancy ≤3 years prior to registration, with the exception of non-melanotic skin cancer or carcinoma-in-situ of the cervix; history of myocardial infarction or unstable angina ≤6 months prior to registration, New York Heart Association classification II, III or IV congestive heart failure; significant vascular disease; pregnancy, nursing, and/or to unwillingness to employ adequate contraception during the study and for up to 6 months after bevacizumab treatment has ended.
Study assessments
PFS was defined as the time from start of the study therapy to documentation of disease progression or date of last known disease evaluation, which comes first. For PFS, patients who died without documentation of progression were considered to have had tumor progression at the time of death unless there was documented evidence that no progression occurred before death. TTP was defined as the time from start of study therapy to documentation of disease progression. For TTP, patients who died without documentation of progression were censored at the time of their last disease evaluation. Patients who discontinued follow-up without documented progression were censored at the end of therapy. Response assessment was performed according to the RANO criteria.17 Duration of response analysis only included patients who had an objective response and was defined as the time from the first report of an objective response to the time of subsequent progression. Radiographic responses were verified by Central Review. Time to Treatment Failure (TTF) was defined as the time from start of study therapy to the end of treatment for any reason. Adverse events (AEs) were assessed per the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0). QOL endpoints were assessed every other cycle via the Functional Assessment of Cancer Therapy-Brain (FACT-Br)18 and the Folstein mini mental state examination (MMSE) 19 questionnaires.
Correlative analysis explored associations between Src family kinase expression/activation of downstream signaling pathways and outcomes. Expression was determined by immunohistochemistry (IHC) in pre-treatment tumor samples.
Tumor immunohistochemical analysis
Paraformaldehyde-fixed brain tumor tissues were embedded in paraffin and the tissue was then cut into 5 μm sections, mounted on charged slides, and processed by the Mayo Clinic Cancer Biology Histology Facility for hematoxylin and eosin (H and E) staining and IHC. Staining for YES and CD31 were performed using mouse monoclonal antibodies (C-10, Santa Cruz Biotechnology, Dallas, TX) and (JC70A, Agilent DAKO, Santa Clara, CA) respectively. Rabbit monoclonal antibodies for LYN (C13F9) and VEGF-R2 (55B11), as well as a rabbit polyclonal against phospho (p) Y416 Src were obtained from Cell Signaling Technology (Danvers, MA). All slides were counterstained with hematoxylin.
High-resolution digital images of the stained slides were obtained by scanning the slides on an Aperio AT2 slide scanner (Leica Biosystems, Vista, CA), and tumors were then annotated using Aperio ImageScope software. Antibody positivity in the tumors was determined by applying the Aperio IHC Positive Pixel Count (v9) algorithm, which uses color deconvolution to separate the DAB (positive) from hematoxylin (negative) signals, to count the percent of positively stained pixels present in each sample. All biomarker IHC values were reported on a 0–1 continuous scale, with values closer to 1.0 indicating higher levels of activation.
Statistical analysis
The Pocock and Simon dynamic allocation procedure,20 which balances the marginal distributions of the stratification factors (age [>70 versus ≤70 years] and performance status [0 versus 1 or 2]) across the two arms, was applied. Statistical considerations regarding sample size and study power were based on the primary endpoint, PFS6. The study required 111 evaluable patients (74 in arm A and 37 in arm B) in order to provide 85% power to detect a 20% difference in PFS6 between the two arms, with a type-I error rate of 0.15. The analysis was based on a modified intention-to-treat principle with all eligible patients who began treatment being evaluable for the arm to which they were randomized. The PFS6 rates for each arm were determined by the number of evaluable patients alive and progression-free at 6 months divided by the number of evaluable patients respectively for each arm. One interim analysis was performed once 50% of patients were accrued and followed for at least 6 months. Confidence intervals for PFS6 were estimated using the Clopper-Pearson Method. Objective response rates per arm where compared and confidence intervals calculated using Fisher’s exact test. The distributions of PFS, TTP, TTF, OS, and duration of response were estimated using Cox proportional hazards models (adjusting for age, gender, and baseline performance score), plotted via the Kaplan-Meier (K-M) method and compared between arms using log-rank tests.
Number and severity of AEs were summarized by descriptive statistics. Best objective response rates and grade ≥3 AEs between the 2 arms were compared using a Chi-Square or Fisher’s Exact test.
Statistical analysis of correlative assays
PFS and OS were assessed for differences between patients with and without biomarker IHC values using K-M analysis and log-rank p-values. The amount of similarity between the biomarker IHC values for VEGF.R2, pSRC, CD31, LYN, and YES were assessed for correlations between all pairs of biomarkers using Spearman-correlation analysis.
Each univariate IHC value was searched for the best cut-point value for PFS, and separately for OS, using two methods. First, to evaluate for the interaction where the level of the biomarker IHC value would be associated with a PFS or OS advantage in just the bevacizumab plus dasatinib-treated patients, who were used as a training set to determine the best cut-point for each of the five biomarkers. These cut-points were then applied to the biomarker IHC values for all patients. The resulting four groups based on the combination of treatment arm and biomarker high and low values (high versus low expression, respectively) were used in K-M analyses for each biomarker for PFS and OS, separately. Second, and in a similar fashion, all patient data regardless of treatment arm were used to determine the best overall univariate expression value cut-point for each biomarker separately for PFS and OS. The resulting high versus low biomarker groups and separately the four groups using the combination of biomarker expression group and treatment arm were analyzed using K-M analysis.
The best expression-value cut-points for PFS, and separately for OS, were chosen as the cut-point value that had the lowest log-rank p-value in K-M analysis. In cases of cut-points with the same minimum p-value, the cut-point that allowed for maximizing the number of patients in the smallest group was chosen. Relevant cut-points required at least 8 patients in the smallest group.
Recursive partitioning analysis (R-software function “rpart”)21, 22 was used to evaluate the biomarker IHC values combined with treatment arm and the stratification factors age and performance score to determine if any subgroups existed with PFS, and separately, OS advantages. Subgroups of patients identified via rpart were collapsed into larger main-groups, and PFS and OS K-M analysis was used to evaluate for PFS or OS advantages.
Statistical analysis of Quality of life
The FACT-Br and MMSE were assessed every 2 cycles (every 4 weeks). Generalized linear mixed-effect repeated-measures models were used to conduct longitudinal data analysis in order to assess for differences between patients on arm A vs. arm B in QOL over time as measured by the overall FACT-Br score and separately for the MMSE score.
All analysis was performed with either the software SAS linux-version 9.4 or R version 3.2.3.
Results
Patient demographics and baseline characteristics
In the phase I portion of the study, 16 patients were enrolled. Three patients each were enrolled on dose levels 0 and 2, 4 patients were enrolled on dose 1 (one patient was replaced for MTD determination due to having no DLTs before disease progression prior to the end of cycle 2), and 6 patients were enrolled in cohorts of 3 on dose level 3. Only one DLT was observed in the second cohort of 3 on dose level 3. This patient experienced a grade 3 creatinine increase considered to be at least possibly related to treatment. The MTD was determined to be dose level 3: 10 mg/kg i.v. bevacizumab q2wks plus 100 mg dasatinib p.o. twice daily. Other toxicities reported in these 16 phase I patients included one patient each reporting the following grade 3 AEs considered to be at least possibly related to study treatment: lymphopenia, fatigue, rash/desquamation, diarrhea, heartburn/dyspepsia, and hypophosphatemia. Five of the 16 patients (31.2%) achieved an objective response. Three of these were in dose level 2 and two patients in dose level 3.
In the phase II portion, 128 patients were randomized (Figure 1). Of these 128, 121 were evaluable for PFS6 assessment, with 83 patients treated with bevacizumab in combination with dasatinib (arm A), and 38 patients treated with bevacizumab alone (arm B). Five patients did not begin treatment (4 on arm A and 1 on arm B) and two patients were deemed ineligible after randomization (1 on each arm). At the time of the data freeze for this analysis all patients had progressed and there were three patients still alive on arm A with survival time censored at 33, 34, and 39 months. Demographics and baseline characteristics are summarized in Table 1. Overall, demographics and patient characteristics were balanced across arms, with no statistical differences detected.
Figure 1:
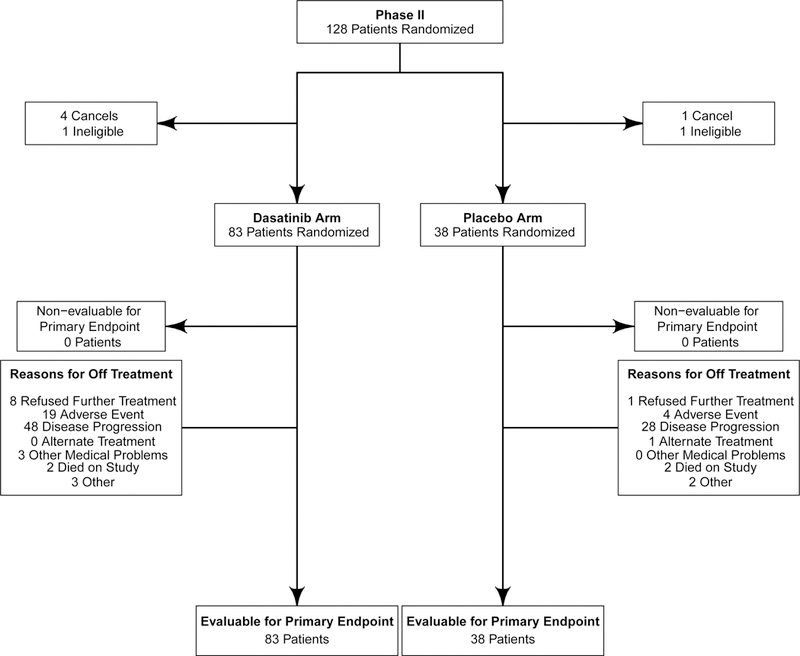
CONSORT diagram
Table 1:
Patient demographics and baseline characteristics.
| Phase I Bevacizumab + Dasatanib (N=16) |
Arm A Bevacizumab + Dasatinib (N=83) |
Arm B Bevacizumab + Placebo (N=38) |
Phase II Total (N=121) |
|
|---|---|---|---|---|
| Age Group | ||||
| > 70 | 0 | 5 (6.0%) | 1 (2.6%) | 6 (5.0%) |
| ≤ 70 | 16 (100%) | 78 (94.0%) | 37 (97.4%) | 115 (95.0%) |
| Age | ||||
| Mean (SD) | 48.1 (13.8) | 56.8 (10.9) | 52.5 (14.1) | 55.5 (12.1) |
| Median (range) | 46.5 (28–68) | 58.0 (29.0–79.0) | 56.5 (18.0–71.0) | 57.0 (18.0–79.0) |
| Gender, n (%) | ||||
| Female | 3 (18.8%) | 28 (33.7%) | 16 (42.1%) | 44 (36.4%) |
| Male | 13 (81.3%) | 55 (66.3%) | 22 (57.9%) | 77 (63.6%) |
| ECOG Performance Score, n (%) | ||||
| 0 | 3 (18.8%) | 16 (19.3%) | 7 (18.4%) | 23 (19.0%) |
| 1 | 7 (43.8%) | 40 (48.2%) | 19 (50.0%) | 59 (48.8%) |
| 2 | 6 (37.5%) | 27 (32.5%) | 12 (31.6%) | 39 (32.2%) |
| Corticosteroid Therapy at Study Entry, n (%) | ||||
| Missing | 0 | 14 | 9 | 23 |
| Yes | 12 (75%) | 41 (59.4%) | 21 (72.4%) | 62 (63.3%) |
| Baseline FACT-BR Scores | ||||
| N | - | 64 | 26 | 90 |
| Mean | - | 141.01 | 135.09 | 139.30 |
| SD | - | 37.43 | 46.83 | 23.34 |
| Baseline MMSE Scores | ||||
| N | - | 56 | 28 | 84 |
| Mean | - | 25.43 | 26.04 | 25.63 |
| SD | - | 9.48 | 8.12 | 5.15 |
ECOG, Eastern Cooperative Oncology Group.
Efficacy
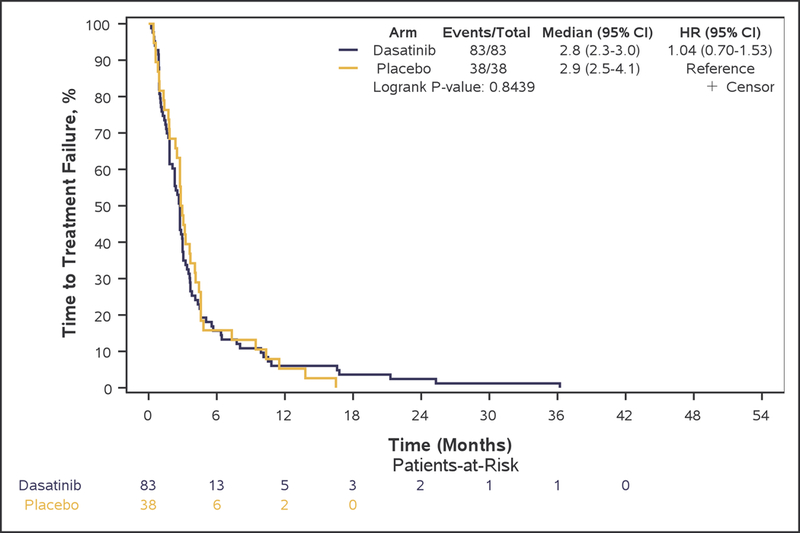
Although PFS6 rate was numerically higher in bevacizumab plus dasatinib-treated patients (arm A: 28.9% [95% CI, 19.5–40.0%]), compared with patients treated with single-agent bevacizumab (arm B: 18.4% [95% CI, 7.7–34.4%], this difference did not reach statistical significance (p=0.22), and the primary study goal was not met. Median OS (7.3 vs. 7.7 months, adjusted-HR=0.96; 95%CI 0.64–1.43, p=0.83), PFS (3.2 vs. 3.2 months, adjusted-HR=0.79; 95%CI 0.53–1.19, p=0.27), TTP (4.1 vs. 3.6 months, adjusted-HR=0.82; 95%CI 0.55–1.23, p=0.33), and TTF (2.8 vs. 2.9 months, adjusted-HR=1.01; 95%CI 0.68–1.51, p=0.95) were also not statistically different between arms (Figure 2: A, B, C, and E respectively).
Figure 2:
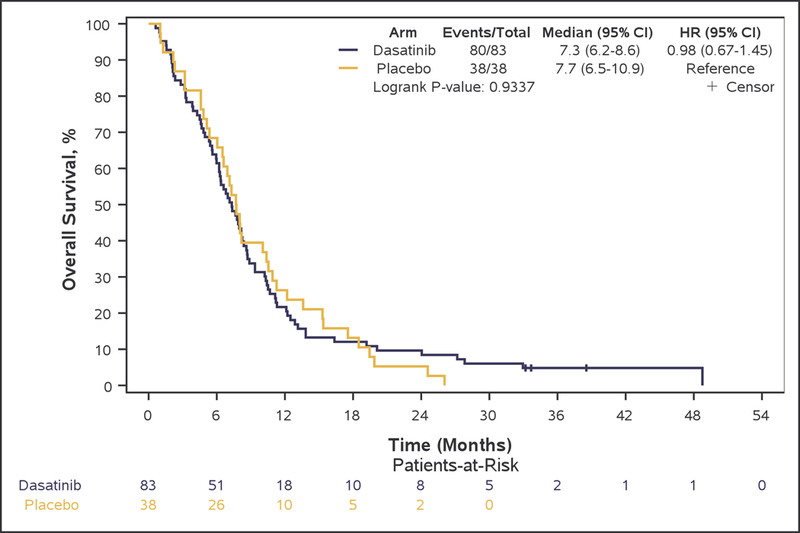
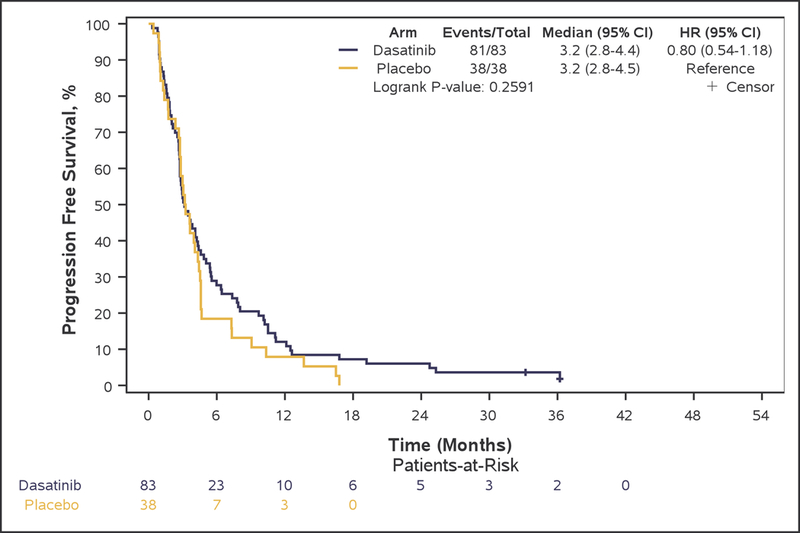
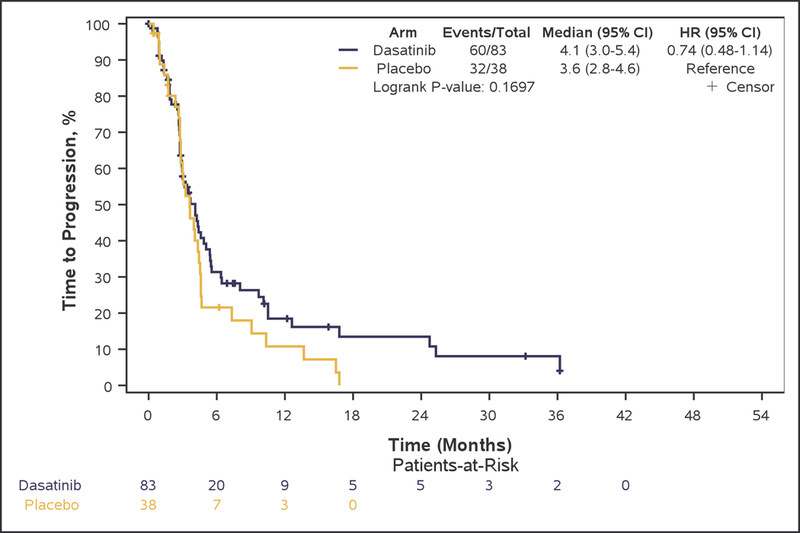
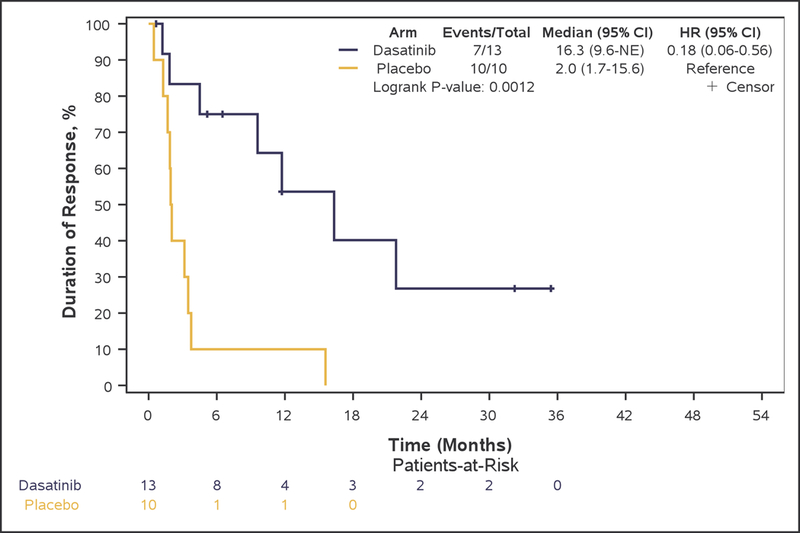
Kaplan-Meier curves of overall survival (A), progression-free survival (B), and time to progression (C), duration of response (D), and time to treatment failure (E) in Phase II patients.
An objective response was achieved in 13 (15.7%, 95%CI 8.6–25.3%) patients in arm A and in 10 (26.3%, 95%CI 13.4–43.1%) patients in arm B (Fisher’s exact test, p=0.21). Median duration of response was 16.3 months with the addition of dasatinib to bevacizumab versus 2.0 months with bevacizumab only therapy (adjusted-HR=0.27, 95%CI 0.09–0.82, p=0.021) (Figure 2D). Stable disease (SD) was the best response in 48 (57.8%) and 22 (57.9%) patients in arm A and B, respectively.
Safety
Most common AEs in both arms combined, considered to be at least possibly related to treatment (toxicities) are summarized in Table 2 and Figure 3. The most frequent treatment-related AEs (all grades and at least possibly related) include fatigue (62.0%), anemia (52.9%), thrombocytopenia (44.6%), and diarrhea (41.3%).
Table 2:
Overview of most common (≥5% patient in either arm) grade ≥3 toxicities, at least possibly related to treatment
| Adverse event, n (%) | Arm A Bevacizumab + Dasatinib (N=83) |
Arm B Bevacizumab + Placebo (N=38) |
|---|---|---|
| Hematologic | ||
| Lymphopenia | 8 (9.6%) | 1 (2.6%) |
| Non-hematologic | ||
| Hypertension | 6 (7.2%) | 5 (13.2%) |
| Fatigue | 10 (12.0%) | 0 |
| Diarrhea | 5 (6.0%) | 0 |
| Hypophosphatemia | 12 (14.5%) | 1 (2.6%) |
Figure 3:
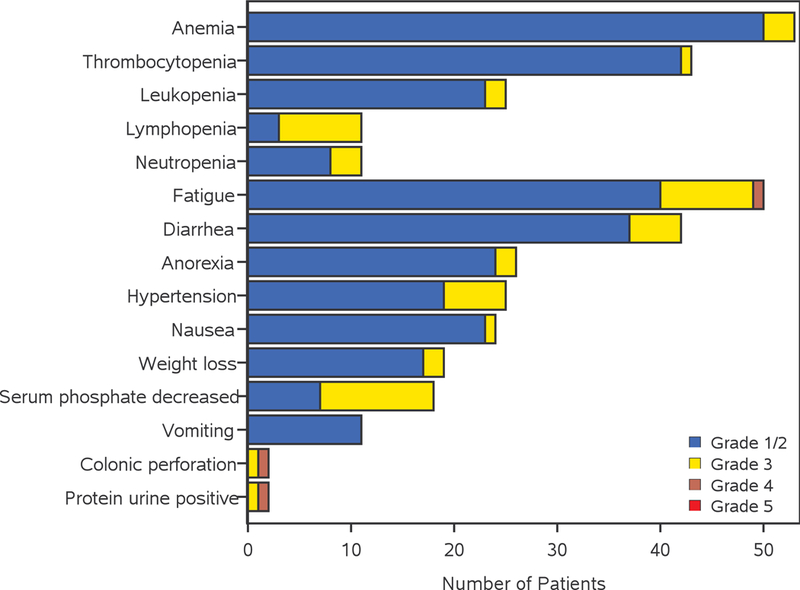
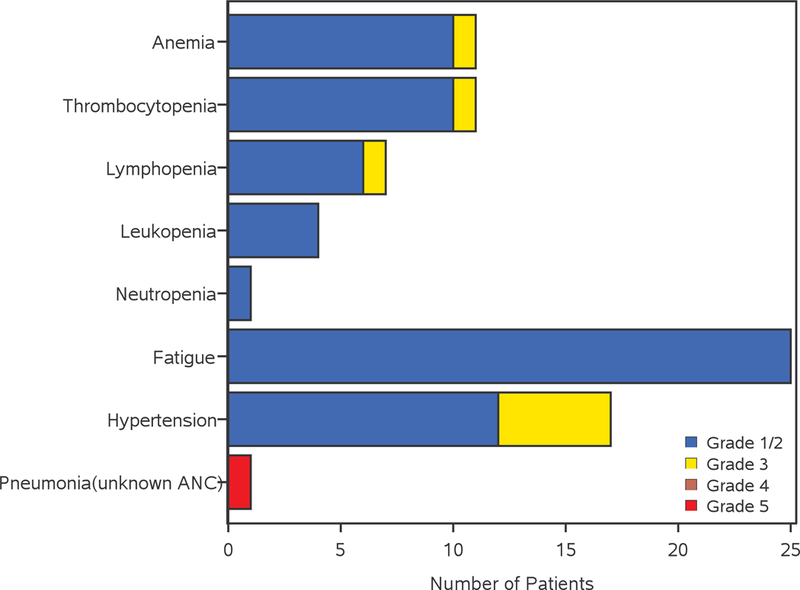
Adverse events possibly related to treatment in the bevacizumab/dasatinib arm (A) and bevacizumab arm (B). AEs are included when 2 or more patients have grade 3 or 4 AEs, or 10 or more patients have grade 1 or 2 AEs which are deemed to be at least possibly related to treatment.
Overall, the incidence of grade ≥3 toxicities was slightly higher but not statistically significant in arm A vs. arm B (47.0% vs. 36.8%, respectively, Fisher’s exact-test p=0.33.). This remained the case when considering grade ≥3 hematologic toxicities (15.7% in arm A and 7.9% in arm B; p=0.39) and grade ≥3 non-hematologic toxicities (44.6% in arm A and 34.2% in arm B; p=0.33) separately. Common (≥5% of patients in either arm) grade ≥3 toxicities are shown in Table 2. Lymphopenia was the most frequent grade ≥3 hematologic toxicity (10% in arm A, 3% in arm B), whereas most common non-hematologic toxicities were hypophosphatemia (15% in arm A, 0% in arm B) and fatigue (12% in arm A, 0% in arm B). Four patients died during the study (2 in arm A and 2 in arm B). One death was considered possibly related to treatment. This patient, who was in the bevacizumab-only arm, experienced grade 5 pneumonia with unknown ANC.
Quality of life
At least one post-baseline FACT-Br assessment was obtained for 67 (80.7%) patients on arm A and 29 (76.3%) on arm B. The median number of post-baseline FACT-Br assessments per patient were 3 (range 1 – 32) and 3 (range 1 – 12) for arms A and B respectively. For the MMSE Assessment, at least one post-baseline assessment was obtained for 63 (75.9%) patients on arm A and 30 (78.9%) on arm B. The median number of post-baseline MMSE assessments per patient were 3 (range 1 – 33) and 3 (range 1 – 17) for arms A and B respectively. Combination treatment with bevacizumab and dasatinib did not result in a difference in quality of life as assessed either via the overall FACT-Br score (p=0.79) or MMSE (p=0.23) over time.
Correlative analysis
In total, 80 of the 121 patients were tested by IHC values for VEGFR2, Y416.SRC (pSRC), CD31, LYN, and YES. Most of the pairs of IHC values had a significant low-level correlation (Spearman-rho: range (0.50 – 0.35, p-values: range 0.002-<0.001). Only CD31 versus VEGFR2 (Spearman rho=0.21, p=0.065) and CD31 versus pSRC (Spearman rho=0.22, p=0.055) did not show a statistically significant correlation between the levels of the biomarker IHC values within baseline tissue. There were no significant interactions for the level of PFS or OS advantage for patients receiving bevacizumab plus dasatinib that were dependent on the level of the biomarker IHC values. In univariate cut-point analysis, combining all patients regardless of treatment arm, identified that patients with higher values of pSRC (cut-point above 0.043) had a PFS disadvantage (HR=2.23 [95%CI: 1.23, 4.04, p=0.007]) whereas patients with higher values of CD31 (cut-point above 0.023) or LYN (cut-point above 0.007) had a PFS advantage (HR=0.57 [95%CI: 0.35, 0.95,p=0.03] and HR=0.45 [95%CI: 0.28, 0.74, p=0.001], respectively).
Univariate cut-point analyses for OS resulted in no significant relevant cut-points with OS advantage. The rpart PFS analysis returned seven subgroups of patients based on the values of LYN, pSRC, and CD31. Partition 1 and 2 included patients with higher values of LYN, and either low values of pSRC, or medium values of pSRC combined with low values of CD31, respectively. All other patients were in partitions 3–7. When combined, the patients in partitions 3–7 had a PFS disadvantage (HR=5.18 [95%CI 2.74, 9.78, p<0.001]), which appeared to be regardless of treatment received.
The OS rpart analysis returned seven subgroups of patients based on the values of all five biomarkers: CD31, LYN, pSRC, VEGFR2, and YES. Partitions 1 and 2 were patients with either medium to high values of CD31 and low values of pSRC, or medium values of CD31, high values of pSRC, combined with high values of LYN. All other patients were in partitions 3–7. When combined, the patients in partitions 3–7 have a OS disadvantage (HR=3.06 [95%CI: 1.72, 5.45, p<0.001]); this disadvantage appeared to be regardless of treatment received.
The two partitions with the survival advantage in both the PFS and OS rpart analysis had common themes for the biomarker IHC values. The rpart partition-1 from PFS and OS included patients with low values of pSRC, while the rpart partition-2 included patients with high values of LYN, higher or medium values of SRC, combined with medium to low values of CD31.
Discussion
The combination of bevacizumab with dasatinib in our trial did not improve outcomes significantly in patients with recurrent GBM, compared with bevacizumab treatment alone. Although the combination had an acceptable tolerability profile in this patient population and there was a numerical improvement in the PFS6 rate in the combination arm, the prespecified efficacy threshold was not met, with a PFS6 of 28.9% for bevacizumab plus dasatinib versus 18.4% for bevacizumab alone (p=0.22). Of note, despite the fact that the objective response rate was comparable between the two arms (15.7% in Arm A vs 26.3% in Arm B), the median duration of response was significantly longer in responding patients who received the bevacizumab/dasatinib combination (16.3 vs 2.0 mo) suggesting a possible benefit conferred by dasatinib to responding patients.
Although the PFS6 with bevacizumab is inferior in this study, compared to the 42.6% in the BRAIN trial,8 this likely reflects the difference in the patient population (community-based versus treated in academic centers, performance status, steroid use, etc.), and the response criteria employed. It is of note that a number of other recent trials evaluating bevacizumab in recurrent GBM, including CABARET, BELOB and EORTC 26101,11, 23, 24 have observed PFS6s consistent with our results.
Over the last 10 years, a number of single arm phase II studies have investigated bevacizumab in combination with various agents for recurrent GBM, including irinotecan, cetuximab, carboplatin, erlotinib and sorafenib, without demonstrating an improvement in clinical efficacy over single-agent bevacizumab treatment.10, 25–31 More recently, the CABARET trial reported a PFS6 of 15% with bevacizumab in combination with carboplatin compared with 18% with bevacizumab alone, with a median OS of 6.9 months in the combination arm versus 7.5 months in the monotherapy arm (HR:1.18, 95% CI: 0.82 – 1.69, p=0.38); these data also indicated that the combination was not clinically beneficial.23 Following the randomized non-comparative BELOB trial, which showed promising PFS6 and OS9 with the bevacizumab/lomustine combination (42% and 59%, respectively,11 EORTC 26101, investigated whether the combination of bevacizumab and lomustine produced a definitive OS benefit, compared with monotherapy of either agent in patients with recurrent GBM at first progression.24 A PFS6 rate of 30% was reported with bevacizumab in combination with lomustine, compared with 17% with bevacizumab alone, with a PFS of 4.2 vs 1.5 months, respectively; however, median OS was 9.1 months in the combination arm versus 8.6 months in the lomustine arm (HR:0.95, 95% CI: 0.74–1.21, p=0.650).24 Therefore, while the data indicated a prolonged PFS in the combination arm, there was no survival advantage to the combination; of note 35.5% of single agent lomustine treated patients went on to receive bevacizumab at progression.
Initial encouraging data from the ReACT phase II study, which investigated the combination of rindopepimut, a vaccine that generates a specific immune response against tumors expressing EGFRvIII (approximately 30% of GBMs), with bevacizumab showed a PFS6 of 27% with bevacizumab in combination with rindopepimut, compared with 11% with bevacizumab alone.32 However, the follow-up data have been disappointing.33 The phase II METMAB study investigated the combination of bevacizumab with onartuzumab, the monovalent mesenchymal epithelial transition factor (MET) inhibitor, since aberrant MET expression has been reported in GBM.34 No evidence of clinical benefit in patients with recurrent GBM was observed.35
Recent clinical research investigating dasatinib monotherapy in recurrent GBM reported a PFS6 of only 6%, despite efforts to enrich the patient population and to increase the dose of dasatinib.36 The lack of clinical benefit was considered to be potentially a result of the different molecular profiles of dasatinib targets between archival tumor resected at GBM diagnosis and the profile at disease recurrence; it was also noted that response to dasatinib in GBM may require driver mutations in dasatinib targets. The strategy of targeting SFK signaling has been studied recently in culture and orthotopic xenograft models. The results indicate that individual SFKs (Src, Fyn, Yes and Lyn) differ significantly in their importance for glioma growth and motility, and the efficacy of dasatinib therapy may depend on the relative expression of particular SFKs.37 Notably, depletion of LYN resulted in increased tumor growth and reduced overall survival in these orthotopic patient-derived GBM xenografts, suggesting a tumor suppressor function for LYN in GBM.37 Consistent with this, higher levels of LYN expression correlated with both a PFS6 and OS advantage in our study, regardless of treatment. Conversely, high level of pSRC correlated with PFS6 disadvantage, regardless of treatment, arguing that increased activation of SFKs contributes to the aggressive nature of GBM.
In conclusion, while the results of our trial signal that the combination of dasatinib and bevacizumab does not lead to improved clinical outcomes in patients with recurrent GBM, there was a trend of improvement of PFS6 with use of dasatinib, which did not reach statistical significance, and statistically significant improvement in the duration of response with the addition of dasatinib. As such, selective SFK inhibition, possibly employing different inhibitors, may still be a promising avenue to pursue, given the high SFK activity in GBM and associations with patient outcomes.
Acknowledgments
The authors thank the patients who participated in this clinical trial and their families, the study investigators for their contributions, the National Cancer Institute/Cancer Therapy Evaluation Program (NCI/CTEP), and BMS and Genentech for trial support. Editorial and medical writing assistance was provided by Shilu Amin, PhD, TRM Oncology, The Hague, The Netherlands, funded by the Mayo Clinic, Rochester, MN, USA.
Funding Statement: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under the Award Number UG1CA189823 (Alliance for Clinical Trials in Oncology NCORP Grant) U10CA180821, U10CA180882, and U10CA180833. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The following institutional networks participated in this study:
Cancer Alliance of Nebraska, Omaha, NE, Gamini Soori; Carle Cancer Center NCI Community Oncology Research Program, Urbana, IL, Kendrith Rowland, UG1CA189861; Coborn Cancer Center at Saint Cloud Hospital, Saint Cloud, MN, Donald Jurgens; Colorado Cancer Research Program NCORP, Denver, CO, Keren Sturtz, UG1CA189805; Dana-Farber / Partners CancerCare LAPS, Boston, MA, Harold Burstein, U10CA180867; Dartmouth College - Norris Cotton Cancer Center LAPS, Lebanon, NH, Konstantin Dragnev, U10CA180854; Dayton NCI Community Oncology Research Program, Dayton, OH, Howard Gross, UG1CA189957; Delaware/Christiana Care NCI Community Oncology Research Program, Newark, DE, Gregory Masters, UG1CA189819; Edwards Comprehensive Cancer Center, Huntington, WV, Maria Tria Tirona; Essentia Health NCI Community Oncology Research Program, Duluth, MN, Bret Friday, UG1CA189812; Hawaii Minority Underserved NCORP, Honolulu, HI, Jeffrey Berenberg, UG1CA189804; Heartland Cancer Research NCORP, Decatur, IL, James Wade, UG1CA189830; Iowa-Wide Oncology Research Coalition NCORP, Des Moines, IA, Robert Behrens, UG1CA189816; Mayo Clinic LAPS, Rochester, MN, Steven Alberts, U10CA180790; MedStar Georgetown University Hospital, Washington, DC, Filipa Lynce; Metro Minnesota Community Oncology Research Consortium, Saint Louis Park, MN, Daniel Anderson, UG1CA189863; Michigan Cancer Research Consortium NCORP, Ann Arbor, MI, Philip Stella, UG1CA189971; Montana Cancer Consortium NCORP, Billings, MT, Benjamin Marchello, UG1CA189872; Mount Sinai Hospital, New York, NY, Lewis Silverman; Mount Sinai Medical Center, Miami Beach, FL, Michael Schwartz; Nevada Cancer Research Foundation NCORP, Las Vegas, NV, John Ellerton, UG1CA189829; New Hampshire Oncology Hematology PA-Hooksett, Hooksett, NH, Douglas Weckstein; Rapid City Regional Hospital, Rapid City, SD, Kathryn Arrambide; Rhode Island Hospital, Providence, RI, Howard Safran; Rush University Medical Center, Chicago, IL, Melissa Larson; Sanford NCI Community Oncology Research Program of the North Central Plains, Sioux Falls, SD, Preston Steen, UG1CA189825; Toledo Clinic Cancer Centers-Toledo, Toledo, OH, Rex Mowat; UC San Diego Moores Cancer Center, La Jolla, CA, Barbara Parker; University of Nebraska Medical Center, Omaha, NE, Apar Ganti; Wake Forest University Health Sciences, Winston-Salem, NC, Heidi Klepin; Washington University - Siteman Cancer Center LAPS, Saint Louis, MO, Nancy Bartlett, U10CA180833; Wichita NCI Community Oncology Research Program, Wichita, KS, Shaker Dakhil, UG1CA189808; and Wisconsin NCI Community Oncology Research Program, Marshfield, WI, Anthony Jaslowski, UG1CA189956.
Suriya A. Jeyapalan is a member of the speakers’ bureau for Sigma-Tau. Jan C. Buckner has received travel and accommodation expenses from Genentech/Roche. David Schiff serves on data safety committees for Orbus and Monteris. He serves as a consultant without personal compensation to Cavion. Evanthia Galanis serves as Advisory Board member to Vyriad (compensation to Mayo) and Celgene Corporation / KIYATEC (personal compensation); general consulting with MedImmune, Inc., F. Hoffman La Roche, Ltd (compensation to Mayo), Tactical Therapeutics, Inc., Oncorus (personal compensation); and has clinical trial funding with MedImmune, Inc.; Denovo Biopharma; Tracon; Genentech; Bristol-Myers Squibb (Mayo).
Footnotes
Conflict of Interest
The remaining authors declare no potential conflicts of interest.
ClinicalTrials.gov Identifier:NCT00892177
References
- 1.Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;17 Suppl 4: iv1-iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilbert MR, Wang M, Aldape KD, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31: 4085–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Taillibert S, Kanner A, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA. 2017;318: 2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seystahl K, Wick W, Weller M. Therapeutic options in recurrent glioblastoma--An update. Crit Rev Oncol Hematol. 2016;99: 389–408. [DOI] [PubMed] [Google Scholar]
- 5.Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D. Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol. 2005;15: 297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kargiotis O, Rao JS, Kyritsis AP. Mechanisms of angiogenesis in gliomas. J Neurooncol. 2006;78: 281–293. [DOI] [PubMed] [Google Scholar]
- 7.Cohen MH, Shen YL, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist. 2009;14: 1131–1138. [DOI] [PubMed] [Google Scholar]
- 8.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27: 4733–4740. [DOI] [PubMed] [Google Scholar]
- 9.Kreisl TN, Kim L, Moore K, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27: 740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moller S, Grunnet K, Hansen S, et al. A phase II trial with bevacizumab and irinotecan for patients with primary brain tumors and progression after standard therapy. Acta Oncol. 2012;51: 797–804. [DOI] [PubMed] [Google Scholar]
- 11.Taal W, Oosterkamp HM, Walenkamp AM, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 2014;15: 943–953. [DOI] [PubMed] [Google Scholar]
- 12.Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology. 2008;70: 779–787. [DOI] [PubMed] [Google Scholar]
- 13.de Groot JF, Fuller G, Kumar AJ, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010;12: 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huveldt D, Lewis-Tuffin LJ, Carlson BL, et al. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS One. 2013;8: e56505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15: 4589–4599. [DOI] [PubMed] [Google Scholar]
- 16.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4: 470–480. [DOI] [PubMed] [Google Scholar]
- 17.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28: 1963–1972. [DOI] [PubMed] [Google Scholar]
- 18.Weitzner MA, Meyers CA, Gelke CK, Byrne KS, Cella DF, Levin VA. The Functional Assessment of Cancer Therapy (FACT) scale. Development of a brain subscale and revalidation of the general version (FACT-G) in patients with primary brain tumors. Cancer. 1995;75: 1151–1161. [DOI] [PubMed] [Google Scholar]
- 19.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12: 189–198. [DOI] [PubMed] [Google Scholar]
- 20.Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31: 103–115. [PubMed] [Google Scholar]
- 21.Breinman L, Friedman JH, Oshen RA. Classification and regression trees. CRC Press; 1984. [Google Scholar]
- 22.Therneau T, Atkinson B, Ripley B. rpart: Recursive partitioning and regression trees. Available from URL: https://CRAN.R-project.org/package=rpart 2015]. [Google Scholar]
- 23.Field KM, Simes J, Nowak AK, et al. Randomized phase 2 study of carboplatin and bevacizumab in recurrent glioblastoma. Neuro Oncol. 2015;17: 1504–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wick W, Gorlia T, Bendszus M, et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N Engl J Med. 2017;377: 1954–1963. [DOI] [PubMed] [Google Scholar]
- 25.Desjardins A, Reardon DA, Coan A, et al. Bevacizumab and daily temozolomide for recurrent glioblastoma. Cancer. 2012;118: 1302–1312. [DOI] [PubMed] [Google Scholar]
- 26.Galanis E, Anderson SK, Lafky JM, et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a north central cancer treatment group trial. Clin Cancer Res. 2013;19: 4816–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasselbalch B, Lassen U, Hansen S, et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol. 2010;12: 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reardon DA, Desjardins A, Peters KB, et al. Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naive, recurrent glioblastoma. J Neurooncol. 2012;107: 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Metronomic chemotherapy with daily, oral etoposide plus bevacizumab for recurrent malignant glioma: a phase II study. Br J Cancer. 2009;101: 1986–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sathornsumetee S, Desjardins A, Vredenburgh JJ, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010;12: 1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vredenburgh JJ, Desjardins A, Herndon JE 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25: 4722–4729. [DOI] [PubMed] [Google Scholar]
- 32.Reardon DA, Schuster J, Tran DD. ReACT: Overall survival froma randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J Clin Oncol. 2015;33: [abst 2009]. [Google Scholar]
- 33.Reardon DA, Desjardins A, Schuster J. ReACT: Long-term survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. Soc for Neuro Oncol. 2015: [abstr IMCT-08]. [Google Scholar]
- 34.Huang M, Liu T, Ma P, et al. c-Met-mediated endothelial plasticity drives aberrant vascularization and chemoresistance in glioblastoma. J Clin Invest. 2016;126: 1801–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cloughesy T, Finocchiaro G, Belda-Iniesta C, et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O(6)-Methylguanine-DNA Methyltransferase Biomarker Analyses. J Clin Oncol. 2017;35: 343–351. [DOI] [PubMed] [Google Scholar]
- 36.Lassman AB, Pugh SL, Gilbert MR, et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 2015;17: 992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewis-Tuffin LJ, Feathers R, Hari P, et al. Src family kinases differentially influence glioma growth and motility. Mol Oncol. 2015;9: 1783–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]