

Abstract
Background: Sorafenib is an oral multi-kinase inhibitor that inhibits hepatocellular carcinoma (HCC) via the Ras/Raf/MAPK pathway. However, sorafenib loses effectiveness because most tumors acquire drug resistance over time. As the PI3K/AKT/mTOR signaling pathway is also activated abnormally in HCC, we evaluated the effect of sorafenib, in combination with a dual PI3K/mTOR inhibitor, BEZ235, on HCC cell proliferation and survival in vitro. Materials and methods: Biological phenotypes were analysed in HCC cell lines, parental and sorafenib-resistant HepG2 cells (HepG2 and HepG2R), treated with sorafenib or BEZ235, alone or in combination. HCC cellular proliferation and apoptosis were investigated, and perturbations of the Ras/Raf/MAPK and PI3K/AKT/mTOR signaling/survival pathways were evaluated by western blot analysis. Results: BEZ235 enhanced sorafenib inhibition of cellular proliferation, migration, and promotion of apoptosis in HepG2 and HepG2R cells. The combined effects were associated with inhibition of phosphorylation of AKT, mTOR and S6K in the PI3K/AKT/mTOR pathway, whereas the combination of sorafenib and BEZ235 did not significantly alter the Ras/Raf/MAPK pathway compared with the effect of sorafenib alone. Conclusion: Sorafenib/BEZ235 combination has potent anti-HCC cell activity. This anti-tumor activity is most likely multi-factorial, mainly involving PI3K down-regulation and AKT, mTOR and S6K dephosphorylation. Combined inhibition of PI3K/AKT/mTOR and Ras/Raf/MAPK pathways enhances sorafenib inhibition of HCC. The results of these in vitro studies suggest that trials of combined sorafenib and BEZ235 in the treatment of HCC should be considered.
Keywords: Sorafenib, BEZ235, hepatocellular carcinoma, PI3K/AKT/mTOR pathway, HepG2 cells
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common primary malignancy and the third leading cause of cancer-related death worldwide [1]. In most patients, the disease is diagnosed at an advanced stage. In the recent ten years, targeted therapy has provided a novel alternative for advanced HCC [2,3].
In most advanced HCCs, the Ras/Raf/MAPK signaling pathway is activated as a result of stimulation by growth factors, such as epidermal growth factor, hepatocyte growth factor, and insulin-like growth factor [4]. Sorafenib, which inhibits multi-target molecules, including Raf kinase, platelet-derived growth factor receptor, vascular endothelial growth factor receptor, and c-Kit [5], inhibits HCC cell proliferation and angiogenesis by blocking the Ras/Raf/MAPK signaling pathway [6]. The US Food and Drug Administration approved sorafenib for treatment of advanced HCC in 2007 [7].
Despite improvement in the overall survival of HCC patients treated with sorafenib, the median survival time of those with advanced disease has been only about 3-5 months. Furthermore, some patients have had tumor progression during sorafenib therapy and a very low response rate (2-3%). Most advanced HCCs acquire resistance to sorafenib, resulting in tumor growth or distant metastasis over time [8,9]. Thus, exploring combined strategies to improve the therapeutic effect of sorafenib on HCC is high priority.
The development and proliferation of HCC involve several signaling pathway perturbations, of which the Ras/Raf/MAPK and the PI3K/AKT/mTOR pathways are critical [10]. There are different activation levels of PI3K/AKT/mTOR pathways in most sorafenib-resistant HCCs. The constitutive activation of phosphatidylinositol 3-kinase (PI3K), AKT and the mammalian target of rapamycin (mTOR) confers drug resistance to many types of cancer, including HCC. The downstream protein of mTOR signaling, S6K, is aberrantly activated in 40%-50% of HCC cases and regulates basic cellular processes, including mRNA translation, cell-cycle progression, and cell proliferation, migration, and survival [11,12]. Activated S6K often leads to crosstalk on the Ras/Raf/MAPK signaling pathway, thus diminishing the inhibitory effect of sorafenib on HCC, with resultant resistance of HCC to the drug.
Recently, inhibitors of the PI3K/AKT/mTOR pathway have been developed [13]. BEZ235, an imidazoquinoline derivative, inhibits the kinase activities of PI3K, mTORC1 and mTORC2, and its anti-tumor efficacy has been demonstrated in preclinical models [14,15]. In this study, we assessed the effect of BEZ235 in combination with sorafenib on parental and sorafenib-resistant HepG2 cell lines (HepG2 and HepG2R). Our results support the view that sorafenib combined with BEZ235, by inhibiting the PI3K/AKT/mTOR signaling pathway, may be more effective in treating HCC than is sorafenib alone.
Materials and methods
Cell lines, chemicals and antibodies
Human HCC HepG2 cell line was obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). Sorafenib-resistant cell line, HepG2R, was induced by continuous treatment of HepG2 cells with sorafenib up to 10 μmol/L to imitate acquired resistance. Both cell lines were cultured in RPMI-1640 medium plus 10% fetal bovine serum (FBS) and maintained in a 5% CO2 incubator at 37°C. Sora-fenib was purchased from Bayer HealthCare AG Pharmaceuticals (Berlin, Germany); BEZ235 was obtained from Med Chem Express (Monmouth Junction, USA). All antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA).
MTT assay
Cell viability was measured with MTT Assay in accordance with the manufacturer’s instructions. Cells were seeded into 96-well plates at 4×104 cells/mL in 100 μl of RPMI-1640 plus 10% FBS and cultured for 48 h in a 5% CO2 incubator at 37°C. The cells were treated with various concentrations of agents for 12 h, 24 h or 48 h. Carrier dimethyl sulfoxide was used as a control. Ten microliters of MTT (5 mg/mL) were added to each well and incubated for 4 h. After aspiration, 100 μl of dimethyl sulfoxide were added. The optical density was read at 490 nm in a microplate.
Colony-formation assay
Cells were seeded in six-well plates at 1000 cells/well and cultured for 24 h. The cells were treated with various concentrations of drugs for another 24 h. The drug-containing medium was aspirated and replaced with drug-free medium, and the cells were cultured for about 10 days. Cell colonies were fixed with paraformaldehyde and stained with crystal violet solution. Cell colonies were counted, and the number cultured with drugs was compared with the number cultured without drugs.
Monolayer wound-healing assay
Cells were planted in six-well plates and incubated in RPMI-1640 containing 10% FBS until they reached confluence. The cell monolayer was scratched with a sterile 10-μl tip to create a cell-free zone. After the cells were washed with PBS three times, serum-free medium containing agents was added to the wells. The scratched fields were recorded with a light microscope immediately after injury and at 12-h intervals.
Cell-cycle analysis
The cell cycle was analyzed with flow cytometry. Cells were planted in 12-well plates at 1×105 cells per well and incubated in RPMI-1640 containing 10% FBS for 24 h followed by treatment with drugs for additional 24 h. The cells were pretreated with 0.25% trypsin-EDTA, then dissociated and fixed with 700 μl cold 75% ethanol at 4°C for 1 h. The cells were incubated with RNase (100 ng/mL) at room temperature for 30 min, then stained with 500 μl propidium iodide (PI, 50 ng/mL) for 1 h at 25°C. Dye was removed, and the cells were resuspended with 300 μl of PBS for cell-cycle analysis by flow cytometry (BD FACSCalibur, USA).
Immunofluorescence analysis
To detect cell apoptosis, cells were seeded on glass slides pretreated in 24-well plates at 1×105 cells per well and incubated for 24 h, followed by drug treatment for an additional 24 h. After washing with PBS, the cells were incubated with 1 μl fluorescence DNA stain (DAPI), 5 μl AnnexinV-FITC and 5 μl propidium iodide at room temperature for 15 min in the dark. The staining was visualized with a fluorescence microscope.
Western blot analysis
Total protein was extracted from cells lysed with radioimmunoprecipitation buffer (RIPA, Beyotime Biotechnology, Shanghai, China) with addition of a protease inhibitor cocktail (Beyotime Biotechnology, Shanghai, China) for 30 min at 4°C. After centrifugation for 20 min at 16,000 rpm at 4°C, the protein concentration of the supernatant was measured with BCA200 protein assay kit (Biosharp Life Science, Hefei, China) and equalized before loading to the gel. Thirty micrograms of cellular protein per lane were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto a polyvinylidene fluoride membrane (Millipore, USA). After blocking with Tris-buffered saline/Tween 20 containing 5% fat-free milk for 1 h, the membranes were incubated with primary antibody, according to the manufacturers’ recommendations, overnight at 4°C, then with the secondary antibody for 1 h at room temperature. Protein bands were visualized with a chemiluminescence detection kit (Thermo), and images were captured with a scanner using Quality One software (Bio-Rad).
Statistical analysis
All analyses were performed with the software SPSS version 18.0. Data were presented as mean ± SD. All experiments were performed in triplicate. Group differences were calculated by t test or one-way ANOVA. Significance was defined as P<0.05.
Results
BEZ235 increased sorafenib inhibition of HepG2 and HepG2R cell viability
As the PI3K/AKT/mTOR pathway is usually upregulated in HCC cells treated with sorafenib and sorafenib-resistant HCC cells, we analyzed the effects of sorafenib combined with BEZ235 on the viability of HepG2 and HepG2R cells with MTT assay. IC50s of sorafenib on HepG2 and HepG2R cells were tested (Figure 1A). Cell-growth inhibition was significant: cell viability decreased in a dose- and time-dependent manner when cells were incubated with sorafenib, BEZ235 or combination of the two drugs. In comparison with sorafenib, BEZ235 had a greater effect: BEZ235 (0.25 μM) reduced the cell viability by 47% after 24 h, whereas sorafenib (4 μM) reduced viability by only 20%. The combination of sorafenib (4 μM) and BEZ235 (0.25 μM) reduced cell viability further, by 40%, 54% and 83% after 12 h, 24 h and 48 h, respectively (Figure 1B). Similar results were obtained with HepG2R cells when BEZ235 combined with sorafenib was used simultaneously (Figure 1C). All these results suggest that BEZ235 enhances the sorafenib inhibition of HCC cells.
Figure 1.
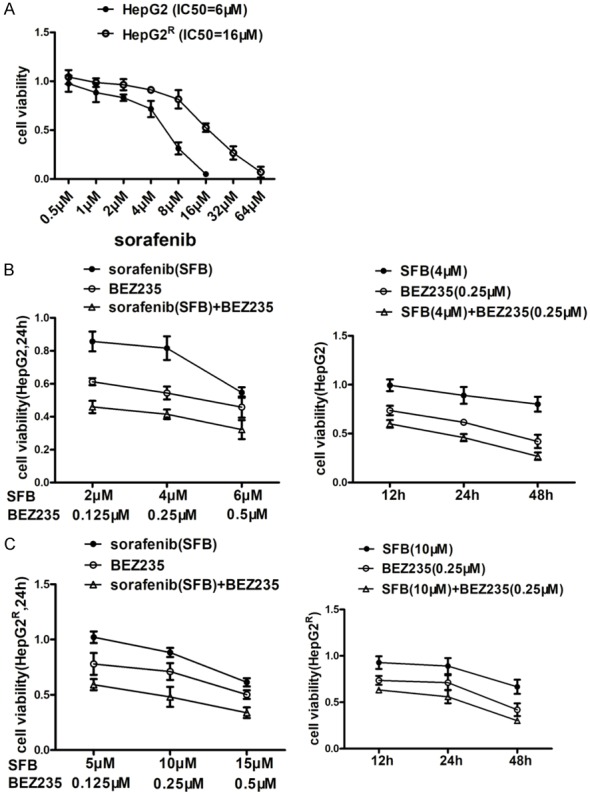
Cell viability of HepG2 and HepG2R cells incubated with sorafenib (SFB), BEZ235, or a combination of sorafenib and BEZ235, measured with MTT assay. A. HepG2 and HepG2R cells were treated with various concentrations of drugs for 24 h and IC50 was calculated as described by Chou et al by MTT assay. The IC50 of sorafenib on HepG2 and HepG2R cells was 6 μM and 16 μM, respectively. B. Left: In comparison with sorafenib, BEZ235 monotherapy and two drugs in combination had a greater effect in reducing the cell viability after 24 h. Also, there was a dose-dependent decrease of cell viability of HepG2 cells treated with sorafenib, BEZ235 or the combination of sorafenib and BEZ235 for 24 h. Right: In comparison with sorafenib (4 μM), BEZ235 (0.25 μM), or combination of sorafenib and BEZ235 decreased cell viability of HepG2 cells for 12 h, 24 h and 48 h. C. The viability of HepG2R cells treated with sorafenib, BEZ235 or in combination was like that of HepG2 cells. Thus, BEZ235 increases sorafenib inhibition of both HepG2 and HepG2R cell viability in a dose- and time-dependent fashion. Compared with control and single-drug treatment, the two drugs in combination were significantly (P<0.05). Data shown are the mean ± SD from three independent experiments.
BEZ235 sensitizes sorafenib-resistant proliferation of HCC single cells
To further assess the effect of sorafenib combined with BEZ235, we performed colony formation assays with the HepG2 and HepG2R cell lines. As illustrated in Figure 2, sorafenib single therapy did not significantly decrease HepG2 and HepG2R cell colony formation compared with control values (P>0.05), whereas BEZ235 did. Sorafenib plus BEZ235 had a greater inhibitory effect than did BEZ235 alone (**P<0.01), and both BEZ235 monotherapy and BEZ235 plus sorafenib had a greater inhibitory effect than did sorafenib alone (**P<0.01). These results suggest that BEZ235 has a sorafenib-sensitizing effect on HepG2 and HepG2R cells (reduces sorafenib-induced HCC cell resistance), leading to inhibition of cell growth and the promotion of cell death.
Figure 2.
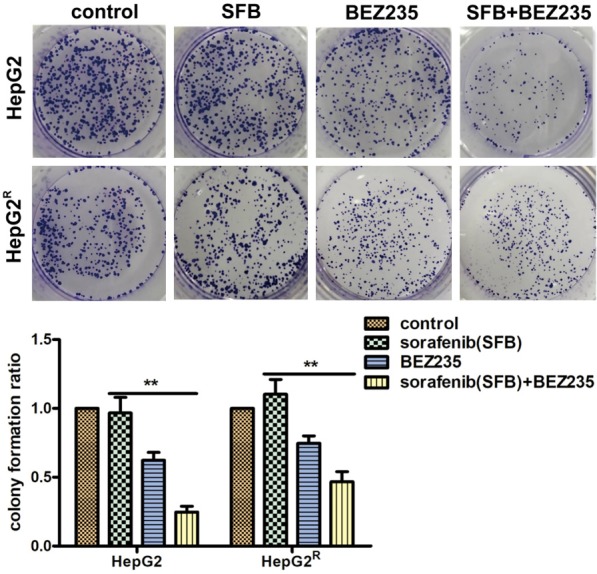
Colony formation assay was carried out to assess the proliferation of HepG2 and HepG2R single cells treated with sorafenib (HepG2, 1 μM; HepG2R, 2 μM) and BEZ235 (0.0625 μM) monotherapy or in combination for 24 h. After the drug-containing medium was aspirated and replaced drug-free medium, the cells were further cultured for about 10 days. Cell colonies were fixed with paraformaldehyde and stained with crystal violet solution. The number of colonies in HepG2 and HepG2R cells after sorafenib mono-treatment was not statistically different from the control number (P>0.05). In contrast, BEZ235 as single agent or in combination with sorafenib, significantly reduced the number of HepG2 and HepG2R colonies compared with the numbers when treated with sorafenib alone (**P<0.01). Data shown are the mean ± SD from three independent experiments.
BEZ235 increased sorafenib inhibition of HepG2 and HepG2R cell migration
We incubated cells with sorafenib (HepG2, 4 μM; HepG2R, 10 μM) with or without BEZ235 (0.25 μM) to investigate the effect of two drugs combined on HepG2 and HepG2R cell migration. The results of wound healing assays (Figure 3) revealed no significant difference between the cell migration area of HepG2 and HepG2R cells treated with sorafenib and that of the control cells in 12 h (P>0.05). BEZ235 treatment increased the wound size of HepG2 to 78% (12 h) and 68% (24 h) and that of HepG2R to 76% (12 h) and 73% (24 h). Combination treatment with sorafenib plus BEZ235 increased the wound size of HepG2 to 92% (12 h) and 87% (24 h) and that of HepG2R to 90% (12 h) and 86% (24 h). All data were compared to that of control cells.
Figure 3.

Cell motility measured with wound-healing assay. The cell monolayer was cultured in serum-free medium containing sorafenib (HepG2, 4 μM; HepG2R, 10 μM), BEZ235 (0.25 μM), or the agents combined after scratch with a sterile 10-μl tip. The scratched fields were recorded with a light microscope after injury and at 12-h intervals. A. Compared with control values, the wound size of HepG2 cells treated with sorafenib (4 μM) for 12 h was not significantly different (P>0.05). BEZ235 (0.25 μM), as a single agent and in combination with sorafenib (4 μM), decreased the migration of HepG2 cells at 12 h and 24 h. B. Similar results were observed with HepG2R cells. (**P<0.01; and *P<0.05). Data shown are the mean ± SD from three independent experiments.
BEZ235 enhanced sorafenib blocking of cell-cycle progression
To investigate whether sorafenib combined with BEZ235-induced inhibition of cell proliferation was associated with cell-cycle dysregulation, we analyzed the effect of BEZ235 on cell-cycle progression in sorafenib-treated HepG2 and HepG2R cells based on DNA content by propidium iodide staining of flow cytometry analysis. With HepG2 cells treated with sorafenib and BEZ235 individually, there was an increase in the percentage of cells in the G0/G1 phase (76%, 85%) compared with the percentage in controls. With HepG2R treated with sorafenib and BEZ235 individually, cells in the G0/G1 phase were 63% and 64%, respectively, compared with control values. With combined sorafenib and BEZ235 treatment, the percentages of HepG2 and HepG2R cells in the G0/G1 phase were 90% and 82%, respectively, compared with control values. Collectively, these results are evidence that BEZ235 enhanced sorafenib blocking of cell-cycle progression (Figure 4A).
Figure 4.

Analysis of the effect of combined sorafenib and BEZ235 treatment on the cell cycle, as determined with flow cytometry and western blot. A. HepG2 and HepG2R cells stained with propidium iodide after treatment with sorafenib (HepG2, 4 μM; HepG2R, 10 μM), BEZ235 (0.25 μM), or combined agents for 24 h. Cell-cycle distribution was analyzed by flow cytometry. Compared with control values, sorafenib and BEZ235 blocked the cell cycle at the G0/G1 phase, and blocking was greater with the two drugs combined (P<0.05). B. Total protein was extracted from HepG2 and HepG2R cells after treatment with sorafenib (HepG2, 4 μM; HepG2R, 10 μM), BEZ235 (0.25 μM), or combined agents for 24 h. Western blot analysis revealed that the key G1-phase regulatory proteins, cyclin D1 and p-Rb, and their upstream molecule, p-GSK3β, were also down-regulated. (**P<0.01; and *P<0.05), which arrested cells at G0/G1 phase. Data shown are the mean ± SD from three independent experiments.
The proteins cyclin D1 and p-Rb govern G1-to-S phase progression, and down-regulation of them leads to G1-phase arrest [16]. Thus, we examined the expression of endogenous cyclin D1, p-Rb, and their upstream regulatory protein GSK3β. As indicated in Figure 4B, p-GSK3β, cyclin D1 and p-Rb values were reduced in the HepG2 and HepG2R cell lines treated with BEZ235 and BEZ235 plus sorafenib. These data indicate that BEZ235 enhanced sorafenib arrest at G0/G1 mainly through the down-regulation of p-GSK3β and the key G1-phase regulatory proteins cyclin D1 and p-Rb.
BEZ235 enhanced the pro-apoptotic effect of sorafenib on HCC cells
To assess the ability of BEZ235 plus sorafenib to induce HCC cell apoptosis, the pro-apoptotic effects of the various treatments were examined with AnnexinV-FITC/propidium iodide staining and western blot. After 24 h of drug treatment, sorafenib and BEZ235 individually induced apoptosis in HepG2 and HepG2R cells, and the combination of two drugs had a greater effect (**P<0.01). The apoptosis rates of each group of HepG2 cells were 3%, 42%, 25%, and 80%, and the apoptosis rates of HepG2R were 5%, 26%, 42% and 73%, as suggested by the absence of bright red-stained cells (propidium iodide + cells) (Figure 5A). Western blot revealed that the amounts of caspase-7, caspase-3, caspase-9 cleavages, and the pro-apoptosis related protein, Bad, (but not Bax or Bak), were increased in HepG2 and HepG2R cells by treatment with the two drugs combined compared with the amounts with sorafenib or BEZ235 alone (Figure 5B). Over-expression of Bad, a key regulatory protein in the mitochondrial apoptotic pathway, leads to apoptosis by suppressing the activity of anti-apoptotic protein Bcl-xL, and further promotes the release of cytochrome C and cascade activation of caspase-9, caspase-3, and caspase-7 [17,18]. These results document that the pro-apoptotic effect of BEZ235 combined with sorafenib is superior to single treatment with these drugs in HCC cells.
Figure 5.

Immunofluorescence and protein analysis of the pro-apoptotic effect of sorafenib and BEZ235 on HepG2 and HepG2R cells. A. Annexin V-FITC/propidium iodide staining and histograms showing the percentage of apoptotic cells in HepG2 and HepG2R treated with sorafenib (HepG2, 4 μM; HepG2R, 10 μM), BEZ235 (0.25 μM), and in combination for 24 h. Sorafenib and BEZ235 individually induced apoptosis of HepG2 and HepG2R cells compared with the control, and the combination of two drugs had a greater effect than did the agents alone (**P<0.01). B. Total protein was extracted from HepG2 and HepG2R cells treated with sorafenib (HepG2, 4 μM; HepG2R, 10 μM), BEZ235 (0.25 μM), and in combination for 24 h. The amounts of apoptosis-related proteins were determined with western blot. The combination of sorafenib and BEZ235 promoted pro-apoptotic proteins caspase-9, caspase-3, and caspase-7 activation and up-regulated pro-apoptotic protein Bad, but not Bax or Bak. (***P<0.001; **P<0.01; and *P<0.05). Data shown are the mean ± SD from three independent experiments.
Effects of sorafenib and BEZ235 treatment on PI3K/AKT/mTOR signaling in HCC cells
To determine whether the effects of combined sorafenib and BEZ235 treatment are mediated by inhibition of the PI3K/AKT/mTOR pathway in HCC cells, we detected molecules of PI3K/AKT/mTOR signaling after 24 h-drug treatment, using western blot assay. As shown in Figure 6A, sorafenib promoted AKT and S6K phosphorylation but did not significantly affect phosphorylation of other proteins in the PI3K/AKT/mTOR pathway. BEZ235, in combination with sorafenib, suppressed PI3K and reduced p-AKT, p-mTOR, and downstream p-S6K in HepG2 and HepG2R cells compared with sorafenib or BEZ235 single treatment (Figure 6A). Interestingly, sorafenib also promoted c-Raf, MEK, ERK and p90RSK phosphorylation when no growth factors were present. However, BEZ235 had no effect on the key enzyme of Ras/Raf/MAPK signaling pathway (Figure 6B). Similar results were obtained in HepG2 and HepG2R cells.
Figure 6.
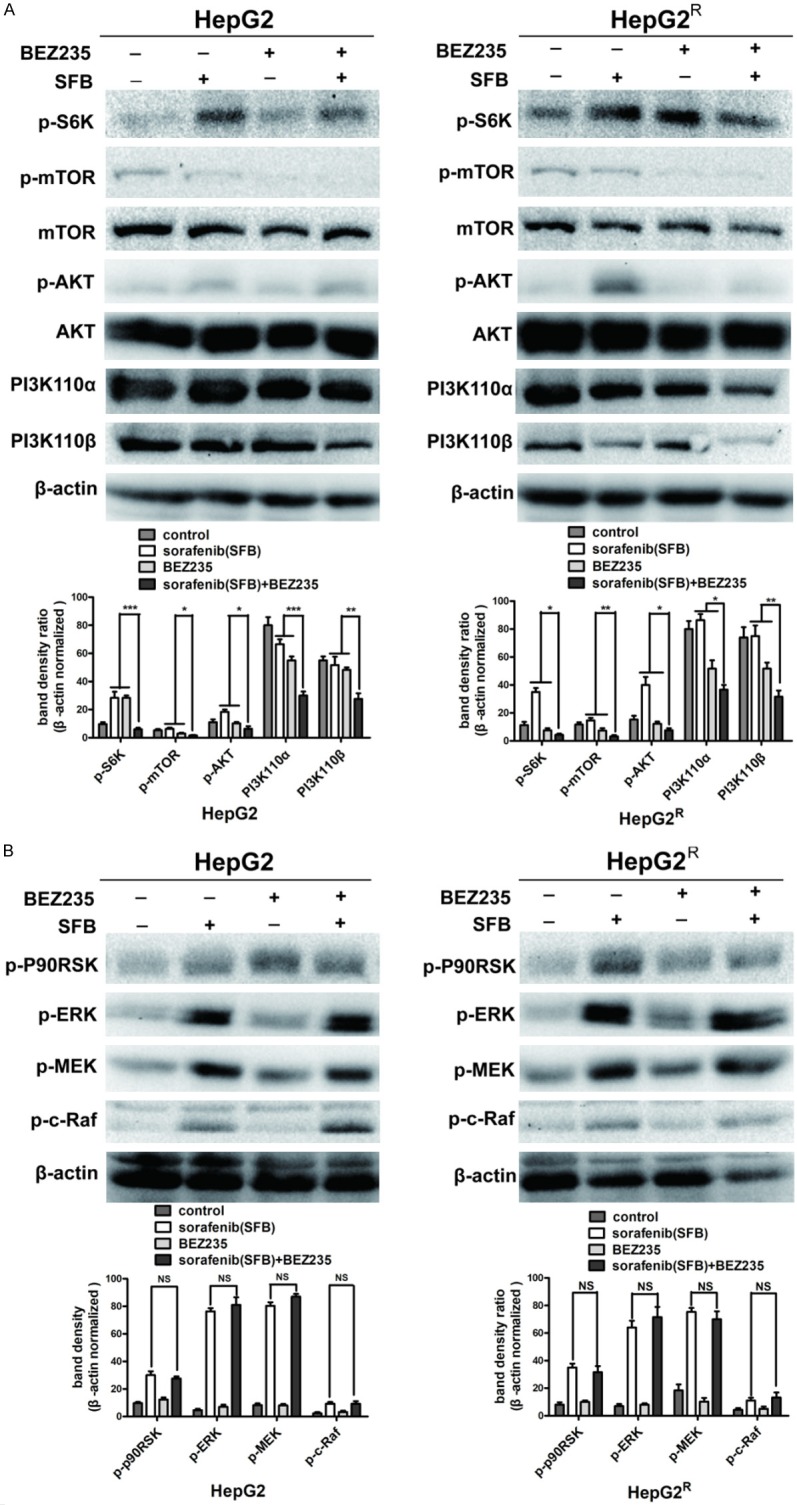
Protein analysis of PI3K/AKT/mTOR and Ras/Raf/MAPK pathways in HepG2 and HepG2R cells treated with sorafenib (HepG2, 4 μM; HepG2R, 10 μM), BEZ235 (0.25 μM), or the two drugs in combination for 24 h. A. Western blot analysis reveals that sorafenib promoted AKT and S6K phosphorylation in the PI3K/AKT/mTOR pathway in both HepG2 and HepG2R cells, which indicated that sorafenib monotherapy activated PI3K/AKT/mTOR signaling pathway. BEZ235, in combination with sorafenib, down-regulated PI3K, inhibited mTOR phosphorylation and sorafenib-induced phosphorylation of AKT and S6K. B. In both HepG2 and HepG2R cells, sorafenib promoted c-Raf, MEK, ERK and p90RSK phosphorylation in the absence of growth-factor stimulation, which may be a mechanism of cell self-protection and drug resistance. However, compared with sorafenib monotherapy, BEZ235 together with sorafenib did not decrease phosphorylation of sorafenib-induced key enzymes of the Ras/Raf/MAPK signaling pathway. Taken together, sorafenib and BEZ235 in combination treatment suppressed the PI3K/AKT/mTOR but not the Ras/Raf/MAPK pathway. (***P<0.001; **P<0.01; and *P<0.05). Data shown are the mean ± SD from three independent experiments.
Discussion
In this study, we described the anti-tumor effect of BEZ235 in combination with sorafenib in the HCC cell lines HepG2 and HepG2R. The anti-proliferative and pro-apoptotic activities of BEZ235 and sorafenib were increased in the cells when used in combination compared with activities when the drugs were used individually. Similarly, the inhibition of cell motility was greater when the two drugs were used in combination. Colony-formation and cell-motility assays also revealed that sorafenib induced HepG2 drug resistance, and colony proliferation and migration were enhanced more in HepG2R cells than in HepG2 cells. We speculate that this difference may be the result of sorafenib’s greater promotion of AKT phosphorylation in HepG2R cells than in HepG2 cells, as indicated in Figure 6A. BEZ235, a dual PI3K/mTOR inhibitor, dramatically inhibited cell motility and colony formation, especially when combined with sorafenib. Collectively, our observations suggest that BEZ235 developed anti-tumor effects with sorafenib, and the PI3K/AKT/mTOR pathway is important in HCC cell biological activities and drug resistance.
We also found that BEZ235 promoted sorafenib induction of apoptosis of HepG2 and HepG2R cells by upregulating Bad and activating caspase-9, caspase-3 and caspase-7. Thus, we speculate that the pro-apoptotic effect was achieved via the mitochondrial apoptotic pathway, even though it had no effect on the pro-apoptotic proteins Bax or Bak. The mitochondrial pro-apoptotic protein Bad is regulated by Ras/Raf/MAPK and PI3K/AKT/mTOR signaling pathways [18-20]. Activation of Ras/Raf/MAPK and PI3K/AKT/mTOR pathways could block Bad expression and further activate Bcl-xL to inhibit the mitochondrial apoptotic pathway. Sorafenib and BEZ235 suppress Ras/Raf/MAPK and PI3K/AKT/mTOR pathways, resulting in Bad activation and thus promotion of the mitochondrial apoptotic pathway.
Sorafenib, a multi-target kinase inhibitor, mainly inhibits Raf kinase to block tumor cell proliferation [21,22] (Figure 7). Interestingly, we found that sorafenib increased c-Raf, especially MEK and ERK phosphorylation, which we suspect may be a mechanism of cell self-protection. Western blot assay also showed that sorafenib enhanced AKT and S6K phosphorylation in the PI3K/AKT/mTOR signal pathway, which indicated crosstalk between Ras/Raf/MAPK and PI3K/AKT/mTOR signaling pathways and can explain sorafenib-induced HepG2 drug resistance [23,24]. The dual PI3K/mTOR inhibitor BEZ235, in combination with sorafenib treatment strongly downregulated PI3K and reduced phosphorylation of AKT, mTOR, and S6K.
Figure 7.

Diagram of Ras/Raf/MAPK and PI3K/AKT/mTOR signaling pathways. Binding of growth factors to receptors activates cascading activation of PI3K and Ras signaling pathways and promotes cell survival, proliferation, and migration. Activation of downstream proteins P90RSK and S6K down-regulates Bad expression and transposition to mitochondrial membranes, which promotes anti-apoptotic protein Bcl-xL expression and inhibits Cytochrome C release. Cytochrome C release can lead to cascading activation of caspase-9, caspase-3, and caspase-7 and result in cell apoptosis. In terms of cell-cycle regulation, GSK3β reduced the expression of cyclin D1, thereby inhibiting Rb phosphorylation and blocking cell-cycle progress. Phosphorylated AKT inhibits its activity by phosphorylating GSK3β and eventually promotes cell-cycle progression. Sorafenib (SFB), as well as BEZ235, targets multiple molecules to inhibit cell proliferation, migration, and cell-cycle progression and to promote cell apoptosis.
Many studies have reported that the Ras/Raf/MAPK pathway is activated in HCC cells [25-27]. Thus, sorafenib in advanced HCC has a significant impact on tumor progression and patient survival [28]. However, we had found that sorafenib loses effectiveness because the tumors develop drug resistance. One of the most important mechanisms for this outcome is crosstalk between signaling pathways, especially activation of PI3K/AKT/mTOR pathway [29,30]. mTORC2 phosphorylates AKT to enhance tumor cell survival and proliferation, and it also induces resistance to mTOR inhibitors [31,32]. BEZ235 is a dual PI3K/mTOR inhibitor that not only inhibits mTORC1 and mTORC2, but also blocks upstream of mTOR, resulting in a stronger inhibition of the PI3K/AKT/mTOR signal pathway (Figure 7).
We appreciate that our study was limited to in vitro study of HCC cells. Nonetheless, we believe that the results are promising enough to justify trials of combined sorafenib and BEZ235 in the treatment of HCC. Our future studies will be focused on in vivo research to validate in vitro results.
Conclusions
Sorafenib or BEZ235 treatment of HepG2 and HepG2R HCC cells differentially activated and inhibited the key kinases in the Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. This observation underscores the importance of the two-drug combination treatment of HCC cells in vitro. Application of the two drugs together further inhibited HCC cell proliferation and survival by suppressing the PI3K/AKT/mTOR pathway. Our results suggest a novel combination treatment strategy for HCC that deserves clinical study.
Acknowledgements
The National Natural Science Fund of China (NO. 81872017, NO. 81572431), Anhui Provincial Science and Technology program (NO. 1604a0802094), University Natural Science Research Project of Anhui Province (NO. KJ2018ZD011, KJ2019A0305, KJ2019A0093) and Huainan Science and Technology Project (NO. 2017B41) funded this research.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.de Rosamel L, Blanc JF. Emerging tyrosine kinase inhibitors for the treatment of hepatocellular carcinoma. Expert Opin Emerg Drugs. 2017;22:175–190. doi: 10.1080/14728214.2017.1336538. [DOI] [PubMed] [Google Scholar]
- 3.Kudo M. Targeted and immune therapies for hepatocellular carcinoma: predictions for 2019 and beyond. World J Gastroenterol. 2019;25:789–807. doi: 10.3748/wjg.v25.i7.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Psyrri A, Arkadopoulos N, Vassilakopoulou M, Smyrniotis V, Dimitriadis G. Pathways and targets in hepatocellular carcinoma. Expert Rev Anticancer Ther. 2012;12:1347–1357. doi: 10.1586/era.12.113. [DOI] [PubMed] [Google Scholar]
- 5.Tuchen M, Wilisch-Neumann A, Daniel EA, Baldauf L, Pachow D, Scholz J, Angenstein F, Stork O, Kirches E, Mawrin C. Receptor tyrosine kinase inhibition by regorafenib/sorafenib inhibits growth and invasion of meningioma cells. Eur J Cancer. 2017;73:9–21. doi: 10.1016/j.ejca.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Keating GM. Sorafenib: a review in hepatocellular carcinoma. Target Oncol. 2017;12:243–253. doi: 10.1007/s11523-017-0484-7. [DOI] [PubMed] [Google Scholar]
- 7.Parsons HM, Chu Q, Karlitz JJ, Stevens JL, Harlan LC. Adoption of sorafenib for the treatment of advanced-stage hepatocellular carcinoma in oncology practices in the united states. Liver Cancer. 2017;6:216–226. doi: 10.1159/000473862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38:614–622. doi: 10.1038/aps.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ray EM, Sanoff HK. Optimal therapy for patients with hepatocellular carcinoma and resistance or intolerance tosorafenib: challenges and solutions. J Hepatocell Carcinoma. 2017;4:131–138. doi: 10.2147/JHC.S124366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhat V, Srinathan S, Pasini E, Angeli M, Chen E, Baciu C, Bhat M. Epigenetic basis of hepatocellular carcinoma: a network-based integrative meta-analysis. World J Hepatol. 2018;10:155–165. doi: 10.4254/wjh.v10.i1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang X, Zhou S, Tao X, Wang J, Wang F, Liang Y. Targeted delivery of docetaxel via Pi-Pi stacking stabilized dendritic polymeric micelles for enhanced therapy of liver cancer. Mater Sci Eng C Mater Biol Appl. 2017;75:1042–1048. doi: 10.1016/j.msec.2017.02.098. [DOI] [PubMed] [Google Scholar]
- 12.Gedaly R, Angulo P, Hundley J, Daily MF, Chen C, Koch A, Evers BM. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res. 2010;30:4951–4958. [PMC free article] [PubMed] [Google Scholar]
- 13.Barra F, Evangelisti G, Ferro Desideri L, Di Domenico S, Ferraioli D, Vellone VG, De Cian F, Ferrero S. Investigational PI3K/AKT/mTOR inhibitors in development for endometrial cancer. Expert Opin Investig Drugs. 2019;28:131–142. doi: 10.1080/13543784.2018.1558202. [DOI] [PubMed] [Google Scholar]
- 14.Calero R, Morchon E, Martinez-Argudo I, Serrano R. Synergistic anti-tumor effect of 17AAG with the PI3K/mTOR inhibitor NVP-BEZ235 on human melanoma. Cancer Lett. 2017;406:1–411. doi: 10.1016/j.canlet.2017.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Jin T, Zhu K, Piao Y, Quan T, Quan C, Lin Z. PI3K/mTOR dual inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A synergistically exert anti-tumor activity in breast cancer. Oncotarget. 2017;8:11937–11949. doi: 10.18632/oncotarget.14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong JY, Park SH, Park HJ, Lee SK. Anti-proliferative effect of 15,16-dihydrotanshinone I through cell cycle arrest and the regulation of AMP-activated protein kinase/Akt/mTOR and mitogen-activated protein kinase signaling pathway in human hepatocellular carcinoma cells. J Cancer Prev. 2018;23:63–69. doi: 10.15430/JCP.2018.23.2.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi MD, Shiao CK, Lee YC, Shih YW. Apigenin, a dietary flavonoid, inhibits proliferation of human bladder cancer T-24 cells via blocking cell cycle progression and inducing apoptosis. Cancer Cell Int. 2015;15:33. doi: 10.1186/s12935-015-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351:41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 19.Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009;21:871–877. doi: 10.1016/j.ceb.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 20.Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 21.Wei JC, Meng FD, Qu K, Wang ZX, Wu QF, Zhang LQ, Pang Q, Liu C. Sorafenib inhibits proliferation and invasion of human hepatocellular carcinoma cells via up-regulation of p53 and suppressing FoxM1. Acta Pharmacol Sin. 2015;36:241–251. doi: 10.1038/aps.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 23.Yokoi K, Kobayashi A, Motoyama H, Kitazawa M, Shimizu A, Notake T, Yokoyama T, Matsumura T, Takeoka M, Miyagawa SI. Survival pathway of cholangiocarcinoma via AKT/mTOR signaling to escape RAF/MEK/ERK pathway inhibition by sorafenib. Oncol Rep. 2018;39:843–850. doi: 10.3892/or.2017.6153. [DOI] [PubMed] [Google Scholar]
- 24.Han P, Li H, Jiang X, Zhai B, Tan G, Zhao D, Qiao H, Liu B, Jiang H, Sun X. Dual inhibition of Akt and c-Met as a second-line therapy following acquired resistance tosorafenib in hepatocellular carcinoma cells. Mol Oncol. 2017;11:320–334. doi: 10.1002/1878-0261.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang XL, Lyu YG, Xie D, Li AM, Liang Y, Zheng DH. Therapeutic effect of sorafenib-loaded TPGS-b-PCL nanoparticles on liver cancer. J Biomed Nanotechnol. 2018;14:396–403. doi: 10.1166/jbn.2018.2529. [DOI] [PubMed] [Google Scholar]
- 26.Parikh ND, Marshall VD, Singal AG, Nathan H, Lok AS, Balkrishnan R, Shahinian V. Survival and cost-effectiveness of sorafenib therapy in advanced hepatocellular carcinoma: an analysis of the SEER-Medicare database. Hepatology. 2017;65:122–133. doi: 10.1002/hep.28881. [DOI] [PubMed] [Google Scholar]
- 27.Galuppo R, Maynard E, Shah M, Daily MF, Chen C, Spear BT, Gedaly R. Synergistic inhibition of HCC and liver cancer stem cell proliferation by targeting RAS/RAF/MAPKand WNT/β-catenin pathways. Anticancer Res. 2014;34:1709–1713. [PMC free article] [PubMed] [Google Scholar]
- 28.Tang X, Chen L, Li A, Cai S, Zhang Y, Liu X, Jiang Z, Liu X, Liang Y, Ma D. Anti-GPC3 antibody-modified sorafenib-loaded nanoparticles significantly inhibited HepG2 hepatocellular carcinoma. Drug Deliv. 2018;25:1484–1494. doi: 10.1080/10717544.2018.1477859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen D, Soh CK, Goh WH, Wang H. Design, synthesis, and preclinical evaluation of fused pyrimidine-based hydroxamates for the treatment of hepatocellular carcinoma. J Med Chem. 2018;61:1552–1575. doi: 10.1021/acs.jmedchem.7b01465. [DOI] [PubMed] [Google Scholar]
- 30.Tan W, Zhu S, Cao J, Zhang L, Li W, Liu K, Zhong J, Shang C, Chen Y. Inhibition of MMP-2 expression enhances the antitumor effect of sorafenib in hepatocellular carcinoma by suppressing the PI3K/AKT/mTOR pathway. Oncol Res. 2017;25:1543–1553. doi: 10.3727/096504017X14886444100783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jhanwar-Uniyal M, Amin AG, Cooper JB, Das K, Schmidt MH, Murali R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv Biol Regul. 2017;64:39–48. doi: 10.1016/j.jbior.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Jia QA, Kadel D, Zhang XF, Zhang QB. Targeting mTORC1/2 complexes inhibit tumorigenesis and enhance sensitivity to 5-flourouracil (5-FU) in hepatocellular carcinoma: a preclinical study of mTORC1/2-targeted therapy in hepatocellular carcinoma (HCC) Med Sci Monit. 2018;24:2735–2743. doi: 10.12659/MSM.907514. [DOI] [PMC free article] [PubMed] [Google Scholar]