

Abstract
p110α is an important subunit of phosphatidylinositol-3-kinases (PI3Ks) encoded by PIK3CA. Though PI3Ks are known as crucial regulators of cellular growth and proliferation, the function of PIK3CA mutations in fulvestrant resistance remains elusive. Thus, this study aimed to investigate the roles of PIK3CA mutations in fulvestrant resistance and tumor progression. Using circulating tumor DNA (ctDNA) from four fulvestrant-resistant patients, we found three of them were PIK3CA mutated, one with a novel PIK3CA mutation (p.R115P). In vitro experiments further evaluated the functions of those mutations and underlying mechanisms. We identified PIK3CA mutations could not only confer to fulvestrant resistance, but also promote cell proliferation and migration. Inhibition of corresponding activated pathways could effectively suppress cell growth.
Keywords: Fulvestrant resistance, estrogen receptor-positive breast cancer, PIK3CA mutation
Introduction
Currently, the mortality of breast cancer has still remained relatively high compared with others, despite of early diagnosis and comprehensive treatment [1]. Highly heterogeneous, different subtypes of breast cancer have different biological behaviors and clinical prognosis, among which estrogen receptor (ER)-positive breast cancer is the most common type and accounts for 65%-75% of breast cancer [2-4]. ER positivity is also the rationale that antiestrogen therapeutics were developed. Binding to ER, estradiol forms estradiol/ER complex, which mediates gene transcription via receptor dimerization and nuclear translocation. What is more, through non-genomic pathway, the complex can also activate mitogen-activated-protein-kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/AKT so as to promote cell growth [5,6].
Targeting ER, endocrine therapy includes selective ER modulators (SERMs, e.g., Tamoxifen), aromatase inhibitors (AIs, e.g., Letrozole) and selective ER downregulators (SERDs, e.g., Fulvestrant) [7-9]. However, along with benefits, the resistance in ER-positive breast cancer to these agents is inevitable, which drives tumor progression [10]. Whereas mechanisms concerning SERMs and AIs resistance have been widely studied, those of fulvestrant resistance are still waited to be elucidated [11,12].
Based on the recent progress on circulating DNA (ctDNA) testing, we had four fulvestrant-resistant patients sequenced and found three of them carrying PIK3CA mutations [13]. As previous studies have shown, PI3K pathway might be implicated in fulvestrant resistance. Subsequent to growth factor binding and activation of receptor tyrosine kinases (RTKs), phosphatidylinositol 4,5-bisphosphate (PIP2) is phosphorylated by PI3K to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3), thus to recruit pleckstrin homology (PH) domain-containing proteins, such as phosphoinositide-dependent kinase 1 (PDK1) and AKT, so as to activate multiple downstream targets. P110α, encoded by PIK3CA, is a key component of PI3K pathway [14,15]. Interestingly, PIK3CA mutation occurs frequently in tumors and is found closely associated with tumor progression [16]. Nevertheless, the relation between fulvestrant resistance and PIK3CA is still not clear.
Thus, in this study, we explored the functions of PIK3CA mutations and their roles in generating resistance to fulvestrant. Furthermore, this study also sought to identify the strategy to treat fulvestrant-resistant breast cancer with mutant PIK3CA.
Patients and methods
Patients and samples collection
In total, four blood samples were collected from patients treated in Nanjing Jinling Hospital from 2015 to 2016. Regardless of previous treatment, ER-positive breast cancer patients who progressed during the treatment of fulvestrant were eligible. The study protocol was approved by the Ethics Committee of Jinling Hospital. All patients gave their informed consent prior to inclusion in the study. Specimens and all experimental procedures were handled and performed in accordance with the approved guidelines. Blood samples (5-10 mL) were collected from each patient when disease progressed.
Sample processing
The whole blood samples were collected in EDTA tubes and then centrifuged at 1000 g for 15 minutes within 1 hour after collection. Plasma and blood cells were collected into separate tubes and stored at -80°C. After DNA extraction and quantification, samples were further sequenced for genes associated with endocrine resistance.
Cell lines, reagents and lentivirus transfection
Breast cancer cell line MCF-7 was purchased from the Chinese Academy of Science Committee type culture collection cell bank (Shanghai, China) in 2017. Cells were cultured in DMEM (Gibco, USA) containing 10% fetal bovine serum (Gibco, USA) and 1% penicillin/streptomycin. Then, cells were incubated in a humidified atmosphere with 5% CO2 at 37°C. Fulvestrant, BKM120 and Palbociclib were purchased from MedChem Express. To identify the function of PIK3CA mutations p.R115P, p.N345K and p.E542K, we transfected MCF-7 cells with recombinant lentivirus of wild-type PIK3CA or mutant PIK3CA (p.R115P, p.N345K or p.E542K), which were purchased from Applied Biological Materials (ABM) (Zhenjiang, China) and confirmed by DNA sequencing. A nonspecific control was also purchased from ABM. Cells were harvested for further study after 72 hours of transfection.
Cell survival assay
Cell viability was measured using Cell Counting Kit-8 (CCK-8) (MedChem Express, China). Briefly, cells were seeded into a 96-well plate at a density of 5 × 103 cells/well with 6 repeats for each condition. After 24 hours, the cells were treated with fulvestrant or BKM120 with or without Palbociclib for another 72 or 24 hours. Then, the supernatants were removed and 100 μl medium with 10 μl CCK-8 was added into each well of the plate and incubated at 37°C. After 2 hours, the absorbance value (OD) of each well was measured at 450 nm using an ELX-800 spectrometer reader (Bio-Tek Instruments, Winooski, USA).
Colony formation assay
Cells transfected with wild-type or mutant PIK3CA and/or treated with medication were diluted and seeded into six-well plates at a density of 500 cells per well. After being incubated in a CO2 incubator at 37°C for 14 days, cells were fixed with 100% methanol and stained with 0.5% crystal violet. Colonies larger than 1 mm were manually counted. These experiments were performed at least three times.
Apoptosis and cell cycle assays
Cells transfected with wild-type or mutant PIK3CA and/or treated with medication were incubated for 24 or 72 hours, then harvested by trypsinization (no EDTA) and washed three times with phosphate-buffered saline (PBS). For apoptosis analysis, the cells were resuspended in 500 μl of 1 × binding buffer and stained with 5 μl of Annexin V-APC and 5 μl of 7-AAD for 15 minutes at room temperature in the dark. For cell cycle analysis, cells were washed with PBS and fixed in 70% ethanol overnight at -20°C, then fixed cells were resuspended in PBS and stained by PI/RNase for 30 minutes in the dark. A flow cytometer (Becton-Dickinson) was used to evaluate the apoptotic rates and cell cycle distribution in each sample. Each sample was tested in triplicate.
Wound healing assay
Cells were seeded in six-well plates and incubated to generate confluent cultures. Using 200 μl sterile pipette tips, wounds were scratched in the cell monolayer and rinsed with PBS. Subsequently, the cells were cultured in serum-free medium for 48 hours. The migration of the cells was photographed at time 0 and 48 hours.
Western blotting
The whole protein was extracted by RIPA buffer supplemented with protease and phosphatase inhibitors. 20 μg cell lysates were loaded per lane and resolved by sodium dodecylsulfate-polyacrylamide (SDS-PAGE) electrophoresis and blotted onto polyvinylidene fluoride (PVDF) membranes. Following 2-hour blockade with 5% skim milk in tris-buffered saline/0.1% tween-20, the membranes were incubated with the primary antibodies overnight at 4°C and with a horseradish peroxidase-conjugated secondary antibody (1:10000) for 2 hours the next day. Antibodies for detecting p110α (#4249), Akt (#4685), phospho-Ser473 Akt (#4060), Cyclin D1 (#2978), β-Actin (#4967) were from Cell Signaling Technology. Antibodies for detecting mTOR (ab32028), phospho-Ser2481 mTOR (ab137133), Rb (ab181616) and phospho-Ser780 Rb (ab47763) were from Abcam. Results from at least two separate experiments were analyzed.
Statistical analysis
All the in vitro experiments were performed in triplicate. One-way ANOVA and t-test were performed using GraphPad Prism version 7 (GraphPad Prism, San Diego). A P value of < 0.05 was considered statistically significant.
Results
Investigation of PIK3CA mutations in circulating tumor DNA
We collected the blood samples from four patients with fulvestrant-resistant ER-positive breast cancer to sequence common mutations in a panel of genes involved in endocrine therapy resistance (Figure 1A, 1B). As a result, it was identified that PIK3CA mutations were the most common mutations found in circulating tumor DNA (ctDNA) with three in four being PIK3CA mutation carriers (Figure 1B). While two of them carried previously described mutations p.N345K and p.E542K, one was found to carry a novel mutation which has not been reported in the breast cancer dataset of the COSMIC database (p.R115P). The corresponding altered nucleobases were shown in Figure 1C. By using Polyphen-2, we predicted the potential functional impacts, brought by the mutations, on protein. It showed that all three mutations were “probably damaging”, indicating a further analysis was needed (Figure 1D).
Figure 1.

Screening and functional analysis of endocrine resistance related mutations. A. The chart of mutations identification in ctDNA. ctDNA, circulating tumor DNA. B. Genetic mutations identified in the ctDNA of patients resistant to fulvestrant. C. Specific mutation sites of detected mutant PIK3CA. D. Potential effects brought by identified candidate mutations on protein function, as predicted by PolyPhen-2. E. Distribution of PIK3CA mutations among different subtypes of breast cancer in database.
Subsequently, we analyzed the distribution of PIK3CA mutations in different breast cancer subtypes and found that PIK3CA were mostly mutated in ER-positive breast cancer and clustered in exons 9 and 20, with the most frequently mutations being E545K and H1047R (Figure 1E).
Functional analysis of the PIK3CA mutations in vitro
In order to investigate the effects of PIK3CA mutations found in patients, MCF-7 cells were transfected with wild-type PIK3CA, PIK3CA mutations p.R115P, p.N345K and p.E542K respectively. Subsequently, we performed cell viability to explore the effects of such mutations in cell sensitivity to fulvestrant. By using CCK-8 assay, we found that cells with transfection of PIK3CA mutations exhibited a significant resistance to fulvestrant compared with these with wild-type PIK3CA (Figure 2A). What is more, the results of colony formation showed that the proliferation of wild-type PIK3CA transfected cells were markedly lower than that of cells transfected with PIK3CA mutations (Figure 2B). As for the results of cell apoptosis, compared with control, cells with mutant PIK3CA displayed only a slightly decreased apoptosis rate (Figure 2C). Next, the influence of PIK3CA mutations on cell cycle was analyzed. Interestingly, whereas cells transfected with PIK3CA mutations p.R115P and p.N345K showed a similar distribution of cells in different phases, cells with PIK3CA mutations p.E542K displayed a decreased percentage in G1 phase and a slightly increased percentage of S phase and G2 phase (Figure 2D), implying a G1-to-S phase transition. Furthermore, the wound healing assay illustrated that the PIK3CA mutations might have the potential to promote cell migration, which finally contributed to the metastasis of fulvestrant-resistant breast cancer (Figure 2E).
Figure 2.

Mutant PIK3CA promotes fulvestrant resistance, cell proliferation and migration. A. The effect of fulvestrant on cell growth of ER-positive breast cancer cells with wild-type or mutant PIK3CA was measured by CCK-8. B. Colony formation of cells transfected with wild-type or mutant PIK3CA. C. The representative images of cell apoptosis analyzed by flow cytometry in cells transfected with wild-type or mutant PIK3CA. D. The representative images of cell cycle analyzed by flow cytometry in cells transfected with wild-type or mutant PIK3CA. E. Wound-healing assay in transfected cells were performed. *P < 0.05. n.s., nonsignificant.
Taken together, our results showed that the PIK3CA mutations p.R115P, p.N345K and p.E542K could not only induce an insensitivity to fulvestrant treatment, but also contribute to the promotion of cell growth and cell migration.
Activation of PI3K pathway by PIK3CA mutations
It has been widely recognized that the mutations of PIK3CA could activate downstream PI3K pathway to promote the cell growth. To confirm the activating ability of PIK3CA mutations, we performed western blotting assays for p-mTOR (phosphorylated mTOR) and p-AKT (phosphorylated AKT) in cells transfected with these mutations. In comparison with phosphorylation levels of mTOR and AKT in control group, the expressions of these in cells with PIK3CA mutations p.R115P, p.N345K and p.E542K were remarkably elevated, suggesting an activation of PI3K pathway (Figure 3A). As shown above (Figure 2D), the G1-to-S phase transition was promoted in cells with PIK3CA mutations p.E542K. Thus, we further explored the expression of cell cycle-related key proteins Cyclin D1 and p-Rb (phosphorylated Rb). The results showed that in cells with the mutation, Cyclin D1 and p-Rb were upregulated compared with these of control (Figure 3B). However, although PIK3CA mutations p.R115P and p.N345K seemed not to have strong impacts on cell cycle promotion (Figure 2D), the levels of Cyclin D1 and p-Rb were also increased (Figure 3B). In view of the complexity of cell cycle and mutations, it was speculated that different mutations might involve different proteins in the regulation of cell cycle.
Figure 3.
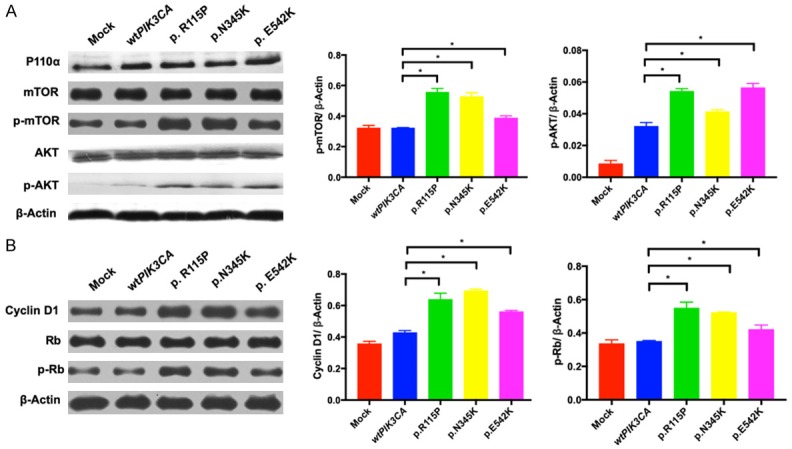
PI3K pathway and downstream cell cycle proteins are activated. A. Western blotting of PI3K pathway proteins p110α, mTOR, p-mTOR, AKT and p-AKT were performed in wild-type or mutant PIK3CA ER-positive breast cancer cells. B. Western blotting analysis of cell cycle proteins Cyclin D1, Rb and p-Rb in wild-type or mutant PIK3CA cells. β-Actin expression was used as the loading control. *P < 0.05.
Briefly, the confirmed PIK3CA mutations could further promote cell proliferation by activating downstream signaling pathway such as PI3K pathway and altering cell cycle proteins.
PI3K inhibitor plus CDK4/6 inhibitor inhibits growth of cells transfected with mutant PIK3CA
Given the activation of both PI3K pathway and its downstream cell cycle proteins in PIK3CA-mutant cells, as well as previous studies which demonstrated that PI3K blockade alone was not as potent as initially assumed [17,18], we hypothesized that the combination of PI3K inhibitor with CDK4/6 inhibitor might exhibit a stronger inhibition on cell proliferation than PI3K inhibitor alone. Therefore, we subsequently performed a dose matrix of PI3K inhibitor BKM120 and CDK4/6 inhibitor Palbociclib in all three mutant PIK3CA-transfected cells to confirm the synergistic activity. It was observed that single PI3K inhibition was relatively ineffective in mutant cells, while the combination of PI3K inhibitor and CDK4/6 inhibitor successfully suppressed cell growth in a low concentration (Figure 4A). What is more, following treatment of BKM120 alone or with Palbociclib, the results of colony formation showed that the combination could significantly inhibit cell proliferation (Figure 4B). To explore the role the combined agents played in cell growth inhibition, cell cycle and apoptosis assays were carried out. As the results showed, we assumed that the combination effect might depend on both apoptosis and cell cycle arrest, ultimately leading to decreased cell viability (Figure 4C, 4D).
Figure 4.

The impacts of combined inhibitors of PI3K and CDK4/6 on PIK3CA mutated cells. A. A dose matrix of PI3K inhibitor BKM120 and CDK 4/6 inhibitor Palbociclib was performed in cell lines transfected with mutant PIK3CA. Viability was assessed one day after treatment. The concentrations and percentages of inhibition at each dose of drug were presented. B. Colony formation of cells transfected with mutant PIK3CA were carried out after treatments of single BKM120 alone or with Palbociclib. C. The representative images of cell apoptosis were analyzed by flow cytometry in cells transfected with wild-type or mutant PIK3CA following medication. D. The representative images of cell cycle were analyzed by flow cytometry in cells transfected with mutant PIK3CA after exposure to BKM120 alone or with Palbociclib for 24 hours. Percentages of cells in G1, S, and G2 phases were quantified. *P < 0.05.
Together, compared with PI3K inhibitor alone, combination of PI3K inhibitor with CDK4/6 inhibitor was more potent in cell proliferation inhibition, apoptosis induction and cell cycle arrest.
Discussion
Potent as endocrine therapy is in treating ER-positive breast cancer, however, the resistance to antiestrogen therapeutics has gradually developed. To further improve the life span and quality for those patients, it is urgent to clarify mechanisms underlying such resistance so as to identify and develop new diagnostic methods and agents. PI3K pathway is a crucial signaling hub for both normal cells and cancer cells, and its implication in fulvestrant resistance has been demonstrated. In spite of this, roles of PI3K alterations, such as mutations of PIK3CA, in conferring resistance to fulvestrant are still unsettled [19,20].
Limited by current conditions, we only found four patients who were resistant to fulvestrant after treatment with fulvestrant. We sequenced endocrine therapy resistance-related genes to identify mutations that might contribute to the insensitivity. As the most commonly mutated genes in four patients, PIK3CA was selected for further research. A retrospective study of 19,784 patients showed that compared with expressions of ER and progesterone receptor (PR) in samples with wild-type PIK3CA, expressions of ER and PR were higher among PIK3CA-mutant patients [21]. What is more, it was demonstrated that alterations of PIK3CA was correlated with persistent ER expression, which might result in the irresponsiveness to antiestrogen agents. According to previous studies, most mutations in PIK3CA could constitutively activate the corresponding kinase and subsequently induce cellular transformation and tumor proliferation [16,22]. Thus, we speculated that it might be via activated PI3K pathway that the detected mutations led to fulvestrant resistance.
In this study, we discovered one novel mutations in PIK3CA exon 1 and two reported mutations in PIK3CA exons 4 and 9. Protein functional assay predicted that these three mutations would impact the functions of protein and deserve a further analysis. By a series of in vitro studies, we confirmed that the mutations were responsible for the insensitivity to fulvestrant and could promote cell growth, colony formation and cell migration. These findings were in agreement with results shown in other tumors and verified the gain of functions of PIK3CA in breast cancer cells. Besides, we also demonstrated that it was due to the activated PI3K/AKT pathway and downstream altered Cyclin D1 and Rb that mutant PIK3CA conferred a resistance to fulvestrant.
Since the activated PI3K pathway was observed in cells transfected with mutant PIK3CA, we conjectured PI3K inhibitors might have a preferential inhibition of these cells. Although some laboratory and clinical studies showed tumors with PIK3CA mutations would benefit more from PI3K inhibitors, some studies showed opposite results [23-25]. What is more, it was revealed that a combination inhibition of PI3K with other inhibitors (e.g., mTORC inhibitor) could strengthen tumor suppression [26]. Considering the increased p-Rb in cells with mutant PIK3CA and efficacy of CDK4/6 inhibitors shown in breast cancer, we then sought to explore the effectiveness of PI3K inhibitor BKM120 with CDK4/6 inhibitor Palbociclib. As expected, CDK4/6 inhibition not only sensitized PIK3CA-mutant cells to PI3K inhibitor, but also exhibited an additional benefit. Interestingly, as shown, the combinatorial effect depended on both cell apoptosis and cell cycle arrest in cells with mutations p.R115P and p.E542K. Different from the previous two types of cells, cells with PIK3CA mutations p.N345K did not show a higher apoptosis rate after being exposed to the combination treatment. These results suggested that though addition of CDK4/6 inhibitor to PI3K inhibition would be more effective than single PI3K inhibitor, different mutations might respond in different mechanisms.
Despite the potency of inhibition of both cell cycle proteins and PI3K, the side effects and toxicities should also be taken into consideration. Fortunately, the toxicity profiles of these two drugs do not cross over much. While the toxicity profiles of PI3K inhibitors are mainly rash, hyperglycemia and diarrhea [27,28], CDK4/6 inhibitor differs, with the most common toxicities being hematologic neutropenia [29-31].
Briefly, this study demonstrated that the mutations of PIK3CA contributed to fulvestrant resistance through the activation of PI3K pathway and alteration of cell cycle proteins. Further, it was also found that combined inhibitors of PI3K and CDK4/6 could successfully suppress PIK3CA mutated cells. While further studies of combined agents would be needed with no doubt, such combination might shed light on the future development direction of combination therapeutics.
Acknowledgements
This study was funded by National Natural Science Foundation of China (Grant 81773102, Grant 81470357) and the Foundation for Clinical Medicine Science and Technology Special Project of the Jiangsu Province (Grant BL2014071).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein L, Lacey JV Jr. Receptors, associations, and risk factor differences by breast cancer subtypes: positive or negative? J Natl Cancer Inst. 2011;103:451–3. doi: 10.1093/jnci/djr046. [DOI] [PubMed] [Google Scholar]
- 3.Chen WY, Colditz GA. Risk factors and hormone-receptor status: epidemiology, risk-prediction models and treatment implications for breast cancer. Nat Clin Pract Oncol. 2007;4:415–423. doi: 10.1038/ncponc0851. [DOI] [PubMed] [Google Scholar]
- 4.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 5.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein-kinase encoded by the Akt protooncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 6.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, Cantley LC. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali S, Buluwela L, Coombes RC. Antiestrogens and their therapeutic applications in breast cancer and other diseases. Annu Rev Med. 2011;62:217. doi: 10.1146/annurev-med-052209-100305. [DOI] [PubMed] [Google Scholar]
- 8.Pietras RJ. Biologic basis of sequential and combination therapies for hormone-responsive breast cancer. Oncologist. 2006;11:704–717. doi: 10.1634/theoncologist.11-7-704. [DOI] [PubMed] [Google Scholar]
- 9.Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- 10.Magnani L, Stoeck A, Zhang X, Lanczky A, Mirabella AC, Wang TL, Gyorffy B, Lupien M. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proc Natl Acad Sci U S A. 2013;110:E1490–9. doi: 10.1073/pnas.1219992110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. 2015;15:261–75. doi: 10.1038/nrc3920. [DOI] [PubMed] [Google Scholar]
- 12.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SK, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih lM, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA Jr. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:1–11. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type-I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988;332:644–646. doi: 10.1038/332644a0. [DOI] [PubMed] [Google Scholar]
- 16.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13:224. doi: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maira SM. PI3K inhibitors for cancer treatment: five years of preclinical and clinical research after BEZ235. Mol Cancer Ther. 2011;10:2016. doi: 10.1158/1535-7163.MCT-11-0792. [DOI] [PubMed] [Google Scholar]
- 18.Vora Sadhna R, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman Elizabeth L, Pollack Sarah F, Liu M, Li X, Lehar J, Wiesmann M, Wartmann M, Chen Y, Cao ZA, Pinzon-Ortiz M, Kim S, Schlegel R, Huang A, Engelman Jeffrey A. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang D, Yang F, Wang Y, Guan X. Mechanisms of resistance to selective estrogen receptor down-regulator in metastatic breast cancer. Biochim Biophys Acta Rev Cancer. 2017;1868:148–156. doi: 10.1016/j.bbcan.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, Iwata H, Harbeck N, Zhang K, Theall KP, Jiang Y, Bartlett CH, Koehler M, Slamon D. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425–439. doi: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- 21.Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19784 diverse solid tumors. JAMA Oncol. 2016;2:1565–1573. doi: 10.1001/jamaoncol.2016.0891. [DOI] [PubMed] [Google Scholar]
- 22.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, Yardley DA, Melichar B, Forero-Torres A, Lee SC, de Boer R, Petrakova K, Vallentin S, Perez EA, Piccart M, Ellis M, Winer E, Gendreau S, Derynck M, Lackner M, Levy G, Qiu J, He J, Schmid P. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:811–821. doi: 10.1016/S1470-2045(16)00106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baselga J, Im SA, Iwata H, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng LM, Hurvitz S, Masuda N, Cortés J, De Laurentiis M, Arteaga C, Jiang Z, Jonat W, Hachemi S, Le Mouhaër S, Di Tomaso E, Urban P, Massacesi C, Campone M. Abstract S6-01: PIK3CA status in circulating tumor DNA (ctDNA) predicts efficacy of buparlisib (BUP) plus fulvestrant (FULV) in postmenopausal women with endocrine-resistant HR+/HER2-advanced breast cancer (BC): first results from the randomized, phase III BELLE-2 trial. Cancer Res. 2016;76 S6-01-S6-01. [Google Scholar]
- 25.Dickler M, Saura C, Richards D, Krop I, Cervantes A, Bedard P, Patel M, Pusztai L, Oliveira M, Ware J. A phase II study of the PI3K inhibitor taselisib (GDC-0032) combined with fulvestrant (F) in patients (pts) with HER2-negative (HER2-), hormone receptor-positive (HR+) advanced breast cancer (BC) J. Clin. Oncol. 2016;34 doi: 10.1158/1078-0432.CCR-18-0613. Abstr 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak Ö, Hackl W, Baselga J. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012;23:2399–2408. doi: 10.1093/annonc/mds011. [DOI] [PubMed] [Google Scholar]
- 27.Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M, Gil-Martin M, Rugo HS, Seggewiss-Bernhardt R, Huang A, Bootle D, Demanse D, Blumenstein L, Coughlin C, Quadt C, Baselga J. Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J. Clin. Oncol. 2018;36:1291–1299. doi: 10.1200/JCO.2017.72.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodon J, Braña I, Siu LL, De Jonge MJ, Homji N, Mills D, Di Tomaso E, Sarr C, Trandafir L, Massacesi C, Eskens F, Bendell JC. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2014;32:670–81. doi: 10.1007/s10637-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 29.Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, Wildiers H, Hudis CA, O’Shaughnessy J, Zamora E, Yardley DA, Frenzel M, Koustenis A, Baselga J. MONARCH 1, a phase ii study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2-metastatic breast cancer. Clin Cancer Res. 2017;23:5218–5224. doi: 10.1158/1078-0432.CCR-17-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turner NC, Huang Bartlett C, Cristofanilli M. Palbociclib in hormone-receptor-positive advanced breast cancer. New Engl J Med. 2015;373:1672–1673. doi: 10.1056/NEJMc1510345. [DOI] [PubMed] [Google Scholar]
- 31.Spring LM, Zangardi ML, Moy B, Bardia A. Clinical management of potential toxicities and drug interactions related to cyclin-dependent kinase 4/6 inhibitors in breast cancer: practical considerations and recommendations. Oncologist. 2017;22:1039–1048. doi: 10.1634/theoncologist.2017-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]