

Abstract
Background
Usher syndrome is the most common form of syndromic retinitis pigmentosa and includes types I, II, and III with varying degrees of hearing loss. We present results of 10 eyes with Usher syndrome treated with autologous bone marrow derived stem cells (BMSC) within the Stem Cell Ophthalmology Treatment Study (SCOTS).
Methods
Preoperative Snellen visual acuities ranged from 20/30−1 to 20/400 with the average pre-operative Snellen acuity approximately 20/85 and the average logarithm of the minimum angle of resolution (LogMAR) acuity 0.635. All eyes had significantly impaired visual fields and patients reported hearing loss as part of this syndromic retinitis pigmentosa. Treatment using the protocols of the SCOTS study using BMSC provided by retrobulbar, subtenons, intravitreal and intravenous injections.
Results
Following treatment, 80% of the Usher eyes showed an improvement in visual acuity. Of the eyes that improved the average increase in visual acuity was 36.4% on LogMAR with improvements ranging from 23% to 94%. The average post-operative change in all treated eyes was a gain of 0.18 LogMAR and an increase in visual acuity of 28.3% on LogMAR. The results showed high statistical significance with P<0.001. Visual fields generally improved. No patient experienced a loss of vision. One patient underwent preoperative and 4-month post-operative audiometry testing which demonstrated improvement. The procedures were performed safely and without complications.
Conclusions
Findings confirm meaningful improvement in visual acuity is possible in Usher syndrome using BMSC protocols developed in the SCOTS study. Statistical significance and safety were established.
Keywords: Usher syndrome, retinitis pigmentosa (RP), bone marrow derived stem cells (BMSCs), visual loss, syndromic retinitis pigmentosa
Introduction
The Stem Cell Ophthalmology Treatment Study (SCOTS) and the follow-on Stem Cell Ophthalmology Treatment Study II (SCOTS2) are clinical studies utilizing autologous bone marrow derived stem cells (BMSCs) for treating various optic nerve and retinal diseases. The diseases addressed typically have no effective treatment for improvement of vision or are progressive in nature. Both studies are Institutional Review Board approved and have been registered with the National Institutes of Health—www.clinicaltrials.gov Identifier NCT 01920867 and NCT 03011541, respectively. The researchers in SCOTS have previously published several papers showing results obtained from treatment in the study including for non-arteritic ischemic optic neuropathy and retinitis pigmentosa (RP) for which statistical significance has been shown.
Usher syndrome is also called Usher-Hallgren syndrome, Hallgren syndrome or Retinitis Pigmentosa-Dysacusis syndrome. It is a type of syndromic RP with vision impairment associated with hearing loss. The reported prevalence is estimated at 1/23,000 in the United States. Approximately 1/6 of patients with RP are diagnosed with Usher syndrome. It is a genetic condition and inherited in an autosomal recessive pattern (1-3).
The appearance of the retina in Usher syndrome shows the same findings as in non-syndromic RP. These include a pale color to the optic nerve called a “waxy disc”, attenuation of retinal vessels, a mottled or variable coloration and midperipheral pigmentation changes called “bone spicules”. Please see Figure 1, photograph obtained from Patient 1.
Figure 1.
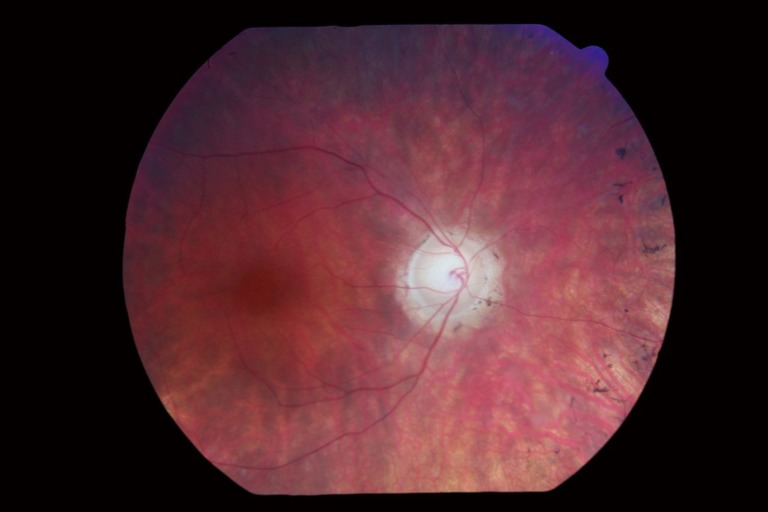
Usher syndrome retinal photograph patient 1.
The syndrome may be sub-classified into 3 main subtypes (Usher I, II, III) in order of decreasing severity of deafness with Usher I and II occurring most commonly (4).
Usher type l may be further subtyped depending on the specific genetic mutations:
❖ IB—MYO7A gene;
❖ IC—USH1C gene;
❖ ID—CDH23 gene;
❖ ID–F—PCDH15 and CDH23 genes;
❖ IF—PCDH15 gene;
❖ IG—SANS gene;
❖ IH—USH1H gene;
❖ IJ—CIB2 gene;
❖ IK—USH1K gene.
Usher type II genetic mutations:
❖ IIA—USH2A gene;
❖ IIC—ADGRV1 gene;
❖ IID—WHRN gene;
❖ IIC—GPR98/PDZD7, from mutation in ADGRV1, also called GPR98 gene.
Usher type III genetic mutations:
❖ IIIA—CLRN1 gene;
❖ IIIB—HARS gene.
Patients with Usher l are generally born deaf and have difficulties in speaking and learning to walk due to vestibular system dysfunction. It is more common in patients of Ashkenazi Jewish heritage and in the French-Acadian group in Louisiana. Visual symptoms from RP are typically noted later in childhood or early adolescence and gradually progress.
Patients with Usher II have a normal vestibular system and manifest reduced hearing but are not fully deaf. The hearing may remain unchanged over time. Visual complaints often manifest from late adolescence to early adulthood.
Patients with Usher III experience progressive hearing and vision loss and approximately half of those affected have vestibular dysfunction.
Treatment of hearing loss is limited to hearing aids and cochlear implants. No standard treatments are available to prevent or improve the visual loss. The medical treatment of hearing loss may include hearing amplification devices and, in appropriate cases, cochlear implants.
Gene based treatment therapies are being developed to treat Usher syndrome type IB (5). Strategies include: (I) gene augmentation using recombinant adeno-associated virus; (II) genome editing by homologous recombinant therapy and; (III) read-through therapy using novel designer aminoglycosides and PTC124. A phase I/IIa dose escalation safety study of a subretinally injected proprietary gene transfer agent in the treatment of Usher syndrome type IB is ongoing (6).
Methods
The SCOTS clinical trial enrolled its first patients in November 2012. It is designed as an open label study to determine the effectiveness of autologous BMSC in treating certain retinopathies and optic neuropathies. Studies are Institutional Review Board approved and reviewed annually and have been registered with the National Institutes of Health: NCT 01920867 and NCT 03011541. All patients meeting eligibility requirements and choosing to enroll receive active treatment without placebo or sham options. To provide the bone marrow stem cell concentrate for injection, bone marrow is aspirated from the posterior iliac crest and processed using a centrifugal cell separator cleared by the United States Food and Drug Administration (FDA). In bilateral disease if both eyes qualify, bilateral treatment is typically offered. In this group of 5 Usher syndrome patients, bilateral treatment was provided allowing a total of 10 eyes for evaluation.
Inclusion criteria for SCOTS states that patients:
Be identified as having damaged retinal or optic nerve tissue which is unlikely to spontaneously resolve or is progressive. Vision should be 20/40 or less when best corrected with spectacles in the eye(s) considered for treatment or that the eye(s) has a diminished visual field (7,8).
Patients cannot have had surgical treatment intended to treat the eye disease within 3 months of the SCOTS procedure and the eye must be stable following such treatments.
Patients taking medication for their eye diseases must be adequately controlled on that treatment but thought unlikely to have vision improvement as a consequence of continued treatment. An example would be a patient with stable intraocular pressures (IOP) on eyedrops, but showing visual field loss.
Patients should have cellular damage with the possibility of vision benefit following the BMSC procedure and be thought to have low risk for any possible harm from treatment in SCOTS.
Patients must not be younger than 18 years old.
Patients with other medical problems should be considered stable by their doctors and all patients must have medical clearance from a primary care physician prior to the procedure. The physician should document that the patient can have the SCOTS procedure and required anesthesia without significant risk to their general well-being.
Exclusion criteria for SCOTS states that:
❖ Patients must be capable of undergoing a sufficient eye examination to adequately document any pathology or they may not be considered for treatment.
❖ Patents must be able and willing to provide the required follow up eye exams with either their own eye doctor or the principal investigator as indicated in the protocol at 1, 3, 6 and 12 months post-treatment or will not be accepted as patients;
❖ Patients should be able to provide informed consent or will not be accepted into the study;
❖ Patients must not be at meaningful risk to their general well-being or visual status if they are to be accepted into the study.
The SCOTS clinical trial offers three treatment arms with options chosen depending on the amount of vision loss, the cause of vision loss and the risks related to the treatment choices. It also considers any medical issues the patient may present. Assuming both eyes meet inclusion criteria, bilateral treatment is offered. The use of autologous bone marrow stem cells eliminates the risk of rejection and therefore no immunosuppression is used following treatment.
The stem cell concentrate used for injection is obtained through the use of an FDA cleared class 2 medical device which uses centrifugation to separate the stem cell concentrate from the obtained bone marrow aspirate material. This stem cell concentrate is routinely assayed and has shown an average total nucleated cell (TNC) count of 1.2 billion cells in the approximately 14–15 cm3 volume. This includes mesenchymal stem cells (MSCs). For local injection of the eye or orbit various approaches are used. Retrobulbar injection consists of 3 cm3 of BMSC posterior to the globe; subtenon injection of 1 cm3 adjacent to the globe, intravitreal injection of 0.05 cm3 within the vitreous through the pars plana, subretinal injection of approximately 0.1 cm3 beneath the retina and intra-optic nerve injection of approximately 0.1 cm3 within the anterior optic nerve. The remaining concentrate is injected intravenously.
The first arm (Arm 1) provides both retrobulbar and subtenon injections to one or both eyes, with intravenous injection following. Patients with mild vision loss or with anatomic or surgical conditions that might impair intravitreal injections, such as silicone oil, might be provided Arm 1 should they comply with inclusion requirements. With the greater experience gained in SCOTS 2, Arm 1 has been found to provide essentially equal benefit as other more complicated procedures with extremely low risk.
The second arm (Arm 2) consists of retrobulbar, subtenon and intravitreal BMSC fraction injections followed by intravenous injection. Typically, patients with greater degrees of vision loss up to 20/200, with or without visual field loss, and satisfying inclusion requirements could be offered Arm 2.
The third arm (Arm 3) may be provided to retinal and optic nerve patients with greater vision loss (visual acuity of 20/200 or worse in one or both eyes). Patients provided Arm 3 typically have poorer vision (less than 20/200). The eye with greater vision loss having a core pars plana vitrectomy and then injection of subretinal or intra-optic nerve concentrate. Arm 3 generally provides Arm 1 or 2 for the eye with greater vision. This is followed by intravenous injection of the remaining BMSC. Monocular patients would not be considered suitable for Arm 3.
After treatment, postoperative eye examinations are required, either with the principal investigator or the patient’s own eye doctor at 1, 3, 6 months and 1 year following treatment in SCOTS. The examination data must be forwarded to the principal investigator and study director.
Complete eye examinations were performed on each patient by the principal investigator (JN Weiss), which included past medical history, past ocular history, record of best vision by both Snellen and Early Treatment Diabetic Retinopathy Study (ETDRS) charts, IOP, and examination of the anterior eye and posterior segment and retina following dilation drops. Fundus photography, ocular coherence tomography (OCT) and visual fields using automated perimetry (depending on the level of the patient’s vision) were performed. Fluorescein angiography was provided if there was concern about neovascularization. A careful review of the medical records available was also performed.
If the patient was unable to correctly identify the 20/400 letter on the Snellen chart, a 20/200 “E” card was presented at ever closer distances until correctly visualized. Eccentric gaze was permitted. If unable to visualize this letter at a distance of 1 foot, or on an ETDRS chart less than 5/200, then hand motion (HM) was recorded (8). If only light could be perceived or not perceived, then this was noted as light perception (LP) or no LP (NLP). For final calculations of differences post operatively, the visual acuity was converted to the LogMAR scale which is the logarithm of the minimal angle of resolution with each 0.1 LogMAR score representing 1 line of visual acuity.
Written informed consent was obtained with each patient receiving careful discussion of potential risks, benefits and the experimental nature of the procedure. All procedures and surgeries were provided by one of the authors (JN Weiss) at a fully licensed outpatient ambulatory surgical center.
Results
Patient 1
A 53-year-old female with a 25-year history of decreased visual acuity and hearing loss secondary to Usher syndrome type II or III. The patient reports that her father and mother have a history of RP. The patient had not undergone genetic testing.
Preoperatively, the best-corrected visual acuity was 20/40+1 in the right eye (OD) and 20/50+1 in the left eye (OS). Following informed consent, the patient underwent SCOTS Arm 1 OD and Arm 2 OS without complication.
The patient returned to her prior ophthalmologist and 2 months postoperatively, the visual acuity had improved to 20/30−2 OD and 20/40−2. There was improvement in visual field testing in both eyes (OU). At 5 months postoperatively, the visual acuity was 20/30 in OU with further improvement in the visual field. Repeat audiology testing at 4 months postoperatively demonstrated an improvement in hearing. The audiologist reported that on audiometer exam there was a 10–15 dB improvement at 250 and 1 kHz in both ears (AU) relative to the preoperative exam using an Aurical audiometer. The speech recognition thresholds (SRT) were in agreement. There was also an improvement in unaided recognition score in right ear (AD) from 76% (90 dB) to 84% (85 dB) and in left ear from 68% (95 dB) to 72% (85 dB).
Patient 2
A 53-year-old woman with a 20-year history of visual and hearing loss secondary to Usher syndrome. The family history was positive for Usher. No genetic testing had been completed.
Preoperatively, the best-corrected visual acuity was 20/80+1, pinhole 20/70−2 OD, and 20/80+1 OS. Mild nuclear sclerosis and posterior subcapsular cataracts, common in Ushers, were noted. Visual fields showed no recordable response on 24−2 automated perimetry. Following detailed informed consent, the patient underwent SCOTS Arm 2 OU without complication.
At the 6-month follow-up examination with her ophthalmologist, the best-corrected visual acuity was 20/80 OD and 20/70 OS with visual field improvement showing some recovery of the central field on 24−2 OU. For this reason, she decided to undergo cataract surgery with intraocular lens implantation sequentially for OU. Immediately following the cataract surgeries, the patient improved with pinhole to 20/30 OD and 20/30−2 OS. Soon after the procedures (OD 2 weeks, OS 1 week) vision with refraction had improved to 20/40− OU.
Patient 3
A 38-year-old male with a 9-year history of decreased visual acuity secondary to Usher type ll. The family history was positive for RP. Genetic testing had not been performed.
The best-corrected visual acuity was 20/40−2 OD and 20/30+1 OS. Following extensive informed consent, the patient underwent SCOTS Arm 2 OU.
At the patient’s 6-month postoperative examination, the visual acuity was 20/25−2 OD and 20/30−1 OS. A small improvement in the visual field was noted.
Patient 4
A 33-year-old male with a 5- to 8-year history of visual loss OU secondary to Usher type III. The family history was positive for RP (sister). No genetic testing was performed.
The best-corrected visual acuity was 20/400 OD and 20/30+1 OS. Following detailed informed consent, the patient elected to undergo SCOTS Arm 2 OD and Arm 1 OS.
The patient was re-examined at the Vanderbilt University Ophthalmology Department comparing their own preoperative visions to the best corrected vision obtained. At 6 months, the patient’s best corrected vision had improved (Vanderbilt) from OD 20/400 to 20/200 and OS from 20/35 to 20/20−. At 1-year post-treatment with the examining Vanderbilt doctor the best corrected visual acuity was OD 20/200 and OS 20/25. The physician and patient were very pleased with the improvement in visual acuity in each eye; electroretinography (ERG) function was similar or slightly better, and visual field improved OU, OD more so than OS. For the visual field of the right eye the mean deviation (MD) improved from −30.82 to −26.48 dB and the pattern standard deviation (PSD) increased from 5.94 to 10.75 dB, likely from the sharper drops from the newly acquired MD. For the visual field of the OS the MD improved mildly from −30.83 to −30.39 dB and the PSD from 6.37 to 5.93 dB. MD is the MD in the deficit in comparison to results from an age-matched normative database and values central points more than peripheral points. PSD is an assessment of the depth of specific losses or focal defects in comparison to adjacent areas.
Patient 5
A 37-year-old Somali male with longstanding visual and hearing loss secondary to Usher type l. The family history is significant for consanguineous parents and a sister with Usher type l. There was no genetic testing.
The best-corrected visual acuity was 0.06 (Snellen 20/333) OD and 0.05 (Snellen 20/400) OS. Following detailed informed consent, the patient elected to undergo SCOTS Arm 1 OU. One month postoperatively the patient’s visual acuity was 0.06 (20/333) OD and 0.1 (20/200) OS. The patient’s family also reported an improvement in his ambulatory ability. Unfortunately, the patient was lost to follow-up.
Bilateral treatment was provided in 5 patients resulting in 10 treatment eyes for analysis. Visual acuities in Snellen and LogMAR notation of eyes pre and post treatment are presented in Table 1. Percent change in acuity was calculated by dividing the average change (delta) in LogMAR acuity from pre-operative vision to post-operative vision by the pre-operative LogMAR visual acuity. LogMAR presents normal 20/20 vision as zero and reduced vision as positive numbers. Therefore, the smaller the LogMAR number the better the visual acuity.
Table 1. Usher patient results.
| No. | Age/sex | Arms | Diagnosis | Pre-Va OD | Pre-Va OS | Post-Va OD | Post-Va OS | OD | OS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Snellen | LogMAR | Snellen | LogMAR | Snellen | LogMAR | Snellen | LogMAR | ΔLogMAR | Preop LogMAR | ΔLogMAR | Preop LogMAR | |||||||||
| Patient 1 | 53 Female | OD 1 OS 2 | II or III | 20/40+ | 0.30 | 20/50+ | 0.40 | 20/30 | 0.20 | 20/30 | 0.20 | +0.10 | Change +33% | +0.20 | Change +50% | |||||
| Patient 2 | 53 Female | OD 2 OS 2 | II or III | 20/70−2 | 0.57 | 20/80+1 | 0.60 | 20/40− | 0.32 | 20/40− | 0.32 | +0.25 | Change +44% | +0.28 | Change +47% | |||||
| Patient 3 | 38 Male | OD 2 OS 2 | II | 20/40−2 | 0.30 | 20/30−1 | 0.20 | 20/25 | 0.10 | 20/30−1 | 0.20 | +0.20 | Change +67% | 0 | Change 0% | |||||
| Patient 4 | 33 Male | OD 2 OS 1 | III | 20/400 | 1.30 | 20/30+1 | 0.18 | 20/200 | 1.00 | 20/20− | 0.01 | +0.30 | Change +23% | +0.17 | Change 94% | |||||
| Patient 5 | 37 Male | OD 1 OS 1 | I | 20/333 | 1.20 | 20/400 | 1.30 | 20/333 | 1.20 | 20/200 | 1.00 | 0 | Change 0% | +0.30 | Change +23% | |||||
Average total: LogMAR preop =0.635; average postop: LogMAR =0.455, ΔLogMAR =+0.18; average change all: eyes treated =+28.3% LogMAR; average change: eyes that improved =+36.4% LogMAR. Pre-Va, preoperative visual acuity; LogMAR, logarithm of the minimum angle of resolution; OD, right eye; OS, left eye.
In this treatment group, 80% of Ushers eyes had an improvement in visual acuity. Of the eyes that improved, the average increase in visual acuity was 36.4% on LogMAR. Individual eye improvement ranged from 23% to 94%. For all eyes treated the average increase of visual acuity was 28.3% on LogMAR. Total average preop LogMAR of all eyes was 0.635. Total average postop LogMAR was 0.455. The average change (delta) in LogMAR was an improvement in acuity of 0.18 for all eyes.
A standard 2 sample t-test was used in statistical calculations. As the sample consisted of acuities of the same eyes measured before and after treatment, a paired (Dependent) t-test was chosen. A two-tailed test was performed; the groups were considered to have near equal variance. For P<0.001 the calculated t exceeded the critical value with 5.0195>4.781 showing high statistical significance. Use of a Welch t-test to account for difference in variance resulted in identical values.
Discussion
RP alone, as well as the syndromic conditions (such as Ushers) that are RP in association with other conditions, are hereditary and complex retinal disorders. The primary issue is abnormalities in the visual pigment regeneration of retinol which initially causes dysfunction of rod photoreceptors. This problem causes nyctalopia, which is visual difficulty experienced by a person in low light conditions, also known as night blindness, and is the first subjective manifestation of retinol photopigment cycling dysfunction. This impairment leads to photoreceptor stress, oxidative damage and subsequent apoptosis of photoreceptors. Because of the location of the rod photoreceptors, this initially causes a loss of peripheral vision in a circular, mid-peripheral pattern called a “ring scotoma”.
As peripheral vision loss spreads, the functioning photoreceptors are progressively confined to an increasing smaller central area, initially with normal or near normal visual acuity as macular photoreceptors are relatively spared. Progressive photoreceptor apoptosis produces the loss of the color sensitive cone photoreceptors (which reside primarily in the fovea) and visual acuity reduction accelerates. Because the remaining photoreceptors are much fewer in number at this point, central vision loss progresses more rapidly to counting fingers (CF), HM and LP. The loss of these remaining photoreceptors ultimately can cause complete blindness or NLP.
Photoreceptor loss throughout the disease process affects the overlying inner and outer nerve plexiform layers, as well as the underlying retinal pigment epithelium and overlying nerve fiber layer. This results in the other pathognomonic findings in RP including the formation of pigmentary changes called bone spicules, loss of optic disc color referred to as optic nerve pallor or a waxy disc, and fluid in the macule called cystoid macular edema. Other pathognomonic findings are cataract formation typically affecting the posterior subcapsular area, and the presence of particles of melanin granules and cells that result in a dust-like appearance within the vitreous.
There are at minimum 60 different genes known to cause RP. Autosomal dominant, autosomal recessive or X-linked recessive can be inheritance patterns. There are hundreds of different potential point mutations depending on the gene affected. This results in a large number of genomic variants for similar phenotypic presentations which leads to a large variety of genetic causes for a similar appearing disease.
RP affects approximately 1 in 2,000 and 1 in 3,000 people in the world (8,9). A number of explanations for RP have been put forth including phototransduction protein synthesis disruptions, structural disruptions or tertiary folding issues with rhodopsin. Rhodopsin gene mutations may cause as much as 40% of RP in autosomal dominant patterns (10). These may include overexpression of the protein or shortening of the protein leading to photoreceptor degeneration (11).
Another potential cause of ocular diseases is epigenetics. Epigenetics is the accessing of genes and their subsequent transcription which is affected by the degree of methylation of the deoxyribonucleic acid (DNA). This may affect the manufacturing of different proteins which may play a role in causing RP or in mitigating the problems caused by an inciting gene. Increases of histone deacetylases (HDAC) activity before photoreceptor impairment has been identified in a murine model of RP (12). Although there are variable roles, HDAC activity has been identified as potentially important for reducing chromatin access (13).
Usher syndrome is responsible for the majority of combined vision and hearing impairment. It is classified into 3 types based on the type of hearing loss and whether vestibular complaints are present. Of sixteen loci associated with Ushers or atypical Ushers, twelve are considered causative and one a modifier. Although the exact cause of the RP remains uncertain, the hearing affect appears related to proteins that are necessary for the hair cell bundles in the cochlea, including their formation, maintenance and activity. The hair bundles are mechanically sensitive portions of the hair cells which ultimately allow for transmission of neurosensory information. The process of turning mechanical stimulation by the sound waves into electrical changes for transmission in the vestibular-cochlear nerves is called mechanoelectrical transduction (14).
Various interventions to alter the inevitable loss of vision in RP have been disappointing. Therapy with 15,000 IU of vitamin A palmitate daily was identified as slightly slowing the progression of Electroretinogram findings but did not affect visual fields or visual acuity (15). Docosahexaenoic acid (DHA) treatment showed a slowing in the loss of visual field function but no difference in the disease course (16). Supplementation with lutein provided increases in the pigmentation of the macule in about 50% of RP patients but no change in their central vision (17). Over a 10-year time period hyperbaric oxygen (HBO2 or HBOT) demonstrated a slowing of expected visual loss by approximately 23%, but no halt in visual loss or an improvement in vision (18). There may be potential for valproic acid as an inhibitor of HDAC for the treatment of RP, but further controlled trials would be required to evaluate any possible benefit (19).
The Argus II retinal prosthesis system uses an eyeglass mounted camera and provides wireless transmission to an epiretinal device to stimulate the nerve layer (20). The device does not replace the photoreceptors and vision quality is limited. It is typically suggested for patients who are near or at total blindness (NLP).
In this paper we present 5 patients with Usher syndrome who underwent treatment in SCOTS and demonstrated improvements of visual function including visual acuity and peripheral vision. One patient underwent preoperative and 4-month post-operative hearing evaluation demonstrated mild improvement in both decibels and word understanding. This is suggestive that the cells responsible for translation of signaling for hearing are impacted in a similar fashion by mutations in the gene. We are unable to determine if this is a consistent finding because other patients did not undergo closely associated preoperative or postoperative hearing evaluations.
We have previously shown that the use of BMSC in another hereditary condition, Leber’s hereditary optic neuropathy (LHON), may be beneficial (21). LHON is a disease caused by various mutations of mitochondrial DNA. In several preclinical studies, it has been demonstrated that transfer of mitochondria can occur from BMSC which may include MSCs and injured or damaged cells. This may result in an improvement of adenosine triphosphate (ATP) production and the reduction of reactive oxygen species (ROS) release, potentially reducing the apoptosis of dysfunctional cells.
The process of mitochondrial transfer has been identified as being via a nanotube like structure. In preclinical work, epithelial cells have been noted to accept mitochondria. Neural tissue including the retinal ganglion cell layer and optic nerve are likely capable of receiving transferred mitochondria.
In another previous paper, the SCOTS protocol demonstrated statistical significance in the improvement or stabilization of vision for patients with non-syndromic RP; 64.7% of patients showed improvement in binocular vision and an average visual improvement of 31% LogMAR over baseline vision (22).
The effects of BMSC on visual acuity and visual fields in Ushers may be similarly explained by identifying their multiple mechanisms of action on target cells. Release of exosomes containing microRNA (miRNA), a small non-coding RNA, can up-regulate or decrease gene expression (23,24). Release of miRNA, exosomes and paracrine factors may improve endogenous cell survival and stimulate further support of photoreceptors and other retinal cells by any residual Muller cells. BMSC can release growth factors such as brain-derived neurotrophic growth factor (BDNF) which can promote neuron survival (25).
Paracrine, or local hormone effects, including neuroprotection from release of nerve growth factor (NGF) can impact cells (26). Other factors, including glial cell line-derived neurotrophic factor (GDNF), may play a role in BMSC treatment of degenerative retinal diseases (27). Ciliary body neurotrophic factor (CBNF) and others which have been shown to offer neuroprotection and could benefit residual photoreceptors and the neuroplexiform and nerve fiber layers. In one of our non-syndromic RP patients treated in SCOTS, BMSC transdifferentiation into neurons was documented histologically, through the recovery of neuronal nuclei (NeuN+) protein, solely found in the nuclei of neurons. All or some of these mechanisms, as well as those still to be identified, may predominate depending on the type of damage present in various retinal pathologies and optic neuropathies.
The treatment provided in SCOTS is not gene therapy. Autologous BMSC are genetically identical to other cells in a particular patient.
However, the pattern of visual loss in RP and Ushers is generally gradual rather than immediate and catastrophic. This may suggest initial compensation for diminished cellular function and resistance to cellular loss during earlier phases of the disease, possibly including responses from retinal Müller cells which are a significant glial component of the retina and can manifest weak neural regenerative efforts (28).
Our findings of improvements across various hereditary retinopathies and optic neuropathies suggests the mechanisms of BMSC action can compensate for residual visual damage caused by deleterious genetic mutations. The use of BMSC may essentially rekindle some of the compensatory and reparative mechanisms displayed in the earlier course of these diseases.
Conclusions
Ushers is one of the syndromic RP disorders causing hearing loss in association with progressive loss of vision.
In reporting 5 patients and 10 eyes with Ushers undergoing treatment using the SCOTS protocol, 80% of eyes were shown to have an improvement in vision. The average increase of visual acuity was 36.4% on LogMAR and improvements ranging from 23% to 94%. In a previous paper regarding SCOTS treatment of non-syndromic RP, improvement in vision was shown and found statistically significant. Of interest is the tangential, mild improvement in hearing in one patient with sufficient pre and post treatment audiometric data. This suggests that, in addition to helping neurosensory retinal function, treatment with BMSC may benefit cochlear hair cell damage as seen in Ushers.
No prior treatment has proven capable of improving acuity in non-syndromic or syndromic RP. SCOTS has now confirmed visual acuity improvement in both RP and Ushers is possible, with meaningful benefit for patients with these otherwise progressive causes of blindness. Eyecare providers now have this option for patients seeking possible treatment for Ushers.
Acknowledgments
None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Studies are Institutional Review Board approved and reviewed annually and have been registered with the National Institutes of Health: NCT 01920867 and NCT 03011541. Written informed consent was obtained with each patient receiving careful discussion of potential risks, benefits and the experimental nature of the procedure.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Mets MB, Young NM, Pass A, et al. Early diagnosis of Usher syndrome in children. Trans Am Ophthalmol Soc 2000;98:237-42; discussion 243-5. [PMC free article] [PubMed] [Google Scholar]
- 2.Vernon M. Usher's syndrome--deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. J Chronic Dis 1969;22:133-51. 10.1016/0021-9681(69)90055-1 [DOI] [PubMed] [Google Scholar]
- 3.Boughman JA, Vernon M, Shaver KA. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis 1983;36:595-603. 10.1016/0021-9681(83)90147-9 [DOI] [PubMed] [Google Scholar]
- 4.Usher syndrome. Genetics Home Reference (GHR). 2016. Available online: http://ghr.nlm.nih.gov/condition/usher-syndrome
- 5.Nagel-Wolfrum K, Baasov T, Wolfrum U. Therapy strategies for Usher syndrome Type 1C in the retina. Adv Exp Med Biol 2014;801:741-7. 10.1007/978-1-4614-3209-8_93 [DOI] [PubMed] [Google Scholar]
- 6.Clinicaltrials.gov Identifier: NCT01505062. Study of SAR421869 in Patients With Retinitis Pigmentosa Associated With Usher Syndrome Type 1B.
- 7.Holladay JT. Proper method for calculating average visual acuity. J Refract Surg 1997;13:388-91. [DOI] [PubMed] [Google Scholar]
- 8.Watt WS. How Visual Acuity is Measured. October 2003 Eye Conditions. Schulze-Bonsel K, Feltgen N, Burau H, et al. Visual acuities "hand motion" and "counting fingers" can be quantified with the freiburg visual acuity test. Invest Ophthalmol Vis Sci 2006;47:1236-40. 10.1167/iovs.05-0981 [DOI] [PubMed] [Google Scholar]
- 9.Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat 2001;17:42-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med 1990;323:1302-7. 10.1056/NEJM199011083231903 [DOI] [PubMed] [Google Scholar]
- 11.Lee ES, Flannery JG. Transport of truncated rhodopsin and its effects on rod function and degeneration. Invest Ophthalmol Vis Sci 2007;48:2868-76. 10.1167/iovs.06-0035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sancho-Pelluz J, Alavi MV, Sahaboglu A, et al. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis 2010;1:e24. 10.1038/cddis.2010.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics 2012;4:5. 10.1186/1868-7083-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta 2015;1852:406-20. [DOI] [PMC free article] [PubMed]
- 15.Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol 1993;111:761-72. 10.1001/archopht.1993.01090060049022 [DOI] [PubMed] [Google Scholar]
- 16.Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol 2004;122:1297-305. 10.1001/archopht.122.9.1297 [DOI] [PubMed] [Google Scholar]
- 17.Aleman TS, Duncan JL, Bieber ML, et al. Macular pigment and lutein supplementation in retinitis pigmentosa and Usher syndrome. Invest Ophthalmol Vis Sci 2001;42:1873-81. [PubMed] [Google Scholar]
- 18.Vingolo EM, Rocco M, Grenga P, et al. Slowing the degenerative process, long lasting effect of hyperbaric oxygen therapy in retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol 2008;246:93-8. 10.1007/s00417-007-0652-z [DOI] [PubMed] [Google Scholar]
- 19.Clemson CM, Tzekov R, Krebs M, et al. Therapeutic potential of valproic acid for retinitis pigmentosa. Br J Ophthalmol 2011;95:89-93. 10.1136/bjo.2009.175356 [DOI] [PubMed] [Google Scholar]
- 20.Ahuja AK, Dorn JD, Caspi A, et al. Blind subjects implanted with the Argus II retinal prosthesis are able to improve performance in a spatial-motor task. Br J Ophthalmol 2011;95:539-43. 10.1136/bjo.2010.179622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiss JN, Levy S, Benes SC. Stem Cell Ophthalmology Treatment Study (SCOTS): bone marrow-derived stem cells in the treatment of Leber's hereditary optic neuropathy. Neural Regen Res 2016;11:1685-94. 10.4103/1673-5374.193251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss JN, Levy S. Stem Cell Ophthalmology Treatment Study: bone marrow derived stem cells in the treatment of Retinitis Pigmentosa. Stem Cell Investig 2018;5:18. 10.21037/sci.2018.04.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández-Messina L, Gutiérrez-Vázquez C, Rivas-García E, et al. Immunomodulatory role of microRNAs transferred by extracellular vesicles. Biol Cell 2015;107:61-77. 10.1111/boc.201400081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kordelas L, Rebmann V, Ludwig AK, et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014;28:970-3. 10.1038/leu.2014.41 [DOI] [PubMed] [Google Scholar]
- 25.Wilkins A, Kemp K, Ginty M, et al. Human bone marrow-derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Res 2009;3:63-70. 10.1016/j.scr.2009.02.006 [DOI] [PubMed] [Google Scholar]
- 26.Huang W, Lv B, Zeng H, et al. Paracrine Factors Secreted by MSCs Promote Astrocyte Survival Associated With GFAP Downregulation After Ischemic Stroke via p38 MAPK and JNK. J Cell Physiol 2015;230:2461-75. 10.1002/jcp.24981 [DOI] [PubMed] [Google Scholar]
- 27.Mead B, Berry M, Logan A, et al. Stem cell treatment of degenerative eye disease. Stem Cell Res 2015;14:243-57. 10.1016/j.scr.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldman D. Müller glial cell reprogramming and retina regeneration. Nat Rev Neurosci 2014;15:431-42. 10.1038/nrn3723 [DOI] [PMC free article] [PubMed] [Google Scholar]