

Abstract
Acute myocardial infarction is one of the leading causes of deaths worldwide. Although ameliorative therapies against ischemic injury have remarkably reduced death rates among patients, they are inevitably complicated by reperfusion injury. Therefore, it is essential to explore other approaches to reduce ischemia/reperfusion injury (IRI). Modulating the levels of nicotinamide adenine dinucleotide (NAD+) is a promising therapeutic strategy against some aging-related diseases. The aim of this study was to determine the role of NAD+ in a swine model of myocardial IRI. Fourteen Bama miniature pigs were subjected to 90 min transluminal balloon occlusion, and then randomly administrated with 20 mg/kg NAD+ or saline before reperfusion. Emission computerized tomography (ECT) was performed immediately and 4 weeks after reperfusion, and the cardiac tissues were analyzed histologically. In addition, the levels of cardiac function markers and the pro-inflammatory cytokines IL-1β and TNF-α were also measured. NAD+ administration markedly reduced myocardial necrosis, enhanced glucose metabolism, and promoted cardiac function recovery. The extent of inflammation was also reduced in the NAD+ treated animals, and corresponded to less cardiac fibrosis and better ventricular compliance. Thus, NAD+ supplementation protected the myocardium from IRI, making it a promising therapeutic agent against acute myocardial ischemic disease.
Keywords: Myocardial, ischemia/reperfusion injury, NAD+, glucose metabolism, necrosis
Introduction
Myocardial infarction (MI) is a leading cause of mortality worldwide. Although several novel therapies targeting ischemic injury have achieved a significant reduction in mortality rates, the very process of myocardia reperfusion results in secondary damage to the myocardium. This ischemia-reperfusion injury (IRI), which can affect almost 50% of the infarcted area, also exerts damaging effects beyond the initial perfusion site [1]. The mechanism of IRI is complex, and pharmacological therapies targeting this pathophysiological process have been ineffective so far [2].
Nicotinamide adenine dinucleotide (NAD+) is a vital enzymatic cofactor that is involved in most metabolic pathways and signal transduction [3], and is a reliable indicator of metabolic efficiency [4]. The major NAD+ dependent enzymes, including sirtuins 1-7 (SIRT1-7), poly (adenosine diphosphate-ribose) polymerases (PARPs) and cyclic ADP-ribose (cADPR, CD38/CD157) SIRTs (SIRT1-SIRT7), respectively function as energy sensors and transcriptional effectors [5], regulators of DNA repair and fat metabolism [6], and as a secondary messenger in Ca2+ signaling [7]. The activity of all these enzymes are tightly controlled by the content of intracellular NAD+. Therefore, modulating NAD+ levels is a potential therapeutic strategy against aging-related diseases like diabetes [8], fatty liver disease [9], cancer [10,11] and neurodegeneration [12,13], which involve dysregulation in the aforementioned enzymes. However, there is insufficient evidence regarding the role of NAD+ in cardiovascular diseases. Since the human heart shows several anatomical similarities with the porcine heart, we investigated the possible therapeutic role of NAD+ in a swine model of myocardial IRI.
Material and methods
Establishment of a porcine model of IRI
Fourteen 3-month old Bama miniature pigs (body weight 17.78±2.45 Kg) were provided by the Shanghai Jiao Tong University School of Agriculture and Biology. The animals were housed and fed according to standard guidelines. All animal experiments were conducted in accordance with relevant ethical standards of the National Institute of Health (NIH), and were approved by the Academic Ethics Committee of Shanghai Chest Hospital of Shanghai Jiao Tong University.
The animals were fasted overnight, and anesthetized with intramuscular injections of 0.6 mg/kg midazolam, 20 mg/kg ketamine and 0.01 mg/kg atropine 30 min before the operation [14]. Sodiumthiopental (5 mg/kg), 0.02 ml/kg/h propofol, 2.5 μg/kg/h sufentanil and 0.1 mg/kg/h pancuronium were then administrated intravenously to maintain the anesthetic state. Supine animals were fixed on the DSA equipment operating table (United States General Electric (China) Co. Ltd., Innova IGS 520), and a 6F sheath was introduced into the femoral artery. After administering 10000 U heparin, a coronary angiography (CAG) was performed. The left anterior descending coronary (LAD) was then occluded for 90 min in the distal lumen by an adequately sized balloon. An ST-T change of the ECG and a follow-up CAG were recorded. Before reperfusion, 20 mg/kg NAD+ (Sigma, St. Louis, Missouri, USA) or 20 ml saline (NS) was administrated through the ear vein as previously described [15]. The animals were defibrillated in case of ventricular fibrillation (VF), and their arterial blood pressure and heart rate were measured continuously.
Emission computerized tomography
Single photon emission computed tomography (SPECT)
Electrocardiography gated myocardial perfusion SPECT was performed immediately after reperfusion and at the 4-weeks follow-up. The animals were placed in a supine position, and 555-740 MBq technetium-99m-sestamibi (99mTc-MIBI; Shanghai Atom Kexing Pharmaceutical, China) was administered intravenously. Scanning was performed after 90 min with a Discovery 670 scanner (GE Medical Systems Inc., Milwaukee, WI, USA), and each image (total 32 in a 64×64 matrix) was acquired for 30 s over a 180° arc at 6° intervals. The data was reconstructed at the horizontal-axis, vertical long-axis and short-axis of the heart.
Positron emission computed tomography (PET)
After SPECT acquisition, PET imaging was performed with a high-spatial-resolution full-ring PET/CT scanner (Truepoint Biography 64, Siemens Healthcare). All scans were attenuation corrected using a low-dose CT scan. A hyperinsulinemic/euglycemic clamp was performed before obtaining the PET images [16] to measure the basal blood glucose levels. Briefly, the animals were intravenously injected with 30 ml of 50% glucose, and when the blood glucose level rose above 8 mM, 2-4 IU insulin was injected. When the blood glucose level decreased to 7-8 mM, 111-148 MBq of 18F labeled fluorodeoxyglucose (18F-FDG; Dongcheng Pharmaceutical, China) was administrated intravenously. The images were acquired and reconstructed after 1 h.
Image analysis
The data were independently reviewed after each scan in a blinded fashion. Manual correction was processed when the mitral valve plane or left ventricular contour was inappropriate for visual interpretation. Standardized quantitative analysis of ECT data was performed with a polar map. The mean signal intensity (MSI) in the respective areas based on a 17-segment model was automatically calculated by QPS software (version 3.1, Cedars-Sinai Medical Center, Los Angeles, CA, USA) as a measure of myocardial perfusion and glucose metabolism [17]. The results were evaluated as per the American Heart Association (AHA) and segmental scoring methods, and the left ventricular average segmental MSI was calculated. MSI<70% indicated a decrease in myocardial perfusion or metabolism. The total perfusion defect (TPD, %) was calculated using the QPS software to evaluate the myocardium at risk area [18].
Regions with perfusion defect but preserved 18F-FDG uptake, or with PET MSI 25% larger than the SPECT MSI, indicated a perfusion-metabolism mismatch (MM, %) with an ischemic but surviving myocardium. A decrease in both 99mTc-MIBI and 18F-FDG uptake indicated a perfusion-metabolism match (M, %) and myocardial necrosis. The MM% and M% in the TPD were calculated automatically using Emory Cardiac Toolbox software (Version 3.3).
The gated SPECT date was analyzed by QGS software (version 3.1, Cedars-Sinai Medical Center, Los Angeles, CA, USA) to calculate the parameters of ventricular function, including LV ejection fraction (EF) and the wall motion (WM) of each segment. The WM score, graded from 0-5, indicated dyskinesis with varying levels of hypokinesis, and segmental WM scores ≥4 were defined as abnormal. Summed motion score (SMS) was also automatically calculated by QGS software. Peak filling rate (PFR) and mean filling rate (MFR) obtained by QGS software were used to evaluate the left ventricular compliance.
Circulating markers
Myocardial enzymes, including troponin I (cTnI) and creatine kinase isozyme (CK-MB) were measured at different time points after reperfusion using a clinical analyzer (AU5811, Beckman Coulter). The concentration of porcine TNF-α and IL-1β were evaluated 24 h after reperfusion using a sandwich ELISA kit (R&D Systems) according to the manufacturer’s protocol.
Histological analysis
The animals were euthanized by injecting pentobarbital sodium, and tissues were collected from the infarcted, non-infarcted (at least 2 mm away from the margin of the infarct) and border regions (just next to the infarcted zone) of the myocardium. The samples were fixed in 10% formalin, and stained with Masson trichrome according to standard protocols. The extent of fibrosis was quantified in the stained tissues in eight fields under 20× magnification using the ImageJ software, and expressed as the percentage of the total section area. The tissue sections were also stained with a commercially available terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) kit (Sigma), and the number of TUNEL+ apoptotic cells were counted in eight fields under 20× magnification.
Statistical analysis
All analyses were performed with SPSS 22.0 (Version 22.0, IBM, Chicago, IL, USA). The data were checked for normal distribution with the Shapiro-Wilks W test. Continuous data was expressed as mean ± standard deviation (SD), and compared using independent samples t-test (Levene homogeneity test of variance and the Mann-Whitney U test were used to calculate the P value). Mann-Whitney U test was used for categorical variables. A 2-sided P-value <0.05 was considered statistically significant.
Results
Animal mortality
Four pigs died before the follow-up duration of 4 weeks - two in the NAD+ group while establishing the IRI model due to persistent VF, and the other two in the control group after reperfusion. The data of these four animals were excluded from the study, leaving 5 pigs each in the NAD+ and NS groups.
NAD+ decreased myocardial necrosis
The cTnI and CK-MB levels within the first 24 h after MI were measured to evaluate the extent of cardiac damage. While cTnI peaked within 2-10 h in the NAD+ group, it took 2-16 hours to rise to maximum levels in the control group, and was significantly higher in the latter (57.68±1.48 ng/ml) compared to the NAD+ group (38.31±3.5 ng/ml) 24 h after MI (P=0.001, Figure 1A). The CK-MB levels were also significantly higher in the control group compared to the NAD+ group (73.12±14.38 ng/ml vs 44.13±9.59 ng/ml, P=0.006, Figure 1B, 1C).
Figure 1.
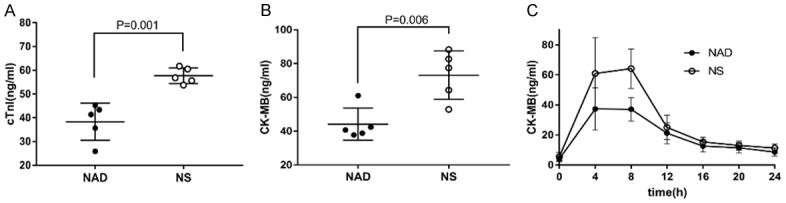
Alteration in the levels of myocardial enzymes after reperfusion. cTnI (A) and CK-MB (B, C) levels were measured at different time points after reperfusion to evaluate the extent of cardiac damage. (A) The cTnI level was significantly higher in the NS group 24 h after MI. (B) Maximum CK-MB level was also higher in the NS group. (C) Time-dependent change in CK-MB levels after MI. NAD+ supplementation reduced myocardial enzyme release.
A decrease in both 99mTc-MIBI and 18F-FDG uptake is indicative of myocardial necrosis. The TPD of both groups after reperfusion were similar (38.40±6.02 vs 39.76±5.61, P>0.05, Figure 2A), but myocardial necrosis was more severe in the control group (12.40±11.10% vs 40.20±10.76%, P=0.004, Figure 2B). At the 4-week follow-up, the TPD of the NS group increased compared to that of the NAD+ treated animals, although the difference was not significant. In addition, the percentage of apoptotic cells in the border zone was also different between the two groups after 4 weeks (71.30±8.96 vs 89.30±13.15, P=0.002, Figure 2C, 2D).
Figure 2.
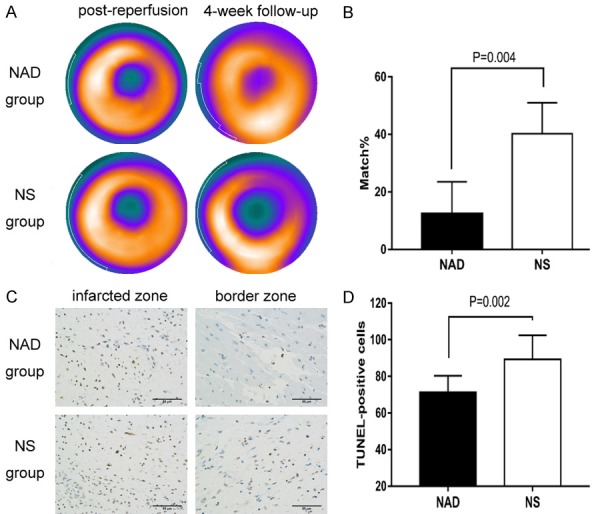
NAD+ supplementation decreased myocardial necrosis. A. The perfusion defect in the NS and NAD+ groups immediately and 4 weeks after reperfusion. B. Reduced 99mTc-MIBI and 18F-FDG uptake indicated a perfusion-metabolism match and myocardial necrosis. The matched regions were smaller in the NAD+ group compared to the control group. C. TUNEL+ stained apoptotic cells in the myocardium 4 weeks after reperfusion. D. The NAD+-treated animals showed fewer apoptotic cells in the myocardium of border zone.
NAD+ improved myocardial metabolism and restored cardiac function
Regions with perfusion defect but preserved 18F-FDG uptake, or with PET MSI 25% larger than the SPECT MSI, were defined as perfusion-metabolism mismatch (Figure 3A), which is indicative of glucose metabolism in the ischemic myocardium. The wall motion was graded from 0-5, which indicated dyskinesis with varying levels of hypokinesis (Figure 3C). NAD+ treatment markedly enhanced glucose metabolism (87.60±11.10% vs 59.80±10.76%, P=0.004, Figure 3B), and restored the cardiac function of these mismatched regions (31 vs 52, Z=-2.611, P=0.009, Figure 3D). However no significant difference was observed in the left ventricular ejection fraction of both groups. After the 4-week follow-up, WM of both groups improved, and no significant difference was then observed due to the slower recovery of the untreated control. A longer follow-up for the animals showing more severe necrosis and fibrosis might result in a significant difference.
Figure 3.
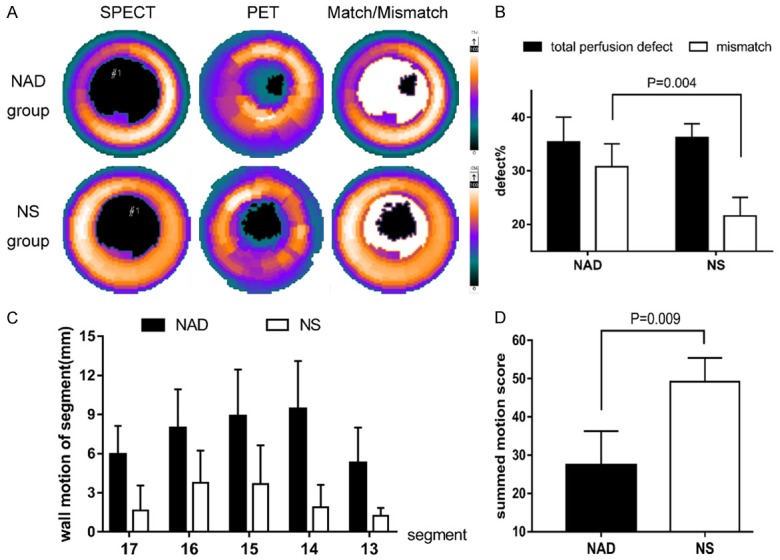
NAD+ improved glucose metabolism and restored cardiac function. A. Polar map showing severely reduced 99mTc-MIBI uptake and normal 18F-FDG uptake indicating a perfusion/metabolism mismatch in the NAD+ group compared to NS group. B. Total perfusion defect and perfusion-metabolism mismatch indicating that NAD+ improved glucose metabolism of ischemic myocardium. C. Polar map (17-segment) showing decreased wall motion in apex, anterior and inferior wall of both groups, and more severe effects in the NS group. D. Semi-quantitative scores of the entire wall motion indicated a better cardiac function of NAD+ group.
NAD+ attenuated inflammation, fibrosis and improved ventricular compliance
NAD+ alleviated the inflammatory response by reducing the levels of TNF-α (30.59±6.56 vs 41.62±5.01 pg/ml, P=0.017, Figure 4A) and IL-1β (34.97±2.18 vs 38.69±1.43 pg/ml, P=0.013, Figure 4B) 24 h after reperfusion. Reduced inflammation in the infarcted area eventually decreases the extent of ventricular fibrosis. After 4-weeks follow-up, the NAD+-treated animals showed significantly lower degree of fibrosis as per Masson staining (2.98±1.28 vs 4.59±1.79%, P=0.019, Figure 4C, 4D), which was consistent with their improved PFR (2.86±0.90 vs 1.50±0.41 EDV/s, P=0.015, Figure 4C) and MFR (1.15±0.39 vs 0.62±0.15 EDV/s, P=0.023, Figure 4C).
Figure 4.
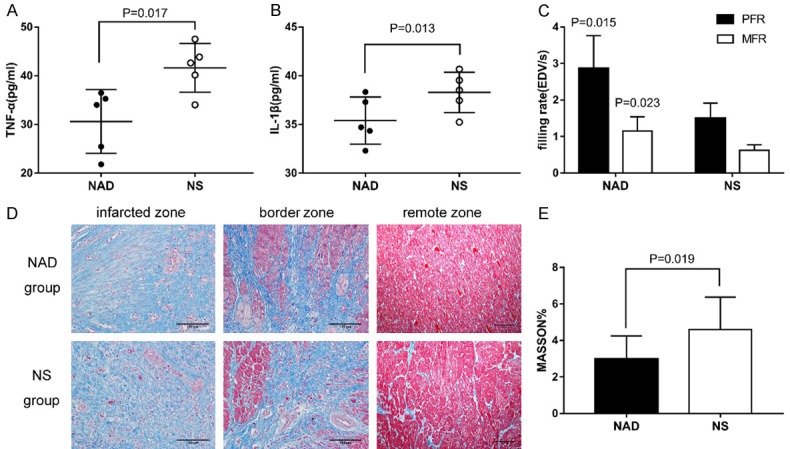
NAD+ modulates inflammation, relieves myocardial fibrosis and improves ventricular compliance. A, B. The levels of inflammatory markers 24 h after reperfusion. C. Improved ventricular compliance of NAD+ group as indicated by PFR and MFR values after 4-weeks follow-up. D. Representative photomicrographs of Masson trichrome-stained sections showing cardiac fibrosis in the infarcted zone, border zone and remote non-infarcted myocardium. Myocardium is stained red and collagen is stained blue. E. Fibrosis of the myocardium 4 weeks after reperfusion, indicating less degree of fibrosis in the remote area of the NAD+ group.
Discussion
Increasing evidence shows vital roles of NAD+ and NAD+-dependent enzymes in ameliorating oxidative stress-induced injuries, preventing different forms of cell death, decreasing DNA damage, enhancing metabolism, protecting mitochondrial fitness, and inhibiting inflammation. Furthermore, the NAD+ levels correlates negatively with the severity of IRI [4]. Both the ischemic and reperfusion processes led to excessive activation of the PARPs, which rapidly depleted NAD+ to only 20%-30% of the baseline levels [19,20], and limited the activity of SIRTs [21]. NAD+ is synthesized de novo through the Preiss-Handler and salvage pathways [22], although the turnover is meagre and slow. Furthermore, IRI significantly inhibits the NAD phosphoribosyltransferase (NAMPT), the rate-limit enzyme of the salvage pathway, thus severely limiting NAD+ replenishment [23,24]. Since exogenous supplementation is known to increase intracellular, including mitochondrial, NAD+ levels [25,26], we hypothesized that administering NAD+ could ameliorate acute IRI. To this end, we established a porcine model of myocardial IRI, and observed significant cardioprotective effects of NAD+ in the animals.
NAD+ reduced myocardial death
NAD+ supplementation significantly prevented apoptosis and decreased myocardial necrosis, which can be attributed to several mechanisms. When exposed to mild to moderate levels of ischemic stress, PARP initiates DNA repair using NAD+ as the substrate to prevent apoptosis; however, the massive DNA damage caused by IRI can constitutively activate PARP and trigger caspase-independent cell death. NAD+ depletion is a necessary intermediary step in this process [27], and NAD+ supplementation restored SITR1 function and prevented PARPs overexpression [28]. In addition, SIRT1 can suppress the activity of p53 and inhibit p53-mediated apoptosis [29]. In our previous study on a rat model of IRI, we observed decreased expression levels of BAX, BCL-XL and caspase-3 in the NAD+-treated animals [15].
NAD+ improved glucose metabolism and restored cardiac function
NAD+ supplementation improved glucose metabolism in the post-ischemic myocardium and thus restored cardiac function. The ischemic myocardium uses glucose as the primary energy source, and studies show that stimulating glucose metabolism during early reperfusion can improve cardiac function following ischemia [30-32]. In addition, since ATP generated by glycolysis is necessary for cytosolic Ca2+ homoeostasis, metabolic recovery can further reduce IRI by preventing Ca2+ overload [33]. NAD+ participates in both cytoplasmic glycolysis and tricarboxylic acid cycle (TAC) in the mitochondria. The production of ATP depends on the intracellular NAD+ levels, and the depletion of cytoplasmic NAD+ by 50% or more would cause glycolytic failure despite excess glucose availability, and lead to cell death [26,34]. Furthermore, since NAD+ cannot be synthesized rapidly, the redox enzymes requiring NAD+ are blocked after IRI, further resulting in metabolic failure. Exogenous NAD+ promotes ATP synthesis not only by directly participating in glucose metabolism, but also by activating SIRTs to upregulate catabolism. SIRTs increase cellular metabolism through multiple pathways, such as increasing glucose uptake, promoting pyruvate entry into the mitochondria for TCA, and up-regulating the activity of different rate-limiting enzymes [35-37].
NAD+ modulates inflammation, relieve myocardial fibrosis and improve ventricular compliance
NAD+ reduced TNF-α and IL-1β levels after ischemic reperfusion, which was followed by a decrease in fibrosis and improved ventricular compliance. IRI triggers a sterile inflammatory reaction, which is associated with expanded infarct size and maladaptive remodeling. Therefore, timely repression of inflammation is critical for effective healing [38]. TNF-α overexpression following MI was observed in both ischemic and normal myocardium, and was sustained in the later stages wherein it promoted fibrosis and extensive remodeling [39,40]. IL-1β plays an important role in the inflammatory response, and its inhibition after MI attenuated cardiomyocyte apoptosis and lowered the incidence of heart failure in both animal models and in clinical trials [41-43]. The positive feedback loop between the two cytokines result in the accumulation of the latter, finally leading to cell death [44,45]. The activation of SIRTs by NAD+ might be responsible for the modulation of the myocardial inflammatory response, since SIRTs inhibit the expression of TNF-α, IL-1β and the downstream transcription factor NF-κB [46,47]. In addition to increasing the ratio of glutathione to oxidized glutathione in mitochondria, SIRTs also upregulate superoxide dismutase (SOD2) activity in the mitochondria to increase oxidative stress resistance [48]. SIRTs and PARPs were closely associated with metabolism, lifespan and aging, and are tightly controlled by the subcellular balance of NAD+. However its over-consumption following IRI limits NAD+-centered energy metabolism and signal transduction. Thus, restoring the NAD+ content by exogenous supplementation protected myocardium from IRI.
Conclusion
Exogenous supplementation of NAD+ protects the myocardium from IRI, and therefore is a potential therapeutic agent against acute MI.
Acknowledgements
The authors thank to the Nuclear Medicine Department of Zhongshan Hospital Affiliated to Fudan University for providing technical support. This study was supported by Shanghai Jiaotong University Grants for Interdisciplinary Research on Medicine and Engineering (YG2014MS74), Research Fund for the Scientific and Technical Project of Shanghai Chest Hospital (2014YZDH20300) and Shanghai Key Laboratory of Clinical Geriatric Medicine (13DZ2260700).
Disclosure of conflict of interest
None.
References
- 1.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibanez B, Heusch G, Ovize M, Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 3.Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gero D, Szabo C. Salvage of nicotinamide adenine dinucleotide plays a critical role in the bioenergetic recovery of post-hypoxic cardiomyocytes. Br J Pharmacol. 2015;172:4817–4832. doi: 10.1111/bph.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai P, Canto C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 7.Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, Vaisitti T, Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 8.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penke M, Larsen PS, Schuster S, Dall M, Jensen BA, Gorski T, Meusel A, Richter S, Vienberg SG, Treebak JT, Kiess W, Garten A. Hepatic NAD salvage pathway is enhanced in mice on a high-fat diet. Mol Cell Endocrinol. 2015;412:65–72. doi: 10.1016/j.mce.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 10.Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 11.Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, He T, Shi X, Gerszten RE, Zhang Y, Yeh JJ, Curry WT, Zhao D, Sundaram S, Nigim F, Koerner MVA, Ho Q, Fisher DE, Roider EM, Kemeny LV, Samuels Y, Flaherty KT, Batchelor TT, Chi AS, Cahill DP. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. 2015;28:773–784. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner C. Boosting NAD to spare hearing. Cell Metab. 2014;20:926–927. doi: 10.1016/j.cmet.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Ding D, Salvi R, Roth JA. Nicotinamide adenine dinucleotide prevents neuroaxonal degeneration induced by manganese in cochlear organotypic cultures. Neurotoxicology. 2014;40:65–74. doi: 10.1016/j.neuro.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Linkenhoker JR, Burkholder TH, Linton CG, Walden A, Abusakran-Monday KA, Rosero AP, Foltz CJ. Effective and safe anesthesia for Yorkshire and Yucatan swine with and without cardiovascular injury and intervention. J Am Assoc Lab Anim Sci. 2010;49:344–351. [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Wang B, Fu X, Guan S, Han W, Zhang J, Gan Q, Fang W, Ying W, Qu X. Exogenous NAD(+) administration significantly protects against myocardial ischemia/reperfusion injury in rat model. Am J Transl Res. 2016;8:3342–3350. [PMC free article] [PubMed] [Google Scholar]
- 16.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 17.Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, Verani MS American Heart Association Writing Group on Myocardial Segmentation and Registration for Cardiac Imaging. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. Int J Cardiovasc Imaging. 2002;18:539–542. [PubMed] [Google Scholar]
- 18.Poulsen RH, Botker HE, Rehling M. Postreperfusion myocardial technetium-99m-sestamibi defect corresponds to area at risk: experimental results from an ischemia-reperfusion porcine model. Nucl Med Biol. 2011;38:819–825. doi: 10.1016/j.nucmedbio.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 21.Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, Auwerx J. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Figueiredo LF, Gossmann TI, Ziegler M, Schuster S. Pathway analysis of NAD+ metabolism. Biochem J. 2011;439:341–348. doi: 10.1042/BJ20110320. [DOI] [PubMed] [Google Scholar]
- 23.Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–491. doi: 10.1161/CIRCRESAHA.109.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 2015;11:535–546. doi: 10.1038/nrendo.2015.117. [DOI] [PubMed] [Google Scholar]
- 25.Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, Chiarugi A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol. 2011;80:1136–1146. doi: 10.1124/mol.111.073916. [DOI] [PubMed] [Google Scholar]
- 26.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- 28.Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–4129. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 30.Hafstad AD, Khalid AM, How OJ, Larsen TS, Aasum E. Glucose and insulin improve cardiac efficiency and postischemic functional recovery in perfused hearts from type 2 diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2007;292:E1288–1294. doi: 10.1152/ajpendo.00504.2006. [DOI] [PubMed] [Google Scholar]
- 31.Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, Kawase Y, Jishage K, Lopaschuk GD. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. 2006;114:1721–1728. doi: 10.1161/CIRCULATIONAHA.106.642009. [DOI] [PubMed] [Google Scholar]
- 32.Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, Dyck JR, Lopaschuk GD. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res. 2012;94:359–369. doi: 10.1093/cvr/cvs129. [DOI] [PubMed] [Google Scholar]
- 33.Jeremy RW, Ambrosio G, Pike MM, Jacobus WE, Becker LC. The functional recovery of post-ischemic myocardium requires glycolysis during early reperfusion. J Mol Cell Cardiol. 1993;25:261–276. doi: 10.1006/jmcc.1993.1033. [DOI] [PubMed] [Google Scholar]
- 34.Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–428. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang S, Jiang B, Zhang T, Liu L, Wang Y, Wang Y, Chen X, Lin H, Zhou L, Xia Y, Chen L, Yang C, Xiong Y, Ye D, Guan KL. Insulin and mTOR pathway regulate HDAC3-mediated deacetylation and activation of PGK1. PLoS Biol. 2015;13:e1002243. doi: 10.1371/journal.pbio.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jing E, O’Neill BT, Rardin MJ, Kleinridders A, Ilkeyeva OR, Ussar S, Bain JR, Lee KY, Verdin EM, Newgard CB, Gibson BW, Kahn CR. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes. 2013;62:3404–3417. doi: 10.2337/db12-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S, Zhao Z, Ke L, Li Z, Li W, Zhang Z, Zhou Y, Feng X, Zhu W. Resveratrol improves glucose uptake in insulin-resistant adipocytes via Sirt1. J Nutr Biochem. 2018;55:209–218. doi: 10.1016/j.jnutbio.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irwin MW, Mak S, Mann DL, Qu R, Penninger JM, Yan A, Dawood F, Wen WH, Shou Z, Liu P. Tissue expression and immunolocalization of tumor necrosis factor-alpha in postinfarction dysfunctional myocardium. Circulation. 1999;99:1492–1498. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- 40.Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, Liu PP. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: regulation by cytokines. Circulation. 2003;107:1046–1052. doi: 10.1161/01.cir.0000051363.86009.3c. [DOI] [PubMed] [Google Scholar]
- 41.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117:2670–2683. doi: 10.1161/CIRCULATIONAHA.107.740233. [DOI] [PubMed] [Google Scholar]
- 42.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, Gelwix CC, Salloum FN, Hastillo A, Dinarello CA, Vetrovec GW VCU-ART Investigators. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010;105:1371–1377. e1371. doi: 10.1016/j.amjcard.2009.12.059. [DOI] [PubMed] [Google Scholar]
- 43.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, Oddi C, Roberts CS, Melchior RD, Mueller GH, Abouzaki NA, Rengel LR, Varma A, Gambill ML, Falcao RA, Voelkel NF, Dinarello CA, Vetrovec GW. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] . Am J Cardiol. 2013;111:1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol. 2014;5:352. doi: 10.3389/fphys.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016;26:249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Busch F, Mobasheri A, Shayan P, Stahlmann R, Shakibaei M. Sirt-1 is required for the inhibition of apoptosis and inflammatory responses in human tenocytes. J Biol Chem. 2012;287:25770–25781. doi: 10.1074/jbc.M112.355420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. 2011;286:9856–9864. doi: 10.1074/jbc.M110.196790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang XX, Wang XL, Tong MM, Gan L, Chen H, Wu SS, Chen JX, Li RL, Wu Y, Zhang HY, Zhu Y, Li YX, He JH, Wang M, Jiang W. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3alpha-dependent antioxidant defense mechanisms. Basic Res Cardiol. 2016;111:13. doi: 10.1007/s00395-016-0531-z. [DOI] [PubMed] [Google Scholar]