

Abstract
Neuroinflammation is being recognized as a hallmark of different neurodegenerative disorders including Alzheimer’s disease. Suppressor of cytokine signaling 3 (SOCS3) is an anti-inflammatory molecule, which is known to inhibit cytokine signaling and inflammatory gene expression in different cells. However, the pathways by which SOCS3 could be upregulated in brain cells are poorly understood. Aspirin is a widely available pain reliever that is showing promise beyond its known pain-relieving capacity. This study underlines the importance of aspirin in upregulating SOCS3 in astrocytes and microglia. Aspirin increased the expression of Socs3 mRNA and protein in mouse astrocytes and BV-2 microglial cells in both a time- and dose-dependent manner. While investigating mechanism, we found that Socs3 gene promoter harbors PPRE and that aspirin upregulated SOCS3 in astrocytes isolated from PPARβ (−/−), but not PPARα (−/−), mice. Accordingly, aspirin increased SOCS3 in vivo in the cortex of wild type and PPARβ (−/−), but not PPARα (−/−), mice. Similarly, aspirin treatment increased astroglial and microglial SOCS3 in the cortex of FAD5X, but not FAD5X/PPARα (−/−), mice. Finally, recruitment of PPARα by aspirin to the proximal, but not distal, PPRE of the Socs3 promoter suggests that aspirin increases the transcription of Socs3 gene via PPARα. This study describes a novel property of aspirin in elevating SOCS3 in glial cells via PPARα and suggests that aspirin may be further considered for therapeutic application in neuroinflammatory and neurodegenerative disorders.
Keywords: Aspirin, SOCS3, Glial cells, PPARα, FAD5X model
Graphical Abstract
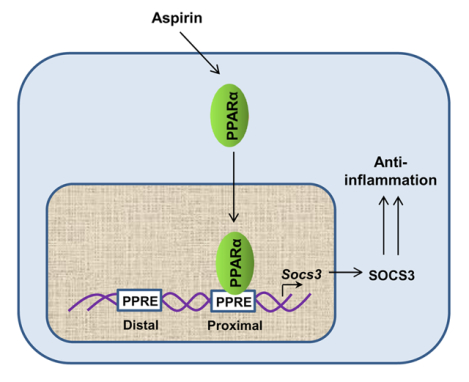
Drugs and mechanisms required to upregulate SOCS3 are poorly understood. Here, we delineate the presence of two consensus peroxisome proliferator response elements (PPRE) in the Socs3 gene promoter. Aspirin, a widely-used drug, stimulates the recruitment of PPARα, but neither PPARβ nor PPARγ, to the proximal PPRE of the Socs3 gene promoter, leading to the upregulation of SOCS3 in glial cells. These results indicate a new mode of action of aspirin.
Introduction
Inflammation is a recognized aspect of neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD) (Chen et al. 2016). Although neurons express inducible nitric oxide synthase (Heneka & Feinstein 2001) and proinflammatory cytokines (Janelsins et al. 2008) under certain conditions, inflammation in the brain is mainly mediated by two distinct glial cell types, astrocytes and microglia (Jha et al. 2012). Astrocytes fulfill numerous roles in the central nervous system and maintain neuronal homeostasis by providing nutrients and by the production of neurotrophic factors (Sofroniew & Vinters 2010). On the other hand, microglia, brain resident macrophages, are involved brain surveillance responding to pathogens and other insults (Kettenmann et al. 2011).
Suppressor of cytokine signaling 3 (SOCS3) is a member of the STAT- induced STAT inhibitor (SSI) family (Carow & Rottenberg 2014). Similar to other members of the SSI family (Cianciulli et al. 2017), SOCS3 is a physiological negative regulator of cytokine signaling. Various proinflammatory cytokines including IL-6 and IFNγ transduce signal via the JAK-STAT pathway. The prototype function of SOCS3 is to negatively regulate the STAT3 pathway by binding to and deactivating the JAK2 kinase, thereby preventing further inflammatory signaling.
Acetylsalicylic acid commonly known as aspirin is a member of the nonsteroidal, anti-inflammatory drugs (NSAIDs) (Reinhart 1999). NSAIDs are widely available and frequently used to treat fevers and pain. Aspirin is effective in the treatment of pain by inhibiting the proinflammatory enzyme cyclooxygenase. While aspirin has been used for millennia as a pain reduction agent modern research is increasingly finding new uses for it. There is emerging research indicating that aspirin has beneficial effects on various cancers, cardiovascular diseases and atherosclerosis (Yeomans 2011). Aspirin is also capable of combating neurodegenerative disorders such as Parkinson’s and Alzheimer’s disease (Choi et al. 2015). Recent research has found that aspirin improves hippocampal plasticity and increases lysosomal activity effectively reducing cerebral plaque load and improving spatial learning and memory (Patel et al. 2018), (Chandra et al. 2018b). We have also demonstrated that low-dose aspirin stimulates dopamine production from dopaminergic neurons (Rangasamy et al. 2018a). Here, we examined whether aspirin could be used to up-regulate the expression of SOCS3 in astrocytes and microglia. Our results demonstrate for the first time that aspirin is capable of elevating the expression of SOCS3 in glial cells via peroxisome proliferator-activated receptor α (PPARα). Moreover, oral aspirin treatment increased SOCS3 in the brain of 5XFAD mice, an animal model of Alzheimer’s disease, via PPARα.
Materials and Methods
Animals
Animal maintaining and experiments were in accordance with National Institute of Health guidelines and were approved (#15–053) by the Institutional Animal Care and Use committee (IACUC) of the Rush University of Medical Center, Chicago, IL. Mice were housed in ventilated micro-isolator cages in an environmentally controlled vivarium (7:00 A.M./7:00P.M. light cycle; temperature maintained at 21–23°C; humidity 35–55%). Animals were provided standard mouse chow and water ad libitum and closely monitored for health and overall well-being daily by veterinary staff and the investigator. This study was not preregistered.
We used 8–10 week old mice from both sexes (n=3 male + n=3 female) for experiments related to Figure 4H. We purchased C57BL/6 mice (RRID: C57BL/6NHsd) from Envigo (Indianapolis, IN) for these experiments. Similarly, 6 month old mice from both sexes (n=3 male + n=3 female) were used for experiments related to Figures 5 and 6. Animals exhibiting mild seizures and tremors were fed and watered through animal feeding needles. However, if any mouse came to the moribund stage, it was decapitated after anesthesia with ketamine/xylazine injectables. Conditions for moribund were as follows: Central nervous system disturbance (Head tilt, Seizures, Tremors, Circling, Spasticity, and Paresis); Inability to remain upright; Evidence of muscle atrophy; Chronic diarrhea or constipation; Rough coat and distended abdomen; Spreading area of alopecia caused by disease; Coughing, rales, wheezing and nasal discharge; Distinct jaundice and/or paleness (anemia); Markedly discolored urine, polyuria or anuria; Frank bleeding from any orifice; Persistent self-induced trauma.
Figure 4. Aspirin increases SOCS3 protein in cultured astrocytes and in vivo in the cortex via PPARα.

WT (A), PPARα (−/−) (B) and PPARβ (−/−) (C) astrocytes were treated with different concentrations of aspirin under serum-free condition for 2 h followed by monitoring the protein level of SOCS3 by Western blot. Actin was run as a loading control. Bands were scanned and values (SOCS3/Actin) presented as relative to control [D, WT; E, PPARα (−/−); F, PPARβ (−/−)]. Results are mean ± SD of three independent cell preparations. *p < 0.05; ***p < 0.001 vs control. Significance of mean between control and aspirin-treated groups was analyzed by a two-tailed paired t-test. WT, PPARα (−/−) and PPARβ (−/−) mice (n=6 in each group; 8–10 week old) were treated with aspirin (2 mg/kg/day) orally for 7 days via gavage (G) followed by monitoring the protein level of SOCS3 in the cortex by Western blot (H). Actin was run as a loading control. Bands were scanned and values (SOCS3/Actin) presented as relative to untreated control in each group (I). Results are mean ± SEM of six mice per group. ***p < 0.001 vs control. Significance of mean between control and aspirin-treated groups was analyzed with a two-tailed paired t-test.
Figure 5. Oral administration of aspirin upregulates SOCS3 in vivo in cortical astrocytes of 5XFAD mice via PPARα.

Six-month old 5XFAD and 5XFAD/PPARα (−/−) mice (n=6 per group) were treated with aspirin (2 mg/kg body wt/d) orally via gavage for 30 d (A) followed by double-labeling of cortical sections for SOCS3 and GFAP (B). Mean Fluorescence Intensity (MFI) of SOCS3 was calculated in two sections (five images per two sections) of each of six mice per group (C). **p < 0.01 vs 5X-Tg control. A two-way ANOVA was performed to test the significance of mean between groups. Treatment (control and aspirin) and genotype (non-Tg, Tg and 5XFAD/PPARα KO) were considered as two independent factors. The descriptive statistics results in F3,20= 19.56 (>Fc =4.65) for genotype and F3,20= 14.18(>Fc =4.65) for treatment. D) SOCS3 positive cells were counted in two sections (five images per two sections) of each of six mice per group. ***p < 0.001 vs 5X-Tg control. Similar two-way ANOVA results in F3,20= 16.33.56 (>Fc =4.65) for genotype and F3,20= 17.17(>Fc =4.65) for treatment.
Figure 6. Oral administration of aspirin upregulates SOCS3 in vivo in cortical microglia of 5XFAD mice via PPARα.

A) Six-month old 5XFAD and 5XFAD/PPARα (−/−) mice (n=6 per group) were treated with aspirin (2 mg/kg body wt/d) via gavage for 30 d followed by double-labeling of cortical sections for SOCS3 and Iba1. GFAP positive (B) and Iba1 positive (C) cells were counted in one section (two images per section) of each of six mice per group. *p < 0.05 vs non-Tg for GFAP; **p < 0.01 vs non-Tg for Iba1. However, a two-way ANOVA with treatment and genotype did not show any significance for either GFAP-positive or Iba1-positive cells across the group since there was not much difference of either GFAP-ir or Iba1-ir cells among 5XFAD, 5XFAD + aspirin, 5XFAD/PPARα (−/−), and 5XFAD/PPARα (−/−) + aspirin-treated groups.
5XFAD [RRID: B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799 Vas/J] and PPARα null (RRID: B6.129S4-Pparatm1Gonz/J) mice were purchased from Jackson Laboratories (Bar Harbor, ME). We received PPARβ null mice from Dr. Frank J. Gonzalez from National Cancer Institute (NIH, Bethesda, MD) (Patel et al. 2018, Roy et al. 2015, Roy et al. 2016). These animals were inbred and subsequent generations were screened by PCR. PPARα null mice (Peters et al. 2000) were maintained as homozygous on the C57BL/6J background. 5XFAD mice mimic some of the features of Alzheimer’s disease including amyloid plaque, neuroinflammation, impairment in memory and learning, etc. (Modi et al. 2015, Patel et al. 2018, Rangasamy et al. 2015). 5XFAD mice were also bred with PPARα (−/−) mice to generate 5XFAD/PPARα (−/−) mice as described before (Corbett et al. 2015, Roy et al. 2015). Following primers were used for genotyping:
APP:
Transgene forward: AGG ACT GAC CAC TCG ACC AG
Transgene reverse: CGG GGG TCT AGT TCT GCA T
Internal positive control forward: CTA GGC CAC AGA ATT GAA AGA TCT
Internal positive control reverse: GTA GGT GGA AAT TCT AGC ATC ATC C
PSEN1:
Transgene forward: AAT AGA GAA CGG CAG GAG CA
Transgene reverse: GCC ATG AGG GCA CTA ATC AT
Internal positive control forward: CTA GGC CAC AGA ATT GAA AGA TCT
Internal positive control reverse: GTA GGT GGA AAT TCT AGC ATC ATC C
PPARα:
Common: GAG AAG TTG CAG GAG GGG ATT GTG
Wild type reverse: CCC ATT TCG GTA GCA GGT AGT CTT
Mutant reverse: GCA ATC CAT CTT GTT CAA TGG C
Treatment of 5XFAD and 5XFAD/PPARα (−/−) mice with aspirin
Age- and sex-matched non-transgenic mice from the same background were used as controls. Six-month old 5XFAD and 5XFAD/PPARα (−/−) mice were used in different treatment groups. Aspirin (RRID: A5376–100G; Sigma, St. Louis, MO) was mixed with 0.5% methyl cellulose (RRID: M1314; Spectrum Chemical, New Brunswick, NJ) and mice were treated with aspirin (2 mg/kg body wt/d) via gavage using gavage needle daily at 10 AM (central time) for 30 days (Fig. 5A). During treatment, no mouse reached the moribund stage (conditions are described above) or died. We also did not use any pain medication during 30 days of aspirin treatment. After 30 d of treatment, mice were anesthetized with ketamine/xylazine injectables followed by perfusion with PBS and paraformaldehyde. During perfusion, animals died.
We employed pseudo randomization for the selection of animals for any group. Here, 6 mice of equal sexes (3 males + 3 females) were used per group and therefore, 30 mice were used for five groups. We did not perform any sample size calculation. Randomization procedure was performed in “QuickCalc” random number generator tool of GraphPad prism software. Animals were identified with the combination of letter and number code as shown below. Random number generator generates the sequence of animals from 1 to 30. 1 = B1, 2 = D1, 3 = A1, 4= C1, 5 = C2, 6 = E1, 7 =B2, 8 = E2, 9 = B3, 10 =A2, 11 = A3, 12 = A4, 13 = D2, 14 = E3, 15 = D3, 16 = D4, 17 = B4, 18 = A5, 19 = E4, 20 = E5, 21 = B5, 22 = A6, 23 = C3, 24 = B6, 25= C4, 26 = E6, 27 = C5, 28 = C6, 29 = D5, 30= D6. A = 5XFAD, B= 5XFAD/PPARα (−/−), C= 5XFAD +Aspirin, D= 5XFAD/PPARα (−/−) +Aspirin, E= Non-Tg. Letter indicates the group number and number indicates the animal number.
Reagents
DMEM/F-12 (RRID: 10–092-CV), Hanks’ balanced salt solution (RRID: 21–022-CM), L-glutamine (RRID: 25–005-CI), 0.05% trypsin (RRID: 25–051-CI), and antibiotic/antimycotic (RRID: 30–004-CI) were obtained from Fisher, Hampton, NH. Fetal bovine serum or FBS (RRID: EF-0500-A) was obtained from Atlas Biologicals (Fort Collins, CO). Molecular Biology grade agarose (RRID: 1613102) was purchased from Bio-Rad (Hercules, CA). Alexa-fluor 488 donkey anti-goat (RRID: 705-546-147), Alexa-fluor 647 donkey anti-rabbit (RRID: 711-605-152), and Alexa-fluor 488 donkey anti-mouse (RRID: 715-545-150) secondary antibodies used in immunostaining were obtained from Jackson ImmunoResearch (West Grove, PA). Sources of primary antibodies used in this study along with their applications and dilutions are listed in Table 1.
Table 1:
Antibodies, sources, applications, and dilutions used in this paper
| Target | Antibody (Clone) | Epitope/Immunogen | Application/Dilution | Source; RRID |
|---|---|---|---|---|
| β-Actin | Mouse monoclonal (AC-15) | a.a. 1–15 of Xenopus laevis β-actin | WB – 1:5000 | Abcam; ab6276 |
| GFAP | Rabbit polyclonal | Synth peptide to cow GFAP | IHC – 1:2000 | Dako; z0334 |
| Iba1 | Goat polyclonal | Synth peptide a.a. 135–147 Human Iba1 (C terminus) |
IHC – 1:500 | Abcam; ab5076 |
| PPARα | Mouse monoclonal (3B6) | Recombinant full-length protein | ChIP – 1:200 | Abcam; ab2779 |
| SOCS3 | Mouse monoclonal | Full length human SOCS3 | WB – 1:3000 IHC – 1:3000 |
Abcam; ab14939 |
WB, Western blot; IHC, Immunohistochemistry; ChIP, Chromatin immunoprecipitation; GFAP, Glial fibrillary acidic protein; PPARα, Peroxisome proliferator-activated receptor alpha
Isolation of Primary Mouse Astroglia
Astroglia were isolated from mixed glial cultures following the procedure of (Giulian & Baker 1986) as described before (Ghosh & Pahan 2012, Khasnavis & Pahan 2012). Briefly, cerebral tissues collected from 2- to 3-d-old mouse pups were homogenized with glass mortar, triturated, passed through mesh, trypsinized, centrifuged, and mixed glial cells plated in DMEM/F-12 containing 10% fetal bovine serum. On day 9, the mixed glial cultures were washed three times with DMEM/F-12 and subjected to a shake at 240 rpm for 2 h at 37°C on a rotary shaker to isolate microglia. Similarly, on day 11, cells were shaken at 180 rpm for 18 h to remove oligodendroglia. By immunofluorescence assay, the attached cells homogeneously expressed glial fibrillary acidic protein (GFAP) (Roy et al. 2015, Ghosh et al. 2015, Modi et al. 2013). These cells were more than 96% pure astrocytes and the percent purity was determined as described by us previously (Jana et al. 2007).
BV-2 microglial cells
Mouse BV-2 microglial cells (kind gift from Virginia Bocchini of University of Perugia) were also maintained in DMEM/F-12 as described by us in many studies (Ghosh et al. 2007, Ghosh et al. 2009, Rangasamy et al. 2018b).
Semi-quantitative RT-PCR Analysis
To remove any contaminating genomic DNA, total RNA was digested with DNase (RRID: 18047019; ThermoFisher Scientific). Semi-quantitative RT-PCR was carried out as described earlier (Jana et al. 2013, Mondal et al. 2009, Brahmachari & Pahan 2007). Briefly, 1 μg of total RNA was reverse transcribed using oligo(dT)12–18 (RRID: 18418012; ThermoFisher Scientific) as primer and Promega MMLV reverse transcriptase (RRID: PR-M1701; ThermoFisher Scientific) in a 20 μL reaction mixture as follows: cDNA synthesis at 50°C for 30 min; reverse transcriptase inactivation at 95°C for 10 min. The resulting cDNA was appropriately-diluted, and diluted cDNA was amplified for 38 cycles (denaturation of templates at 95°C for 30 s, annealing of primers at 52°C for 45 s, initial extension of primers at 72°C for 2 min, and final extension of primers at 72°C for 10 min) using Promega PCR master mix (RRID: PRM7505; ThermoFisher Scientific) and following primers.
Socs3 (mouse):
Sense 5′-CGCCTCAAGACCTTCAGCTC-3′
Antisense 5′-CTGATCCAGGAACTCCCGAA-3′
Gapdh (mouse):
Sense 5′-GGTGAAGGTCGGTGTGAACG-3′
Antisense 5′-TTGGCTCCACCCTTCAAGTG-3′
Amplified products were electrophoresed on a 1.5% agarose gels and visualized by ethidium bromide staining.
Real-time PCR Analysis
It was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems) as described earlier (Chandra et al. 2018a, Khasnavis & Pahan 2012). The mRNA expressions of respective genes were normalized to the level of GAPDH mRNA. Data were processed by the ABI Sequence Detection System 1.6 software and analyzed by analysis of variance.
Western blotting
Western blotting was conducted as described before with modifications (Corbett et al. 2012, Modi et al. 2016). Briefly, cells were scraped in 1X RIPA buffer, protein was measured using Bradford reagent and sodium dodecyl sulfate (SDS) buffer was added and electrophoresed on NuPAGE® Novex® 4–12% Bis-Tris gels (Life Technologies, Grand Island, NY) and proteins transferred onto a nitrocellulose membrane (Bio-Rad) using the Thermo-Pierce Fast Semi-Dry Blotter. The membrane was then washed for 15 min in TBS plus Tween 20 (TBST) and blocked for 1 h in TBST containing BSA. Next, membranes were incubated overnight at 4°C under shaking conditions with primary antibodies (Table 1). The next day, membranes were washed in TBST for 1 h, incubated in secondary antibodies (Donkey anti-mouse 700, Licor, RRID: 926–68022, Dilution# 1:10,000; Donkey anti-mouse 800, Licor, RRID: 926–32212, Dilution# 1:10,000) for 1 h at room temperature, washed for one more hour and visualized under the Odyssey® Infrared Imaging System (Li-COR, Lincoln, NE).
Immunofluorescence Analysis
Analysis was performed as described (Brahmachari et al. 2009, Ghosh & Pahan 2012). Briefly, coverslips containing 100–200 cells/mm2 were fixed with (100%) methanol (RRID: A412–500; ThermoFisher Scientific) followed by treatment with cold ethanol and two rinses in phosphate-buffered saline (PBS). Samples were blocked with 3% bovine serum albumin (BSA) (RRID: A9418–10G; Sigma, St. Louis, MO) in PBS/Tween 20 (PBST) for 30 min and incubated in PBST containing 1% BSA, anti-SOCS3 and anti-GFAP antibodies (Table 1). After three washes in PBST (15 min each), slides were further incubated with Alexa Fluor 488- or Alexa Fluor 647-conjugated AffinityPure Donkey secondary antibodies (Jackson ImmunoResearch) for 2 hr at room temperature. Then the samples were washed with PBST for three times, mounted and observed under an Olympus BX-41 fluorescence microscope. Immunostaining of tissue was performed by fixing the brains in 4% paraformaldehyde followed by 30% sucrose. Tissue was sectioned every 40 microns on a Leica Crysotat CM3050 S and kept in cryoprotectant. DAPI was added at the final step of washing of secondary antibodies for staining nuclei.
Cell counting
Counting analysis was performed using the olympus microsuite V software with the help of touch counting module. After acquiring images under 20 X objective lens, images were further analyzed as follows. Before counting cells, entire Image area was calibrated with the help of a rectangular box available in the touch counting panel. Once the area of the image was measured, touch counting program was applied to count number of fluorescent signals. Next, the total number of signals in a given area was divided by the total area of the image and presented as number of cells per square millimeter unit.
Chromatin Immunoprecipitation (ChIP)
Recruitment of PPRE to the Socs3 gene promoter was determined by using the ChIP assay as described (Ghosh et al. 2012). Briefly, 1 × 106 astrocytes were treated with aspirin, and after 30 min of stimulation, cells were fixed, and cross-linked adducts were resuspended and sonicated. ChIP was performed on the cell lysate by overnight incubation at 4 °C with 2 μg of antibodies against PPARα, PPARβ, PPARγ, RNA Polymerase II, IgG, and p300 followed by overnight incubation with protein G-agarose. The beads were washed and incubated with elution buffer. Reversing the cross-linkages and purifying the DNA, precipitates were incubated in a 65 °C incubator overnight and digested with proteinase K. DNA samples were then purified and precipitated, and precipitates were washed with 75% ethanol, air-dried, and resuspended in Tris/EDTA buffer. The following primers were used to amplify the Socs3 promoter region spanning proximal PPRE:
Sense 5′- TGG GCG TCC TCC TAG GGC GGG CAG −3′
Antisense 5′- GCG CGT GAG TGC CCG GAA CCC TCC A −3′
On the other hand, to amplify the region spanning distal PPRE, the following primers were used:
Sense 5′- GCT CCC CCA CCG CGC CGA GAG TA −3′
Antisense 5′- AGC TAC CTG GCC TCA GCT CAT CC −3′
PCR products were electrophoresed on 2% agarose gels.
Blinding and Statistical analysis
All Western blot and immunohistochemical analyses were performed in a blinded manner. Laboratory personnel only knew about ID of each sample. For animal experiments, six mice were included in each group. All cell culture experiments were repeated at least three times. There is no restriction on the availability of data. The D’Agostino-Pearson test (GraphPad Prism 7 software) was used for normality. Statistical analyses were performed with one-way ANOVA with dose or time as a single factor. Descriptive statistics of one-way ANOVA was included in figure legends with pertinent F value, F critical (Fc) value, and degrees of freedom within a group and between groups. While analyzing the significance between two groups, a paired t-test was performed. All statistical analyses were performed using GraphPad Prism 7 software. Two-way ANOVA followed by Tukey’s posthoc analyses were performed for in vivo experiments with treatment and genotype as two effectors. Data represented as mean±SD or mean±SEM as stated in figure legends. A level of p<0.05 was considered statistically significant. No test for outliers was conducted on the data. The sample size was similar to those reported in previous publications (Corbett et al. 2015, Rangasamy et al. 2015) and is mentioned in figure legends.
Results
Aspirin upregulates SOCS3 in primary mouse astrocytes and BV-2 microglial cells:
Our first step was to determine if SOCS3 is upregulated by aspirin. To do this, primary mouse astrocytes were cultured from C57BL/6 mouse pups. The astrocytes were then treated with increasing dosages (1, 2, 5, & 10 μM) of aspirin (Fig. 1A-B). It was found that aspirin upregulates Socs3 mRNA in a dose-dependent manner with maximum increase in Socs3 mRNA observed at 5 μM of aspirin (Fig. 1A-B). Next, we performed a time course study and found that aspirin induced the upregulation of Socs3 mRNA in a time-dependent manner peaking at 2 h and then receding (Fig. 1C-D). Next, we evaluated SOCS3 protein expression by western blotting (Fig. 1E-F) and found that SOCS3 protein levels also increased in a time-dependent manner after aspirin treatment. SOCS3 protein levels increased beginning at 2 h post-treatment with the highest increase seen at 4 h, which then gradually decreased over time (Fig. 1E-F). To confirm these finding from another angle, we also performed immunofluorescence analysis and found increase in SOCS3 expression in GFAP-positive astrocytes by aspirin treatment (Fig. 1G).
Figure 1. Aspirin upregulates SOCS3 mRNA and protein in primary mouse astrocytes.

Astrocytes were treated with different concentrations of aspirin for 2 h under serum-free condition followed by monitoring the mRNA expression of Socs3 by semi-quantitative RT-PCR (A) and real-time PCR (B). One-way ANOVA was adopted to test the significance of mean between groups with dose as a single factor. The resultant statistics is F4, 10 =8.8235 (>Fc =3.47); p<0.005 (=0.002). A paired t-test to analyze the significance of mean between two groups provides ***p= 0.004675(<0.005) and 0.00315 (<0.005) when comparing control with aspirin 1 and 2 μM, respectively. Moreover, 5 μM aspirin showed higher statistical significance with respect to control as evident from p =0.000073 (<0.001). A paired t-test analysis to compare between 2 and 5 μM aspirin showed strong significance with p=0.0135 (<0.05). Cells were treated with 5 μM aspirin under serum-free condition for different time points followed by monitoring the mRNA expression of Socs3 by semi-quantitative RT-PCR (C) and real-time PCR (D). Similarly, ANOVA with time as a single factor results F6, 11 =25.5 (>Fc =3.09); p<0.001(=7.6 *10−6). ***p= 0.0028 (<0.05) when compared control with aspirin 30 min. The protein expression of SOCS3 was examined by Western blot (E). Actin was run as a loading control. Bands were scanned and values (SOCS3/Actin) presented as relative to control (F). A two-tailed paired t-test was performed to test the significance of mean between control and aspirin-treated groups. Results are mean ± SD of three independent cell preparations. **p < 0.01 and ***p < 0.001 vs control. Cells were treated with 5 μM aspirin under serum-free condition for 4 h followed by double-label immunofluorescence for GFAP and SOCS3 (G). Results represent three independent cell preparations.
To understand whether aspirin-mediated effect was specific to astrocytes or not, we also examined the effect of aspirin on SOCS3 level in microglial cells. Similar to astrocytes, aspirin also increased the mRNA expression of Socs3 in BV-2 microglial cells in a dose-dependent manner (Fig. 2A-B). However, in contrast to astrocytes, aspirin exhibited maximum induction of Socs3 mRNA at a dose of 2 μM (Fig. 2A-B). Similarly aspirin treatment also upregulated the protein level of SOCS3 in microglial cells and the maximum induction was seen at 2 μM concentration (Fig. 2C-D). Time-dependent studies indicated maximum upregulation of SOCS3 at 2 or 4 h of aspirin treatment (Fig. 2E-F). The induction of SOCS3 in microglial cells by aspirin was also confirmed by double-label immunofluorescence analysis of Iba1 and SOCS3 (Fig. 2G). Together, these results indicate that aspirin increases the mRNA and protein expression of SOCS3 in astrocytes and microglia.
Figure 2. Upregulation of SOCS3 mRNA and protein by aspirin mouse BV-2 microglial cells.

Cells were treated with different concentrations of aspirin for 2 h under serum-free condition followed by monitoring the mRNA expression of SOCS3 by semi-quantitative RT-PCR (A) and real-time PCR (B). To test the significance between groups, we performed one-way ANOVA with treatment as a single factor. The ANOVA analysis results in F4, 10 = 9.29 (>Fc=3.47); p<0.001 (=0.00026). While performing a paired two-tailed t-test between control and 1, 2 and 5μM aspirin, we observed a significant difference of mean with absolute p value 0.000625, 0.000129 and 0.000871 respectively with overall ***p< 0.001 vs. control. The protein expression of SOCS3 was examined by Western blot (C). Actin was run as a loading control. Bands were scanned and values (SOCS3/Actin) presented as relative to control (D). Results are mean ± SD of three independent cell preparations. One-way ANOVA with treatment as a single factor results in F4, 10 = 6.47 (>Fc=3.47); p<0.001 (=0.00098). A pair-wise t-test analysis results in **p<0.005 (=0.00247) and *p<0.05 (=0.0078) vs. control. Cells were treated with 2 μM aspirin under serum-free condition for different time points followed by monitoring the protein expression of SOCS3 by Western blot (E). Actin was run as a loading control. Bands were scanned and values (SOCS3/Actin) presented as relative to control (F). One-way ANOVA with the time of aspirin treatment as an independent factor results in F5,18 = 17.57 (>Fc= 3.57); p <0.01 (=0.0085). While performing a pair-wise t-test to analyze the significance between groups, we found that *p<0.05 (= 0.0074) 1 h vs. 0 h, **p<0.01 (= 0.000914) 2 h vs. 0 h and **p<0.01 (=0.0067) 4 h vs. 0 h. Cells were treated with 2 μM aspirin under serum-free condition for 4 h followed by double-label immunofluorescence for Iba1 and SOCS3 (G). Results represent three independent cell preparations.
Aspirin increases SOCS3 in primary mouse astrocytes via PPARα:
The next step was to determine the mechanism by which SOCS3 is upregulated by aspirin. Recently we have delineated that aspirin binds to the ligand-binding domain of PPARα (Patel et al. 2018) and that aspirin reduces amyloid plaques from the hippocampus of 5XFAD mice via PPARα (Chandra et al. 2018b). Therefore, the genomic database was searched for the presence of any putative peroxisome proliferator-response element (PPRE) in the promoter of Socs3 gene. It was found that the Socs3 gene promoter harbors two PPRE’s (Fig. 3A). The two PPRE’s are located at −1723 to −1710 bp and −200 to −171 bp upstream of the Socs3 gene coding region, suggesting that one of the three PPARs (PPARα, β/δ and γ) could be involved in aspirin-mediated stimulation of the Socs3 gene. Therefore, the next step was to examine whether or not and which PPAR’s are involved in SOCS3 upregulation. Primary astrocytes isolated from C57BL/6 (WT), PPARα (−/−) and PPARβ (−/−) mice were treated with different concentrations of aspirin for 2 h. It is clearly evident from RT-PCR (Fig. 3B, D & F) and real-time PCR (Fig. 3C, E & G) analyses that aspirin upregulated Socs3 mRNA in WT and PPARβ (−/−), but not PPARα (−/−), astrocytes (Fig. 3B-G). The relative expression of Socs3 was significant at the p < 0.001 level in the C57BL/6 and PPARβ (−/−) astrocytes but we did not observe any change in Socs3 mRNA expression in PPARα (−/−) astrocytes after aspirin treatment. To confirm this finding further, we monitored the expression of SOCS3 protein. Similar to mRNA expression, aspirin treatment upregulated the protein level of SOCS3 in WT (Fig. 4A-B) and PPARβ (−/−) (Fig. 4E-F), but not PPARα (−/−) (Fig. 4C-D), astrocytes. Next, we investigated the role of PPARγ in aspirin-mediated upregulation of SOCS3. We pretreated WT astrocytes with the PPARγ-specific inhibitor GW9662, followed by treatment with aspirin, and determined the mRNA expression of Socs3. Aspirin was able to upregulate SOCS3 levels even in the presence of GW9662 (Fig. 3H-I), suggesting that PPARγ is not required by aspirin for the upregulation of SOCS3. Together, these results indicate that aspirin upregulates SOCS3 via PPARα, but neither PPARβ nor PPARγ.
Figure 3. Involvement of PPARα in the expression of SOCS3.

(A) Map of the Socs3 gene promoter shows the presence of two PPREs at −1723 to −1710 bp (distal PPRE) and −200 to −171 bp (proximal PPRE). Astrocytes isolated from WT (B-C), PPARα (−/−) (D-E) and PPARβ (−/−) (F-G) mice were treated with different concentrations of aspirin for 2 h followed by monitoring the mRNA expression of Socs3 by semi-quantitative RT-PCR (B, D & F) and real-time PCR (C, E & G). Results are mean ± SD of three independent cell preparations. ***p < 0.001 vs control. WT astrocytes preincubated with different concentrations of GW9662 for 1 h were stimulated with aspirin (5 μM). After 2 h of stimulation, the protein level of SOCS3 was monitored by Western blot (H). Actin was run as a loading control. Bands were scanned and values (SOCS3/Actin) presented as relative to control (I). ***p < 0.001 vs control; ns, not significant. Significance of mean between control and aspirin-treated groups was analyzed by a two-tailed paired t-test.
Oral administration of aspirin upregulates SOCS3 in vivo in cortex of wild type and PPARβ (−/−), but not PPARα (−/−), mice:
After having established that aspirin increases levels of SOCS3 mRNA and protein in cultured brain cells via PPARα, it is imperative to determine if the same effect is seen in vivo in the brain. Therefore, WT, PPARα (−/−) and PPARβ (−/−) mice were treated with 2 mg/kg/d of aspirin for 7 d. Figure 4G shows a flow-chart of the study design. The WT C57BL/6 mice receiving aspirin exhibited significant increase in SOCS3 protein as compared to control animals. Similar to cultured astrocytes, we did not observe any increase in SOCS3 protein in vivo in the cortex of PPARα (−/−) mice (Fig. 4H-I). On the other hand, similar to WT mice, aspirin treatment increased the level of SOCS3 in the cortex of PPARβ (−/−) mice (Fig. 4H-I). Again, the increase between aspirin treated and untreated PPARβ (−/−) mice was significant at the p <0.001 level. Therefore, aspirin treatment also increases SOCS3 in vivo in the brain via PPARα.
Oral administration of aspirin upregulates SOCS3 in vivo in the cortex of 5XFAD mice via PPARα:
Since SOCS3 protein levels were increased after aspirin treatment in C57BL/6 mice, next we examined if aspirin treatment upregulates SOCS3 in 5XFAD mice, animals that are impacted by the amyloid β pathology seen in Alzheimer’s disease. Since aspirin increases SOCS3 via PPARα, together with 5XFAD mice, we also used 5XFAD/PPARα (−/−) mice that lack PPARα. Figure 5A shows a graphical time-line of the experimental procedure. After one month of aspirin treatment, cortical sections of 5XFAD, and 5XFAD/PPARα (−/−) mice were immunostained for SOCS3. The aspirin-fed group of 5XFAD mice had increased levels of SOCS3 immunoreactivity as compared to control 5XFAD mice (Fig. 5B). Aspirin-induced SOCS3 was present in both GFAP-positive astrocytes (Fig. 5) and Iba-1-positive microglia (Fig. 6). Mean fluorescence intensity (MFI) was performed to quantify the level of immuno-stained SOCS3 (Fig. 5C). The MFI values difference between the 5XFAD and aspirin-treated 5XFAD groups was significant at p < 0.01. Counting of SOCS3 positive cells (Fig. 5D) also showed that SOCS3 levels were significantly upregulated in the cortex of 5XFAD mice (p< 0.001). On the other hand, aspirin treatment remained unable to increase SOCS3 expression in the cortex of 5XFAD/PPARα (−/−) mice (Fig. 5A-D). This further supports our hypothesis that aspirin induces SOCS3 via the PPARα pathway.
Consistent with examinations of Alzheimer’s patients and associated mouse models, we observed increased number of astrocytes (Figs. 5B & 6B) and microglia (Fig. 6A & 6C) in the cortex of 5XFAD mice as compared to age-matched non-transgenic mice. However, we did not find any significant difference in either GFAP-positive astrocytes (Figs. 5A & 6B) or Iba-1-positive microglia (Fig. 6A & 6C) between 5XFAD and 5XFAD/PPARα (−/−) groups, suggesting that astrogliosis and microgliosis in 5XFAD mice may not be directly dependent on PPARα. Although aspirin treatment stimulated the level of SOCS3 in the cortex of 5XFAD mice, aspirin remained unable to modulate either GFAP-positive astrocytes (Figs. 5B & 6B) or Iba-1-positive microglia (Fig. 6A & 6C) in the CNS of 5XFAD mice. These results suggest that once astrogliosis and microgliosis occur in the CNS of 5XFAD mice, aspirin cannot reduce the number of astroglia and microglia. However, aspirin treatment can increase the expression of anti-inflammatory molecule like SOCS3 in astroglia and microglia.
Aspirin treatment induces the recruitment of PPARα to the Socs3 gene promoter:
Since aspirin upregulated SOCS3 in cultured cells as well as in vivo in the brain via PPARα, next to understand whether PPARα was directly involved in aspirin-mediated transcription of Socs3 gene, we examined the recruitment of PPARα to the Socs3 gene promoter by ChIP assay. Using Mat-Inspector V2.2 search machinery, we found two consensus PPREs in the Socs3 promoter (Fig. 7A). Therefore, we wanted to determine the role of these two PPREs in aspirin-mediated transcription of Socs3. After immunoprecipitation of aspirin-treated astrocytic chromatin fragments using antibodies against PPARα, we were able to amplify 187 bp fragment from the Socs3 promoter (p < 0.001 vs. control) corresponding to the proximal PPRE (Fig. 7B-C). In contrast, we did not find any amplification for either PPARβ or PPARγ (Fig. 7B-C), indicating that aspirin induces the recruitment of PPARα, but neither PPARβ nor PPARγ, to the proximal PPRE of the Socs3 gene promoter. Consistent to the recruitment of PPARα to the proximal PPRE, aspirin was able to recruit RNA polymerase (p < 0.001 vs. control) to the proximal PPRE (Fig. 7B-C. We did not find any amplification with control IgG, indicating the specificity. Next, we examined if aspirin could also induce the recruitment of PPARα to distal PPRE. However, aspirin remained unable to recruit PPARα, PPARβ, PPARγ, or RNA polymerase to the distal PPRE (Fig. 7D-E), suggesting that distal PPRE is not involved aspirin-mediated transcription of the Socs3 gene.
Figure 7. Aspirin induces the recruitment of PPARα to the Socs3 gene promotor.

A) MAP of the proximal PPRE in the Socs3 gene promoter and the region to be amplified for ChIP assay. Primary astrocytes isolated from WT mice were treated with different concentrations of aspirin for 1 h under serum-free condition followed by monitoring the recruitment of PPARα, PPARβ PPARγ, and RNA Polymerase II to the proximal PPRE by ChIP assay (B, PCR; C, real-time PCR). Results are mean ± SD of three independent cell preparations. ***p < 0.001 vs control. Significance of mean between control and aspirin-treated cells was analyzed by a two-tailed paired t-test. D) MAP of the distal PPRE in the Socs3 gene promoter and the region to be amplified for ChIP assay after aspirin treatment. ChIP assay was performed to monitor the recruitment of PPARα, PPARβ PPARγ, and RNA Polymerase II to the distal PPRE (E, PCR). We did not find any amplification.
Discussion
Although glial inflammation has beneficial functions through scavenging of cellular debris in the CNS and recovery of injured CNS by actively monitoring and controlling the extracellular water, pH, and ion homeostasis, once microglia and astrocytes become activated in a diseased brain, sometimes neuroinflammation goes beyond control, and eventually detrimental effects of chronic neuroinflammation override its beneficial effects (Amor et al. 2014). Upon activation, microglia release cytokines and chemokines such as IL-1β, TNFα among others (Smith et al. 2012). It has been shown that microglial IL-1β plays a vital role in activating astrocytes, the major cell type in the CNS, particularly in human (Jana et al. 2005) that in turn produce a number of proinflammatory molecules including nitric oxide and reactive oxygen species (Saha & Pahan 2006). Additionally there is increased infiltration of peripheral monocytes and macrophages that exacerbate the inflammatory state (Varvel et al. 2016). Fortunately, to take care of different inflammatory insults, we have been endowed with SOCS3, which inhibits cytokine signaling in astrocytes, microglia as well as peripheral immune cells such as macrophages (Cao et al. 2018). Therefore, upregulation of SOCS3 is an important area of research as it exhibits a protective response against neuroinflammatory insults. However, drugs and mechanisms involved in the upregulation of SOCS3 are poorly understood.
Salicylic acid has been used for millennia as a treatment for pain and its modern formulary of aspirin has widespread availability and utility for several ailments beyond pain management. Here, we demonstrate that aspirin is capable of upregulating SOCS3 mRNA and protein in cultured brain cells. Although aspirin is a widely-used drug, it exhibits some side effects that are more prominent at higher doses. Therefore, it is important to see that aspirin was more potent in increasing SOCS3 in both astrocytes and microglia at lower doses. While many molecules show therapeutic effect in cell culture models, very few demonstrate efficacy in vivo in the CNS. It has been shown that after peripheral administration, aspirin crosses the blood-brain barrier and enters into the brain (Vasovic et al. 2008). Consistently, data presented in this manuscript clearly establish that oral aspirin is effective in upregulating SOCS3 in vivo in the brain of normal mice as well as mice with Alzheimer’s pathology. Aspirin is already an established anti-inflammatory drug to attenuate pain, fever, etc. The classical mode of action of aspirin is to suppress the production of prostaglandins and thromboxanes via irreversible inactivation of the cyclooxygenase pathway (Mustard et al. 1983). Later on, it has been found that aspirin also inhibits the activation of proinflammatory transcription factor NF-κB via phosphorylation and proteolytic degradation of IκBα, a physiological inhibitor of classical NF-κB heterodimer (Kopp & Ghosh 1994). Here, we delineate a new mode of anti-inflammatory function of aspirin via SOCS3 upregulation. While most of the proinflammatory cytokines function through NF-κB activation, some proinflammatory molecules employ different signaling pathways. One such molecule is T cell-derived cytokine IFNγ, which functions through JAK-STAT pathway. By antagonizing JAK-STAT pathway, SOCS3 is known to suppress IFNγ signaling (Yoshimura et al. 2018). Moreover, SOCS3 is capable of inhibiting inflammatory signaling transduced by IL-12 through possible interaction with IL-12Rβ1/IL-12Rβ2 pathway (Baetz et al. 2004). Therefore, by increasing SOCS3-mediated anti-inflammatory signaling, low-dose aspirin should be able to attenuate neuroinflammatory responses observed in neurodegenerative and demyelinating disorders.
Mechanisms by which Socs3 gene transcription occurs are poorly understood. Peroxisome proliferator-activated receptor (PPAR) α is a transcription factor that regulates fatty acid metabolism. Although liver is the major source of PPARα, recently we have seen that PPARα is constitutively expressed in different brain regions including hippocampus and cortex (Corbett et al. 2015, Roy et al. 2013, Roy et al. 2015, Roy et al. 2016). Several lines of evidence presented in this manuscript clearly suggest that aspirin upregulates the transcription of SOCS3 via PPARα. First, promoter of SOCS3 gene harbors two consensus PPREs. Second, aspirin increases the expression of SOCS3 in astrocytes isolated from WT and PPARβ (−/−), but not PPARα (−/−), mice. Accordingly, oral aspirin upregulates SOCS3 in the CNS of WT and PPARβ (−/−), but not PPARα (−/−), mice. Third, PPARγ-specific antagonist GW9662 remained unable to abrogate aspirin-mediated upregulation of SOCS3. Fourth, oral aspirin treatment upregulated the expression of SOCS3 in vivo in the cortex of 5XFAD, but not 5XFAD/PPARα (−/−), mice. Fifth, aspirin induced the recruitment of PPARα, but neither PPARβ nor PPARγ, to the PPRE of the Socs3 gene promoter in astrocytes. Although there are two PPREs, aspirin engages the one near the transcriptional start site to initiate the transcription of Socs3. This is the first demonstration that PPARα participates in the transcription of Socs3 gene. One study (Wang et al. 2014) has shown that aspirin-triggered Lipoxin A4 further promotes the increased expression of Socs3 mRNA in the spinal cord induced by chronic constriction injury CCI surgery via inhibition of JAK2/STAT3 signaling. However, we have seen that aspirin rapidly increased the expression of SOCS3 in glial cells via direct transcription by PPARα.
How does aspirin activate PPARα? Although aspirin has been being used for more than a century, there was no known receptor for aspirin. Recently we have seen that after aspirin treatment, PPARα rapidly moves from cytoplasm to the nucleus (Chandra et al. 2018b). Moreover, aspirin directly binds to the ligand-binding domain (LBD) of PPARα to cause its activation (Patel et al. 2018). Interestingly, aspirin serves as a ligand of PPARα through binding to the tyrosine 314 residue (Y314) of PPARα LBD (Patel et al. 2018). Aspirin-mediated activation of PPARα was abrogated by mutation of tyrosine 314 to valine (Patel et al. 2018). Therefore, it is possible that aspirin binds to PPARα LBD to activate PPARα and upregulate SOCS3 in brain cells.
In summary, here, we have demonstrated that low-dose aspirin can increase SOCS3 in cultured brain cells and in vivo in the brain via PPARα. Since SOCS3 is a potent anti-inflammatory molecule, our results suggest that low-dose aspirin may be considered to stimulate anti-inflammation in the CNS of patients with neuroinflammatory and neurodegenerative disorders.
Acknowledgements
This study was supported by a merit award from Veteran Affairs (I01BX002174), the Zenith Fellows Award (ZEN-17–438829) from Alzheimer’s Association and a grant (AG050431) from NIH.
Abbreviations:
- SOCS
Suppressor of cytokine signaling
- PPARα
Peroxisome proliferator-activated receptor alpha
- PPRE
Peroxisome proliferator response element
- ChIP
Chromatin immunoprecipitation
- JAK
Janus kinase
- Signal transducer and activator of transcription
- SDS
Sodium dodecyl sulfate
- TBS
Tris-buffered saline
- TBS
TTBS plus Tween 20
Footnotes
Conflict of interest disclosure: None
References
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D and van Noort JM (2014) Inflammation in neurodegenerative diseases--an update. Immunology, 142, 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetz A, Frey M, Heeg K and Dalpke AH (2004) Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem, 279, 54708–54715. [DOI] [PubMed] [Google Scholar]
- Brahmachari S, Jana A and Pahan K (2009) Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. J Immunol, 183, 5917–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmachari S and Pahan K (2007) Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol, 179, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Wang Z and Wan W (2018) Suppressor of Cytokine Signaling 3: Emerging Role Linking Central Insulin Resistance and Alzheimer’s Disease. Front Neurosci, 12, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carow B and Rottenberg ME (2014) SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol, 5, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra G, Kundu M, Rangasamy SB, Dasarathy S, Ghosh S, Watson R and Pahan K (2018a) Increase in Mitochondrial Biogenesis in Neuronal Cells by RNS60, a Physically-Modified Saline, via Phosphatidylinositol 3-Kinase-Mediated Upregulation of PGC1alpha. J Neuroimmune Pharmacol, 13, 143–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Jana M and Pahan K (2018b) Aspirin Induces Lysosomal Biogenesis and Attenuates Amyloid Plaque Pathology in a Mouse Model of Alzheimer’s Disease via PPARalpha. J Neurosci, 38, 6682–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Zhang X and Huang WJ (2016) Role of neuroinflammation in neurodegenerative diseases (Review). Mol Med Rep, 13, 3391–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HW, Tian M, Manohar M, Harraz MM, Park SW, Schroeder FC, Snyder SH and Klessig DF (2015) Human GAPDH Is a Target of Aspirin’s Primary Metabolite Salicylic Acid and Its Derivatives. PLoS One, 10, e0143447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianciulli A, Calvello R, Porro C, Trotta T and Panaro MA (2017) Understanding the role of SOCS signaling in neurodegenerative diseases: Current and emerging concepts. Cytokine Growth Factor Rev, 37, 67–79. [DOI] [PubMed] [Google Scholar]
- Corbett GT, Gonzalez FJ and Pahan K (2015) Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci U S A, 112, 8445–8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett GT, Roy A and Pahan K (2012) Gemfibrozil, a lipid-lowering drug, upregulates IL-1 receptor antagonist in mouse cortical neurons: implications for neuronal self-defense. J Immunol, 189, 1002–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Corbett GT, Gonzalez FJ and Pahan K (2012) Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor alpha: implications for late infantile Batten disease therapy. J Biol Chem, 287, 38922–38935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Jana M, Modi K, Gonzalez FJ, Sims KB, Berry-Kravis E and Pahan K (2015) Activation of peroxisome proliferator-activated receptor alpha induces lysosomal biogenesis in brain cells: implications for lysosomal storage disorders. J Biol Chem, 290, 10309–10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A and Pahan K (2012) Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders. J Biol Chem, 287, 27189–27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Liu X et al. (2007) Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A, 104, 18754–18759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE and Pahan K (2009) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci, 29, 13543–13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D and Baker TJ (1986) Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci, 6, 2163–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT and Feinstein DL (2001) Expression and function of inducible nitric oxide synthase in neurons. J Neuroimmunol, 114, 8–18. [DOI] [PubMed] [Google Scholar]
- Jana A, Modi KK, Roy A, Anderson JA, van Breemen RB and Pahan K (2013) Up-regulation of neurotrophic factors by cinnamon and its metabolite sodium benzoate: therapeutic implications for neurodegenerative disorders. J Neuroimmune Pharmacol, 8, 739–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana M, Anderson JA, Saha RN, Liu X and Pahan K (2005) Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med, 38, 655–664. [DOI] [PubMed] [Google Scholar]
- Jana M, Jana A, Pal U and Pahan K (2007) A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem Res, 32, 2015–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janelsins MC, Mastrangelo MA, Park KM et al. (2008) Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol, 173, 1768–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Jeon S and Suk K (2012) Glia as a Link between Neuroinflammation and Neuropathic Pain. Immune Netw, 12, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M and Verkhratsky A (2011) Physiology of microglia. Physiol Rev, 91, 461–553. [DOI] [PubMed] [Google Scholar]
- Khasnavis S and Pahan K (2012) Sodium benzoate, a metabolite of cinnamon and a food additive, upregulates neuroprotective Parkinson disease protein DJ-1 in astrocytes and neurons. J Neuroimmune Pharmacol, 7, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E and Ghosh S (1994) Inhibition of NF-kappa B by sodium salicylate and aspirin. Science, 265, 956–959. [DOI] [PubMed] [Google Scholar]
- Modi KK, Rangasamy SB, Dasarathi S, Roy A and Pahan K (2016) Cinnamon Converts Poor Learning Mice to Good Learners: Implications for Memory Improvement. J Neuroimmune Pharmacol, 11, 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi KK, Roy A, Brahmachari S, Rangasamy SB and Pahan K (2015) Cinnamon and Its Metabolite Sodium Benzoate Attenuate the Activation of p21rac and Protect Memory and Learning in an Animal Model of Alzheimer’s Disease. PLoS One, 10, e0130398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi KK, Sendtner M and Pahan K (2013) Up-regulation of ciliary neurotrophic factor in astrocytes by aspirin: implications for remyelination in multiple sclerosis. J Biol Chem, 288, 18533–18545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, Roy A and Pahan K (2009) Functional blocking monoclonal antibodies against IL-12p40 homodimer inhibit adoptive transfer of experimental allergic encephalomyelitis. J Immunol, 182, 5013–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustard JF, Kinlough-Rathbone RL and Packham MA (1983) Aspirin in the treatment of cardiovascular disease: a review. Am J Med, 74, 43–49. [DOI] [PubMed] [Google Scholar]
- Patel D, Roy A, Kundu M, Jana M, Luan CH, Gonzalez FJ and Pahan K (2018) Aspirin binds to PPARalpha to stimulate hippocampal plasticity and protect memory. Proc Natl Acad Sci U S A, 115, E7408–E7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD and Gonzalez FJ (2000) Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta). Mol Cell Biol, 20, 5119–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy SB, Corbett GT, Roy A, Modi KK, Bennett DA, Mufson EJ, Ghosh S and Pahan K (2015) Intranasal Delivery of NEMO-Binding Domain Peptide Prevents Memory Loss in a Mouse Model of Alzheimer’s Disease. J Alzheimers Dis, 47, 385–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy SB, Dasarathi S, Pahan P, Jana M and Pahan K (2018a) Low-Dose Aspirin Upregulates Tyrosine Hydroxylase and Increases Dopamine Production in Dopaminergic Neurons: Implications for Parkinson’s Disease. J Neuroimmune Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy SB, Jana M, Roy A et al. (2018b) Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J Clin Invest, 128, 4297–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart WH (1999) [Aspirin]. Ther Umsch, 56, 713–717. [DOI] [PubMed] [Google Scholar]
- Roy A, Jana M, Corbett GT, Ramaswamy S, Kordower JH, Gonzalez FJ and Pahan K (2013) Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor alpha. Cell Rep, 4, 724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Jana M, Kundu M, Corbett GT, Rangaswamy SB, Mishra RK, Luan CH, Gonzalez FJ and Pahan K (2015) HMG-CoA Reductase Inhibitors Bind to PPARalpha to Upregulate Neurotrophin Expression in the Brain and Improve Memory in Mice. Cell Metab, 22, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kundu M, Jana M, Mishra RK, Yung Y, Luan CH, Gonzalez FJ and Pahan K (2016) Identification and characterization of PPARalpha ligands in the hippocampus. Nat Chem Biol, 12, 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN and Pahan K (2006) Signals for the induction of nitric oxide synthase in astrocytes. Neurochem Int, 49, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Das A, Ray SK and Banik NL (2012) Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull, 87, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV and Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol, 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Neher JJ, Bosch A, Wang W, Ransohoff RM, Miller RJ and Dingledine R (2016) Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A, 113, E5665–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasovic V, Banic B, Jakovljevic V, Tomic Z and Milic-Djordjevic V (2008) Effect of aminophylline on aspirin penetration into the central nervous system in rats. Eur J Drug Metab Pharmacokinet, 33, 23–30. [DOI] [PubMed] [Google Scholar]
- Wang ZF, Li Q, Liu SB et al. (2014) Aspirin-triggered Lipoxin A4 attenuates mechanical allodynia in association with inhibiting spinal JAK2/STAT3 signaling in neuropathic pain in rats. Neuroscience, 273, 65–78. [DOI] [PubMed] [Google Scholar]
- Yeomans ND (2011) Aspirin: old drug, new uses and challenges. J Gastroenterol Hepatol, 26, 426–431. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Ito M, Chikuma S, Akanuma T and Nakatsukasa H (2018) Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb Perspect Biol, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]