

Phosphorylation of maize C4-NADP-malic enzyme at a specific Ser residue decreases the binding affinity to the cofactor, reducing the enzymatic activity during the first hours in the light.
Abstract
Evolution of the C4 photosynthetic pathway involved in some cases recruitment of housekeeping proteins through gene duplication and their further neofunctionalization. NADP-malic enzyme (ME), the most widespread C4 decarboxylase, has increased its catalytic efficiency and acquired regulatory properties that allowed it to participate in the C4 pathway. Here, we show that regulation of maize (Zea mays) C4-NADP-ME activity is much more elaborate than previously thought. Using mass spectrometry, we identified phosphorylation of the Ser419 residue of C4-NADP-ME in protein extracts of maize leaves. The phosphorylation event increases in the light, with a peak at Zeitgeber time 2. Phosphorylation of ZmC4-NADP-ME drastically decreases its activity as shown by the low residual activity of the recombinant phosphomimetic mutant. Analysis of the crystal structure of C4-NADP-ME indicated that Ser419 is involved in the binding of NADP at the active site. Molecular dynamics simulations and effective binding energy computations indicate a less favorable binding of the cofactor NADP in the phosphomimetic and the phosphorylated variants. We propose that phosphorylation of ZmC4-NADP-ME at Ser419 during the first hours in the light is a cellular mechanism that fine tunes the enzymatic activity to coordinate the carbon concentration mechanism with the CO2 fixation rate, probably to avoid CO2 leakiness from bundle sheath cells.
INTRODUCTION
The world’s most productive crops perform C4 photosynthesis. The ancestors of these plants evolved a biochemical pump that concentrates CO2 at the site of ribulose-1,5-bis-phosphate carboxylase/oxygenase (Rubisco), leading to lower photorespiratory fluxes and greater photosynthetic efficiency (Furbank and Hatch, 1987). Compared with the ancestral C3 photosynthesis, the C4 pathway allows increased plant productivity in warm habitats due to a more efficient use of nitrogen and water.
The most agronomically important C4 plants, maize (Zea mays), sorghum (Sorghum bicolor), and sugarcane (Saccharum officinarum), belong to the NADP-malic enzyme (ME) C4 photosynthetic subtype. In these plants, CO2 is initially fixed in the mesophyll cells by phosphoenolpyruvate carboxylase (PEPC), leading to the formation of oxaloacetate. Oxaloacetate is then predominantly reduced to malate and transported into the bundle sheath cells (BSCs), where CO2 is released by NADP-ME for its refixation through Rubisco (Hatch, 1987). By this process, CO2 concentration in the BSCs rises to levels up to 1500 μL L−1 (Furbank and Hatch, 1987; Sage et al., 2012).
The transition from C3 to C4 metabolism involved complex alterations to leaf anatomy and biochemistry. A crucial step in the evolution of the C4 photosynthetic pathway was the recruitment of some enzymes through gene duplication of housekeeping isoforms and subsequent neofunctionalization. In the course of its evolution, the catalytic efficiency of the C4-NADP-ME isoform increased and the enzyme acquired regulatory properties that optimized its efficiency in the C4 pathway (Saigo et al., 2013; Alvarez et al., 2019). During the night, when the delivery of malate from the PEPC reaction is stopped, C4-NADP-ME activity is inhibited by malate (Saigo et al., 2004, 2013). This property, exclusive to the C4 isoform, minimizes malate consumption to avoid extreme carbon loss during the night period, which would otherwise result in carbon starvation (Fahnenstich et al., 2007; Zell et al., 2010). At the pH of the stroma in darkness, a proportion of C4-NADP-ME likely loses its quaternary structure and adopts a lower oligomerization state that is less active or even inactive (Iglesias and Andreo, 1990). This likely represents a second level of regulation that ensures that C4-NADP-ME is less active during the night period.
The enzymatic activities of some enzymes of C4 metabolism and also of the Crassulacean acid metabolism (CAM) carbon-concentrating pathway are known to be regulated through posttranslational modifications (PTMs). PTMs provide mechanisms for rapid and reversible control of activity of these proteins in a coordinated fashion during the day and night and in response to light intensity (Walker et al., 2002; Bailey et al., 2007; Chen et al., 2014). The activity of C4-PEPC is regulated through phosphorylation of a Ser residue located at the N-terminal region by a small PEPC kinase (Nimmo, 2003). C4-PEPC is mainly phosphorylated in the light, where it has a higher catalytic activity and is less sensitive to the allosteric inhibition by malate than in darkness (Jiao and Chollet, 1988, 1989, 1990). Transgenic lines of Flaveria bidentis with antisense or RNA interference constructs targeted at the mRNA of the PEPC kinase demonstrated that the kinase is essential for the phosphorylation of C4-PEPC in vivo (Furumoto et al., 2007). In the transgenic lines, C4-PEPC was not phosphorylated in the light; nevertheless, there were no differences in the CO2 and light response of CO2 assimilation rates between these lines and the wild type (Furumoto et al., 2007). These results suggest that phosphorylation of PEPC in the light is not essential for efficient C4 photosynthesis in plants grown under standard greenhouse conditions, but it remains to be determined whether photosynthetic rates are affected under stress conditions.
PEPC of the obligate CAM species Kalanchoë fedtschenkoi is also regulated by phosphorylation (Hartwell et al., 1999; Nimmo, 2000). A specific PEPC kinase phosphorylates CAM-PEPC during the dark period, reducing its sensitivity to feedback inhibition by malate, enabling nocturnal fixation of CO2 into malate (Boxall et al., 2017). C4-pyruvate orthophosphate dikinase (C4-PPDK) activity, by contrast, is not controlled by the light/dark transition per se but rather by the light intensity. In this case, increased phosphorylation takes place with waning light intensity in the evening, leading to a decrease in activity (Chen et al., 2014). C4-PPDK is regulated by reversible phosphorylation of a Thr residue of the active site (Burnell and Hatch, 1985; Chastain et al., 2000) catalyzed by the PPDK regulatory protein (PDRP; Chen et al., 2014). PDRP is bifunctional; it catalyzes both PPDK activation/dephosphorylation and PPDK inactivation/phosphorylation (Burnell and Chastain, 2006; Chastain et al., 2008; Astley et al., 2011). The regulatory phosphorylation of C4-PPDK is an ancient mechanism, as genes encoding PDRP are present in C3 plants and prokaryotes (Agarie et al., 1997; Imaizumi et al., 1997; Wei et al., 2000; Burnell, 2010). In some C4 plants such as Guinea grass (Panicum maximum) and maize, PEP carboxykinase (C4-PEPCK) is phosphorylated in the dark, when the enzyme is less active, and dephosphorylated under illumination (Walker and Leegood, 1996; Walker et al., 2002; Chao et al., 2014). Modifications in the phosphorylation state of PEPCK lead to changes in its sensitivity to adenylates, which would be involved in the activation of the enzyme in the light (Walker et al., 2002). Recently, it was shown that phosphorylation of a Ser residue in maize C4-PEPCK is also dependent on the light regime (Chao et al., 2014).
Little is known about the participation of PTMs in the regulation of the C4-NADP-ME isoform in vivo. Until now, only the effects of redox modulation on the recombinant ZmC4-NADP-ME activity were reported (Alvarez et al., 2012). Oxidation of ZmC4-NADP-ME decreases the catalytic activity and increases the affinity for malate and the cofactor; in combination, these changes produce no significant changes of the catalytic efficiency. The oxidation of Cys192, Cys246, Cys270, and Cys410 may contribute to the changes observed in vitro (Alvarez et al., 2012).
Here, we show a novel PTM, phosphorylation at Ser419 (S419), of ZmC4-NADP-ME, that takes place in vivo after the light is turned on, with a peak at 2 h after the onset of light. The results of the characterization of the recombinant phosphomimetic mutant S419E together with the analysis of the crystal structure of C4-NADP-ME, and molecular modeling and dynamic simulations of wild-type and mutant enzymes, indicate that phosphorylation at Ser419 inactivates ZmC4-NADP-ME because Ser419 is involved in NADP binding at the active site. We postulate that the novel PTM of ZmC4-NADP-ME described here is a mechanism that fine tunes the enzymatic activity, probably to coordinate malate decarboxylation with carbon fixation in BSCs.
RESULTS
Identification of a Phosphorylation Site in ZmC4-NADP-ME
Mass spectrometric identification of phosphorylation sites in complex protein extracts faces several challenges. Phosphopeptides are often of low abundance and often exhibit low mass spectrometry (MS) signals and inadequate fragmentation patterns. To overcome these issues and to be able to identify phosphorylated peptides of ZmC4-NADP-ME in cell lysates obtained from maize leaf macerates, we loaded maize protein extracts onto 4 to 12% NuPAGE gels and electrophoresed them for a short time. Upon in-gel digestion, samples were subjected to a phosphopeptide enrichment step and analyzed by liquid chromatography (LC)-MS using a TripleTOF 6600 mass spectrometer (AB Sciex). The acquired data were analyzed with Protein Pilot 5.0 and PeakView 2.2. This approach was successful in unambiguously detecting the phosphorylation of ZmC4-NADP-ME at a single amino acid residue, Ser419. Figure 1 shows a tandem MS spectrum of the obtained peptide acVWLVDpSK. The quality of the tandem MS data for this peptide enabled the correct assignment of the phosphorylated residue. With the exception for the y1-ion, the entire y-ion series of the peptide was assigned, and the loss of the phosphate group (H3PO4), corresponding to a mass-to-charge ratio of 98, was detected for the whole y-ion series.
Figure 1.

TripleTOF 6600 Tandem MS Data of the Phosphopeptide acVWLVDpSK of ZmC4-NADP-ME.
The detected b (N-terminal, in red) and y (C-terminal, in blue) fragment ions are labeled in the spectrum. Ac denotes N terminus acetylation and pS denotes phosphorylated Ser. Precursor charge: +2; monoisotopic m/z: 484.7265 D (−1.60 milli-mass unit/−3.30 ppm). Confidence (ProteinPilot): 96.4% (confidence threshold for FDR ≤ 1% = 93.7%).
The MS analysis did not identify any phosphorylation event in the other maize NADP-ME isoforms, as the Val414 from the identified phosphorylated peptide is unique to ZmC4NADP-ME (Alvarez et al., 2013). We conclude that a functional role for this phosphorylation site probably represents a C4-trait. We next compared the protein sequences of plastidic photosynthetic and nonphotosynthetic NADP-ME isoforms within the Panicoideae subfamily of the Poaceae. This sequence alignment indicated that the Ser corresponding to amino acid position 419 in the C4-NADP-ME protein sequence is conserved among all NADP-ME sequences analyzed (Supplemental Figure 1).
Production of the Phosphomimetic Mutant and Characterization of the Biochemical Properties
To analyze the effect of phosphorylation of Ser419 on the biochemical properties of ZmC4-NADP-ME, we generated the phosphomimetic mutant S419E by changing the Ser419 to Glu by site-directed mutagenesis. Phosphorylation adds negative charge to amino acids, and thus the negatively charged Glu can be used to mimic the phosphorylated state of a protein (Dissmeyer and Schnittger, 2011). To assess the importance of the amino acid position for the enzymatic activity, we also created the S419A variant by changing Ser419 to Ala, a small nonpolar amino acid.
ZmC4-NADP-ME wild type, S419E, and S419A were heterologously expressed and purified to homogeneity by affinity chromatography. The recombinant proteins, showing in all cases the expected molecular masses (Supplemental Figure 2), were subjected to a comparative analysis of their enzymatic properties. Analysis of the dependence of the enzymatic activity on the pH of the reaction media indicated that while the recombinant wild-type ZmC4-NADP-ME has a pH optimum of 8.0, the S419E variant presents a shift of the pH optimum toward more acidic pHs between 6.5 and 7.0 (Figure 2A). The S419A variant showed a pH optimum of 7.0 to 7.5 (Figure 2A). At the physiologically relevant pH 8.0 of the photosynthetically active stroma, the S419E variant presents only 8.7% of the wild-type activity, while the S419A variant retains 72.3% of the wild-type activity (Figure 2A).
Figure 2.

Biochemical Characterization of Recombinant ZmC4-NADP-ME Versions.
(A) Dependence of the activity, measured using 0.5 mM NADP and 4 mM malate, on the pH of the assay. Values represent the mean ± se of at least four independent enzyme preparations, each measured in triplicate. Vo, initial velocity.
(B) Kinetic parameters at pH 8.0. Kinetic data were best fitted by nonlinear regression analysis. Values represent the mean ± se of at least three independent enzyme preparations, each measured in triplicate.
Analysis of the affinity to NADP indicated that the exchange of the Ser residue for Glu in the S419E variant dramatically decreases the affinity of the mutant for the cofactor, as saturation was not observed even at NADP concentrations as high as 5 mM. The S419A variant presented a 13-fold lower affinity (Km = 159.5 µM) for the cofactor in comparison to the wild type (Km = 12.3 µM; Figure 2B), indicating that Ser419 is essential for the normal operation of the enzyme.
The affinity for the substrate malate at pH 8.0 of the wild type and S419A was measured at saturating NADP concentrations. As we found that S419E is not saturated by NADP even at very high concentrations, the apparent Michaelis constant (Kmapp) was determined at a subsaturating concentration of the cofactor. In comparison to the wild type (Km = 0.19 mM), the apparent affinity for malate decreased by ∼14-fold in the S419E variant (Kmapp = 2.68 mM) and only 2-fold in the S419A variant (Km = 0.47 mM). The turnover number (kcat) was measured at pH 8.0, at saturating levels of malate, and at 0.5 mM NADP. Under these conditions, the S419E variant displayed a sixfold lower catalytic activity (kcatapp = 2.1 s−1) with respect to the wild type (kcat = 12.2 s−1; Figure 2B). By contrast, the kcat of the S419A variant (8.3 s−1) was in the same range as that of the wild type (Figure 2B).
Structural Organization of the Mutant Variants
As the phosphomimetic mutant showed dramatic changes of its kinetic properties in comparison to the wild-type enzyme, we analyzed whether these could be the result of alterations in the structural organization of the engineered protein. We used circular dichroism (CD) to explore the secondary structure organization and analytical ultracentrifugation (AUC) to explore the quaternary composition.
CD measurements at both pH 7.0 and pH 8.0 indicated no significant changes in the secondary structure organization of the mutated variants in comparison to the wild type (Figures 3A and 3B). ZmC4-NADP-ME wild type and the Ser419E and S419A variants presented similar fractions of different types of secondary structures depending on the pH. At pH 8.0, all enzymes possess almost twice the amount of helical structures observed at pH 7.0, an ∼1.5-fold lower amount of β strand structures, an almost unchanged amount of turns, and an only slightly decreased amount of unordered secondary structures (Supplemental Figure 3; Supplemental Table 1). As shown in Supplemental Figure 3 for the case of the wild type, the higher signal at 195 nm together with the more pronounced minimum at 208 nm is indicative of an overall increase in α-helical structure at pH 8.0. The decrease of β strand structure is hardly visible directly in the spectra, since the CD-effect of β strand structure is lower than that of α helix. This is why slight changes are less visible.
Figure 3.

Exploration of the Secondary Structure Organization and Quaternary Composition of Recombinant ZmC4-NADP-ME Versions.
(A) and (B) CD spectra of ZmC4-NADP-ME wild type (WT) and the two mutated versions obtained at pH 7.0 (A) and pH 8.0 (B). Ten accumulations each were collected from 190 to 260 nm for 0.16 mg mL−1 enzyme, at 20°C. Each graph is showing the reconstructed curves obtained by applying CONTIN/LL algorithm for data evaluation as provided by the Dichroweb server. MRE, molar residue ellipticity.
(C) and (D) Continuous sedimentation coefficient distribution of ZmC4-NADP-ME wild type (WT) and the mutated versions at pH 7.0 (C) and pH 8.0 (D). Data were fitted with the ls-g*(S) model in the software package SEDFIT.
A comparison of the analysis presented in Supplemental Table 1 with the secondary structure determined from the crystal structure of ZmC4-NADP-ME (Protein Data Bank ID code: 5OU5; Alvarez et al., 2019) indicates that the total helix content measured by CD increases with the pH from 24% at pH 7.0 to 37% at pH 8.0. The increase in helical structure is achieved mainly at the expense of total β strand conformation, which decreases from 28% at pH 7.0 to 20% at pH 8.0. The change in the fractions of turn and unordered conformations is less pronounced. In comparison, the crystal structure of ZmC4-NADP-ME has 46% β-helical structure and ∼11% β strand conformation. While the change in pH has a striking impact on the conformation of the protein, the structure measured by CD at pH 8.0 is more similar to the crystal structure than the structure measured by CD at pH 7.0. That the structure of a protein in solution differs from its crystal structure is expected; in solution different conformational ensembles exist, while in a crystal a single conformation prevails. Additionally, different environments in solution and in the crystal and the presence or absence of the cofactor might favor different conformations. Either an ordering of the protein as the pH decreases or alterations due to changes in domain interactions are possible causes of the deviation in secondary structure composition obtained by the different methods.
AUC measurements conducted at both pH 7.0 and pH 8.0 (Figures 3C and 3D) confirmed a stable tetrameric state of wild-type ZmC4-NADP-ME at pH 8.0, with a sedimentation coefficient of 11.7 S (Figure 3D). Both mutated variants also assemble as tetramers at pH 8.0, showing sedimentation coefficients of 11.6 S and 11.1 S (Figure 3D). The sedimentation coefficient distribution of all proteins at pH 7.0 showed two peaks, with sedimentation coefficients of ∼11.5 S and 5.4 S (Figure 3C), indicating that at this pH the enzymes exist as a mixture of monomers/dimers and tetramers with predominance of the latter. The results also indicate that the generated mutant variants have no altered quaternary structure at any pH in comparison to the wild type. It can be speculated that the changes in amount of helical and turn components observed in the CD measurements at the different pHs might be related to the occurrence of a dimeric fraction at pH 7.0, as these changes may contribute to the alterations in the interface contacts between the monomers in the tetramer.
Taken together, the results of CD and AUC analyses indicate that the observed changes in the kinetic properties of the mutant variants are not due to changes in the structural organization of the proteins with respect to the wild type at each pH analyzed.
Diurnal Profiles of ZmC4-NADP-ME Phosphorylation State, NADP-ME Activity, and Malate Concentration in Maize Leaves
We quantified ZmC4-NADP-ME total protein amount in leaves of maize at different time points during the day using sequential window acquisition of all theoretical fragment ion spectra (SWATH)-MS. We found that during the day, there are no major fluctuations of the ZmC4-NADP-ME total protein amount (Figure 4A). Following the light-to-dark transition, there is an increase in total ZmC4-NADP-ME protein level (Figure 4A). Despite a decreasing trend over the dark period from Zeitgeber time 16.5 (ZT16.5) to ZT23.5, the relative levels of ZmC4-NADP-ME to the total protein levels (as measured by SWATH-MS) are higher during the whole dark period compared with the light period.
Figure 4.

Measurements of Different Parameters in Extracts of Maize Leaves during a Diurnal Cycle.
(A) Quantification of total ZmC4-NADP-ME protein by SWATH-MS.
(B) Quantification of the phosphopeptide VWLVDpSK, corresponding to the phosphorylation of ZmC4-NADP-ME at S419.
(C) Total NADP-ME activity measured with 1.5 to 2.0 µg of protein extracts.
(D) Malate concentration assayed by GC-MS analysis. Values represent mean ± se of four (see [A] and [B]) or three (see [C] and [D]) biological replicates. The dark period is highlighted in gray. Statistical analyses were performed in all cases (see [A] to [D]) against the first time point in the night (ZT16.5 = 16.5 h). Letters indicate that the value is statistically significant at 0.05 (a), 0.01 (b), and 0.001 (c) levels (the precise P-values are shown in Supplemental Table 3). FW, fresh weight.
Quantification of phosphorylation in Ser419 throughout the day indicated a significant increase of phosphorylation events after the light is turned on, with a peak at ZT2 that accounts for ∼13% of the total ZmC4-NADP-ME protein. This peak is followed by a substantial decrease to phosphorylation levels as low as 3% at ZT4 in the light. These low levels are maintained for the remainder of the day and also during the night (Figure 4B).
Measurements of total NADP-ME activity in extracts of maize leaves indicated a slight, but not significant, increment of the activity between ZT2 and ZT4 in the light, followed by a constant decline during the rest of the light period (Figure 4C). The activity measured in total leaf extracts is the sum of the activity of all maize NADP-ME isoforms. As the C4 isoform is the most highly expressed isoform in photosynthetic tissues with respect to the other isoforms (Detarsio et al., 2008; Alvarez et al., 2013), we conclude that the tendencies observed in the profile of measured NADP-ME activity represent the activity of the C4 isoform.
We next investigated the profile of malate concentration in whole maize leaf extracts during a diurnal cycle (Figure 4D). Similar high malate concentrations were observed during the light period and at ZT16.5 (530 to 670 nmol g−1 fresh weight). These values decreased during the dark period, reaching a value of ∼340 nmol g−1 fresh weight by the end of the night (Figure 4D).
Localization of S419 in the C4-NADP-ME Structure
We recently solved the crystal structures of C4-NADP-ME of maize (ZmC4-NADP-ME; Protein Data Bank ID code: 5OU5) and sorghum (SbC4-NADP-ME; Protein Data Bank ID code: 6C7N), which both display a homo-tetrameric assembly composed of two dimers (monomers A and B and monomers C and D, respectively; Figure 5A; Alvarez et al., 2019). The overall structure of SbC4-NADP-ME is highly similar to that of ZmC4-NADP-ME (root-mean-square deviation 0.81 Å over 1584 Cα-atoms), although parts of monomers C and D (residues 360 to 530) are not unambiguously traceable in the electron density. However, SbC4-NADP-ME monomers A and B are of high quality and clearly show a NADP molecule bound in the active site in each monomer (Figures 5A and 5B; Supplemental Figure 4A).
Figure 5.

SbC4-NADP-ME Crystal Structure and Amino Acids Involved in Cofactor Binding.
(A) Cartoon and ribbon representation of SbC4-NADP-ME. Monomer A (red) and B (blue) with bound cofactor NADP (colored sticks). Large parts of chains C and D are not well resolved by electron density; therefore, we depicted these chains as gray ribbons only to clarify the tetrameric assembly.
(B) Monomer A from SbC4-NADP-ME as surface representation with the bound cofactor NADP (represented by sticks).
(C) Residues involved in cofactor binding are depicted as sticks with labels (distances are omitted for clarity and are listed in Supplemental Table 2).
To elucidate the influence of Ser419 phosphorylation on the biochemical changes observed in C4-NADP-ME, we analyzed the cofactor binding site on the crystal structure of SbC4-NADP-ME in a complex with NADP (Protein Data Bank ID code: 6C7N; Alvarez et al., 2019). The nicotine amide moiety of NADP interacts with the O-atoms of Ser535, whereas both oxygen atoms of the ribose are hydrogen bonded by the N-atoms of Asn492 and the side chain O of Ser491. The phosphate O-atoms show several interactions to the side and main chain atoms of Glu386, Ala387, and Asn331, while the ribose 3′-OH group is hydrogen bonded to the backbone nitrogen of A384. Most interactions are present between the 2′-phosphate of the adenosine moiety and the protein, that is, to the main and side chain atoms of Lys435 and Ser419 (Figure 5C; Supplemental Table 2). These analyses clearly demonstrate that Ser419 plays an important role in NADP binding.
Using the structural information, we also found that Ser419 is located on the solvent-exposed surface of the enzyme. This residue might therefore be spatially available for the interaction with a kinase/phosphatase (Supplemental Figures 3B and 3C). Analysis of known kinase/phosphatase binding motifs associated with the identified phosphorylation site in ZmC4-NADP-ME indicated the presence of a β-adrenergic receptor kinase (BARK) substrate motif DSKGL (Supplemental Figure 1; Amanchy et al., 2007).
NADP Binding Is Less Favorable to the S419E and Phosphorylated Variants Than to Wild-Type ZmC4-NADP-ME
To assess differences in the binding affinity of NADP to wild-type ZmC4-NADP-ME, the variant carrying the phosphomimetic substitution S419E, and the phosphorylated enzyme pS419, we performed 10 independent molecular dynamics (MD) simulations of 100-ns length each on the different complexes. Only moderate structural changes were observed during the MD simulations, in particular in the second halves of the trajectories (Supplemental Figure 5). However, preliminary simulation results showed that, especially for the variant systems, NADP has the tendency to unbind (Supplemental Figure 6). This finding indicates that the affinity of NADP to the S419E and pS419 variants is lower than to the wild type. Furthermore, the higher mobility of the cofactor in the variant’s binding site may explain the approximately sixfold lower turnover rate (kcat) of the S419E variant compared with the wild-type ZmC4-ME (Figure 2), as then obtaining a reactive configuration with malate becomes less likely.
As the NADP unbinding would compromise the comparability of results of effective binding energy calculations (Homeyer et al., 2014), the MD simulations were repeated restraining NADP to the respective binding sites (Supplemental Figure 7). Effective binding energies were then computed by the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method (Gohlke and Case, 2004; Miller et al., 2012; Wang et al., 2016), using the single-trajectory approach on the second halves of the trajectories and considering contributions due to gas-phase energies and solvation free energies but neglecting contributions due to changes in the configurational entropy (see “Methods” for a justification). In line with the preliminary simulation results, the effective energy of binding of NADP toward the S419E and pS419 variants is significantly larger compared with that of NADP binding to the wild type (Figure 6A), indicating that binding to the two variants is much less favorable than to the wild type. This finding agrees with the experimental observation that no saturation concentration of NADP could be reached during biochemical characterization of the S419E variant (Figure 2).
Figure 6.

Effective Free Energies of Binding of NADP.
(A) Effective binding energies computed according to the MM-PBSA approach for wild-type (WT) ZmC4-NADP-ME, the phosphomimetic variant S419E, and pS419. The error bars indicate the se of the mean over 10 individual trajectories. Statistical significance was calculated according to Student’s t test. ΔGeff, effective binding energy.
(B) Contribution of the different energy terms, computed as variant’s (Var) energy term minus the respective energy term of the wild-type (WT) enzyme [ΔΔGeff. = ΔGeff(Var) − ΔGeff(WT)]. Positive terms indicate more favorable binding to the WT enzyme, negative terms to the variant. The error bars show the se of the mean of the differences in the individual terms over the trajectories. ΔΔGeff, relative binding free energy.
(C) Molecular representation of NADP bound to the wild-type (WT) ZmC4-NADP-ME, the phosphomimetic variant S419E, and pS419. Interactions with residue 419 are highlighted, showing hydrogen bonds for the WT enzyme with green dashed lines and charge-charge repulsion for the phosphomimetic and phosphorylated variants with red dashed lines.
Furthermore, a structural decomposition on a per-residue level of the terms contributing to the effective binding energy revealed only minor differences between the variants and the wild type in the van der Waals interactions and nonpolar contributions of the solvation free energy (Figure 6B). By contrast, the variants’ lower binding affinity to NADP is caused by an increase in the electrostatic energy, which is only partially compensated for by a more favorable polar solvation (Figure 6B). These energetic changes coincide on the structural level with a loss of the hydrogen bond between Ser419 and the 2′-phosphate of NADP in the wild type and charge repulsion of the negatively charged opposing groups in the variants (Figure 6C).
DISCUSSION
ZmC4-NADP-ME Is Phosphorylated in Vivo at a Ser Residue That Is Directly Involved in NADP Binding
Through MS, we identified a single phosphorylation event of ZmC4-NADP-ME at Ser419 in maize leaves. Phosphorylation levels increase after the light is turned on, with a peak at ZT2. Through mutational analysis and biochemical characterization of ZmC4-NADP-ME wild type and enzyme variants, we demonstrate that the phosphomimetic mutant S419E has a highly reduced affinity to the cofactor NADP as well as lower catalytic activity compared with the nonphosphorylated enzyme. S419E presented a low residual activity at the stromal pH in the light, indicating that a negative charge at this site of ZmC4-NADP-ME drastically decreases its activity. Analysis of the crystal structures of Zm- and SbC4-NADP-ME indicated that Ser419 is located on the surface of the enzyme, which makes it spatially available for the interaction with a kinase/phosphatase. Moreover, analysis of the crystal structure indicates that Ser419 is directly involved in NADP binding. Molecular dynamics simulations and free energy calculations revealed that the effective energy for NADP binding is less favorable in the phosphomimetic variant (S419E) and the phosphorylated variants than in the wild type. Additionally, the higher mobility of the cofactor in the variants’ binding sites likely leads to binding positions of NADP that are not suitable for a reaction with the substrate. This explains the highly reduced affinity of S419E for NADP and suggests that phosphorylation of Ser419 modulates the activity of ZmC4-NADP-ME by creating unfavorable binding conditions for its cofactor.
Furthermore, we observed conservation of Ser419, as well as of a BARK substrate motif (DSKGL), which includes Ser419, among plastidic photosynthetic and nonphotosynthetic NADP-ME isoforms within the Poaceae (Supplemental Figure 1). This pattern of sequence conservation and the fact that we found that only the C4-NADP-ME isoform is phosphorylated in leaves suggest that the use of reversible phosphorylation of Ser419 to control the activity of NADP-ME in photosynthetic tissues appeared with the evolution of the C4 pathway and should involve a BSC chloroplastic kinase, which might have been co-opted for this specific task.
Phosphorylation of ZmC4-NADP-ME at S419 Is a Mechanism That Fine Tunes the Enzymatic Activity
The operation of the C4 pathway requires a deep synchronization between mesophyll cells and BSCs during a day–night cycle (Bailey et al., 2007). To efficiently concentrate CO2 in BSC chloroplasts, coordination of C4-NADP-ME decarboxylation and Rubisco carboxylation rates is imperative as this will prevent loss of the CO2 released by C4-NADP-ME. During the night, when the photosynthetic pathway is inactive, C4-NADP-ME activity is regulated through two processes: on the one hand, the enzyme is allosterically inhibited by malate; on the other hand, it partially loses its active quaternary oligomerization state (Iglesias and Andreo, 1990; Saigo et al., 2004, 2013; Alvarez et al., 2019). Production of malate in mesophyll cells and its decarboxylation in BSCs must also be coordinated to accommodate the large and rapid flux changes that occur during the day. We show that malate concentrations in total maize leaf extracts tend to decrease during the night and increase after the onset of light, quickly reaching values that are maintained during the whole light period. This profile of malate concentrations in maize leaves is different from that described in some C3 plants such as Arabidopsis (Arabidopsis thaliana), where malate functions as a transient carbon storage molecule; in these C3 plants, malate concentrations increase continuously during the day and decrease during the night (Fahnenstich et al., 2007; Zell et al., 2010). The differences of malate levels between the end of the night and the day in maize leaves is likely mostly due to the activity of the C4-concentrating mechanism.
Apart from the contribution of malate decarboxylation, respiration of CO2 in BSCs also contributes to the carbon-concentrating mechanism (Bellasio and Griffiths, 2014a, 2014b). It is possible that under our conditions of growth, at ZT2, respiration in BSCs is high enough to make an important contribution to CO2 fixation by Rubisco. In such conditions, decarboxylation of malate by a fully active C4-NADP-ME could lead to high CO2 levels in BSCs, resulting in increased wasteful leakage of CO2 to mesophyll cells (Kromdijk et al., 2014). The naturally evolved efficiency of C4 photosynthesis requires the tight regulation of CO2 supply to Rubisco within the BSCs in order to minimize leakiness and associated energy costs (Furbank et al., 1990). As a consequence, a decrease in C4-NADP-ME activity would avoid extra waste of ATP for the operation of the C4 carbon concentration mechanism. The cooperation of the PEPC-driven carbon concentration and respiration-driven mechanisms requires plasticity, as the extent in which each of these mechanisms is involved may depend on changes of environmental conditions. Phosphorylation of ZmC4-NADP-ME at Ser419 during the first hours in the light might thus be a mechanism that fine tunes the enzymatic activity, helping to adjust the use of malate in the BSCs to avoid CO2 leakiness and energy waste. The functional importance of this molecular switch is probably linked to changes in environmental conditions such as photosynthetically active radiation, shade by canopy, and water availability.
Regulation of C4-NADP-ME activity via phosphorylation may also be important in the context of biochemical flexibility of the C4 pathway. Aspartate movement to maize BSCs carries ∼4% of the CO2 for the C3 cycle. Approximately 40% of this C4-acid is decarboxylated by PEPCK (Arrivault et al., 2017); the remaining aspartate is converted to malate and decarboxylated through NAD(P)-ME. There exists a flexible partitioning of C4 decarboxylation activity between NADP-ME and PEPCK in response to environmental conditions in maize (Furbank, 2011; Sharwood et al., 2014). In this context, we hypothesize that phosphorylation of ZmC4-NADP-ME at Ser419 might be a mechanism that rapidly decreases the activity of C4-NADP-ME and thereby switches the use of malate to aspartate according to the metabolic demands imposed by a constantly changing environment.
A currently highly popular goal within the plant scientific community is the improvement of photosynthesis by introducing the C4 concentrating mechanism into C3 plants. Our results are of great importance in this context, as they describe a novel regulation of a C4 enzyme critical for an efficient C4 cycle. Further work should now be directed toward elucidating the environmental conditions in which this regulatory mechanism is required, and if it effectively contributes to the coordination of the C4 and C3 cycles and hence to the metabolic flexibility of C4 plants in response to fluctuating environments.
METHODS
Plant Growth Conditions
For the MS analysis, maize (Zea mays; B73) seeds were germinated for 48 h in the dark at 28°C and then transferred to a soil:turf (1:1) mixture. Plants were grown under the following conditions: light/dark cycles of 16/8 h at 28°C/26°C; light intensity of 450 µmol m−2 s−1 (72 lamps with the reference LUMILUX T8 liters58 W/840; 12 lamps with the reference HALOLUX CERAM 64,401 ECO 100W; 6 lamps with the reference HQI-T 250 W/D PRO). For metabolite analysis and determination of total NADP-ME enzyme activity, plants were grown in Floraton I soil in a Conviron E15 plant growth chamber under the following conditions: light/dark cycles of 16/8 h at 25°C/20°C; light intensity of 700 µmol m−2 s−1 (three Philips Lighting MH1000/U 1000-W lamps; three high-pressure sodium lamps, Sylvania SHP-T/S400, 400 W).
Sample Preparation for LC-MS Analysis
One-third (from leaf tip) of maize leaves (third leaf) from 12-d-old seedlings were harvested, immediately frozen in liquid nitrogen, and stored at −80°C until further analysis. Maize leaves were collected at ZT0.5, ZT2, ZT4, ZT15.5, ZT16.5, ZT20, and ZT23.5, where ZT0 represents lights on and ZT16 represents lights off. At each time point, four pools of five maize leaves were collected. Each pool constitutes one biological replicate. Frozen maize leaves were ground with a mortar and pestle in liquid nitrogen until a fine powder was obtained. Protein extraction was performed directly in lysis buffer (7 M urea, 2 M thiourea, 30 mM Tris, 4% [w/v] CHAPS, 4% [v/v] cOmplete, EDTA-Free 25× [Roche Applied Science], 0.1% [v/v) Pepstatin 1 mM [Roche Applied Science], 1% [v/v] Nuclease Mix 100% [GE Healthcare], 10% [v/v] PhosSTOP 10× [Roche Applied Science]) as described previously (Luís et al., 2016). For the total protein analysis, protein extracts were loaded onto 4% to 12% (w/v) Bis-Tris polyacrylamide gels (NuPAGE, Thermo Fisher Scientific) and electrophoresed for a short time (10 to 15 min). The whole bands containing the entire protein extract (2 to 3 cm long) were excised and in-gel digested with trypsin followed by a peptide desalt with C18 tips (Thermo Fisher Scientific). For the phosphopeptide analysis of NADP-ME, protein extracts were loaded onto the same kind of gels (4% to 12% [w/v] Bis-Tris polyacrylamide gels, NuPAGE), electrophoresed at a constant voltage of 25 V for 10 min, and then for 150 V until the bromophenol blue reached the bottom of the gel. Next, the gel fractions containing the NADP-ME (ranging from just smaller than 63 kD to just larger than 75 kD, molecular weight (Mw) marker, NZYColor Protein Marker II, NZY Tech) were excised, in-gel digested with trypsin, and further lyophilized to dryness. Vacuum-dried peptides were first desalted using C18 tips (Thermo Fisher Scientific) before being subjected to phosphopeptide enrichment using TiO2 beads (TiO2 Mag Sepharose, GE Healthcare) according to manufacturer’s instructions.
LC-MS Using an AB Sciex TripleTOF 6600 Mass Spectrometer
Samples were analyzed using a TripleTOF 6600 mass spectrometer (AB Sciex), coupled to a nanoLC Eksigent 425 system (AB Sciex). Reverse phase HPLC was performed in a trap and elution configuration using a nano cHiPLC trap column (Eksigent ChromXP C18-CL, 3-µm particle size, 120-Å pore size, 0.5 mm × 200 µm i.d., AB Sciex) and an analytical column (Eksigent ChromXP C18-CL, 3-µm particle size, 120-Å pore size, 15 cm × 75 µm i.d., AB Sciex). Samples were loaded into the trap column at a flow rate of 2 μL min−1 for 10 min using 100% solvent A (0.1% [v/v] formic acid in water) and eluted at a flow rate of 300 nL/min using a stepwise gradient: 0 to 1 min, 4.5% B (0.1% [v/v] formic acid in acetonitrile); 1 to 91 min, 29.7% B; 91 to 93 min, 79.2% B; 93 to 108 min, 79.2% B; 108 to 110 min, 4.5% B; 110 to 127 min, 4.5% B. Samples were run in information-dependent acquisition (IDA) mode to perform peptide and protein identification in order to generate a spectral library for the SWATH quantification. This spectral library was created by combining all the IDA.wiff files in unison using ProteinPilot 5.0 (AB Sciex). A UniProt database search was performed using the Paragon algorithm, which is embedded in ProteinPilot software 5.0 (AB Sciex). A Paragon search method was created with the following settings: sample type, identification; cys alkylation, acrylamide; digestion, trypsin; instrument, tripleTOF 6600; special factors, phosphorylation emphasis and gel-based ID; species, none; ID focus, biological modifications; search effort, thorough; detected protein threshold, >0.05. A false discovery rate (FDR) threshold was set to below 1%.
As for the SWATH-MS analysis, four replicates from each one of the seven time points collected were analyzed by LC-MS using the setup described for the IDA runs. SWATH-MS data were acquired with a standard SWATH acquisition method, using a set of 32 overlapping SWATH windows covering the precursor mass range of 400 to 1200 m/z. For each SWATH window, the Q1 transmission width was 26 m/z (containing 1 m/z for the window overlap). At the beginning of each cycle, a 50-ms survey scan (350 to 1250 m/z) was acquired for instrument calibration, and the subsequent SWATH windows were collected from 100 to 1800 m/z for 60 ms, resulting in a cycle time of 2.02 s. Data processing was performed using a SWATH processing plug-in for PeakView 2.2 (AB Sciex). Briefly, peptides were confirmed by finding and scoring peak groups, which are a set of fragment ions for the peptide. Target fragment ions, up to five, were automatically selected according to previously described criteria (Lambert et al., 2013): (1) fragment ions for a selected peptide were ranked according to ion intensity; (2) ions higher in m/z than the y4 fragment ion for each selected peptide were ranked highest; (3) ions within the SWATH isolation window were excluded from selection; (4) if insufficient target ions were found, ions lower than y4 but outside of the SWATH window were chosen; and (5) if there were still insufficient ions, fragment ions from within the SWATH window region were chosen. The peak group confidence threshold was determined based on an FDR analysis using the target-decoy approach, and a 1% extraction FDR threshold was used for all the analyses. Peptides that met the 1% FDR threshold in all the four replicates were retained, and the peak areas of the target fragment ions of those peptides were extracted across the experiments using an extracted-ion chromatogram window of 12 min. ZmC4-NADP-ME protein levels were estimated by summing the areas of all the transitions from all the peptides for a given protein and normalized to the total area (Collins et al., 2013). As for the ZmC4-NADP-ME phosphopeptide (acVWLVDpSK) levels, they were estimated by summing the areas of the transitions obtained for this phosphopeptide and normalized to the sum of the area of all the transitions from all the peptides within the same region (that include Ser419) for each of the time points.
Site-Directed Mutagenesis of ZmC4-NADP-ME
As template for the PCR reaction (first step of the site-directed mutagenesis), the pET32:WT NADP-ME expression construct (Detarsio et al., 2003) was used. Point mutations were introduced into the protein-coding region by performing site-directed mutagenesis with the following back-to-back primers: 5′-TCGTCCACCAGCCAAAC-3′ and 5′-AAAGGGTTTGATTGTTGACTCTCG-3′ (for and rev primers for the introduction of the S419E mutation; resulting construct has been termed pET32a::S419E NADP-ME) and 5′-GCGTCCACCAGCCAAACC-3′ as the for primer and the same rev primer as used in the case of the first mutagenesis for the introduction of the S419A mutation (resulting construct has been termed pET32a::S419A-NADP-ME). All primers used for the mutagenesis procedure were phosphorylated at the 5′ terminus.
Site-directed mutagenesis was performed as follows: usually, five similar 50-μL PCR reactions were performed simultaneously. Obtained DNA was pooled and (after digestion from the agarose gel, if needed) subjected to the self-ligation by using the T4 DNA ligase (Thermo Fisher Scientific). Self-ligation was performed by incubating the mix at 22°C for 1 h. Subsequently, the ligase was inactivated by incubating the reaction mix at 65°C for 10 min. To destroy the methylated wild-type (sample) DNA, the reaction mix after self-ligation was digested with DpnI (Thermo Fisher Scientific) at 37°C for 2 h. After digestion, the DpnI was inactivated by incubation at 80°C for 5 min. The obtained constructs were used for the transformation of the chemically competent Escherichia coli DH5α cells. After plasmid preparation, the success of the site-directed mutagenesis procedure was confirmed by sequencing with three primers covering the whole length of the protein-coding region.
Expression and Purification of Recombinant ZmC4-NADP-ME
The wild type and two obtained mutated pET32:ME constructs were used for the transformation of the E. coli strain Rosetta (DE3, Novagen). For the heterologous protein production, transformed cells were grown in 400 mL of Luria-Bertani medium at 37°C and agitated at 110 rpm in the presence of 100 µg mL−1 ampicillin and 34 µg mL−1 chloramphenicol until an OD600 of 0.6 to 0.8 was reached. To induce protein expression, a final concentration of 1 mM isopropyl-β-d-thiogalactopyranoside was added to the culture, and cells were grown for an additional 20 h under the same conditions. Cells were harvested by centrifugation at 4000g at 4°C for 15 min. Obtained pellets were transferred into 50-mL Falcon tubes and were stored at −20°C until further use. For the protein extraction, pellets were thawed on ice and resuspended in 20 mM Tris-HCl, pH 8.0, containing 500 mM NaCl, 5 mM imidazole, 2 mM phenylmethanesulfonyl fluoride, and a spatula-tip amount of lysozyme; sonicated; and centrifuged at 14,000g for 20 min at 4°C to remove cell debris. The obtained supernatant was used for protein purification using gravity-flow immobilized metal ion chromatography on nickel-nitrilotriacetic acid agarose (Qiagen). Prior to the supernatant loading, the column was equilibrated with 20 mM Tris-HCl buffer containing 500 mM NaCl and 5 mM imidazole. After the supernatant was loaded, columns were washed in four steps with 20 mM Tris-HCl, pH 8.0, and 500 mM NaCl buffers containing increasing concentrations of imidazole (5, 30, 40, and 50 mM) in order to isolate the histidine (His)-tagged NADP-MEs. Elution was performed using four times 500 μL of 20 mM Tris-HCl buffer containing 500 mM NaCl and 300 mM imidazole. Eluted protein from the first elution fraction was used for further kinetic measurements.
For all constructs used in this work, the calculated molecular mass of the expressed protein corresponded to the expected molecular mass of the fusion protein as follows: mature ZmC4-NADP-ME (63.4 kD) plus 17.3 kD encoded by the expression vector.
Protein Quantification, Gel Electrophoresis, and Immunological Detection
Protein concentration was determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific). SDS-PAGE was performed using 12% (w/v) polyacrylamide gels according to Laemmli (1970). Proteins were visualized by staining with Coomassie blue or electroblotted onto a nitrocellulose membrane (Thermo Scientific) for immunological detection. The membranes were incubated for at least 1 h with a 1:7500 dilution of monoclonal anti–His-Tag antibodies conjugated with horseradish peroxidase (anti–His-HRP; GG11-6F4.3.2, isotype igG2b, Miltenyi Biotec). After several washing steps with Tris-buffered saline (50 mM Tris-HCl, pH 7.6, and 150 mM NaCl) with 0.1% (v/v) Tween 20, the membranes were incubated with a 1:2500 dilution of the goat anti-rabbit IgG antibody HRP-conjugate (Merck). The chemiluminescence signal was detected with Immobilon Western Chemiluminescent HRP Substrate (Merck Millipore), with subsequent visualization on an LAS-4000 Mini Luminescent Image Analyzer (GE Healthcare Life Sciences, formerly Fuji).
Determination of Kinetic Parameters
NADP-ME activity was determined using a Synergy HT Biotek Plate Reader system by measuring the formation of NADPH at 340 nm (extinction coefficient = 6.22 mM−1 cm−1) at room temperature. The standard assay medium contained 0.5 mM NADP+, 10 mM MgCl2, 4 mM L-malate, 50 mM Tris-HCl, pH 8.0, and 0.1 to 0.8 µg of enzyme per well in a final volume of 200 µL. The dependence of the activity with the pH of the medium was determined with the standard assay medium using different buffer systems: 50 mM MES, pH 5.5 to 6.5, 50 mM Tricine-MOPS, pH 7.0 to 7.5, and 50 mM Tris-HCl, pH 7.5 to 9.0. The Michaelis constants (Km in case of the wild type and S419A) and Kmapp in case of S419E) of the substrate were determined by varying the concentration of one substrate, while keeping the other components constant at fixed concentrations, as described for the standard assay medium. NADP concentrations were varied between 2 and 150 µM in the case of the wild type, between 10 and 800 µM in the case of S419A, and between 10 and 5 mM in the case of S419E. Malate concentrations were varied between 0.001 and 8 mM in the case of the wild type and S419A and between 0.1 and 10 mM in the case of S419E. As Ser419 was not saturated with NADP at least until 5 mM NADP, and the Km of NADP for S419E could not be estimated. Measurements of enzymatic activity using high NADP concentrations are technically impaired by the high absorption of this compound; thus, the apparent kinetic parameters (kcatapp and Kmapp malate) of S419E were estimated using 0.5 mM NADP. All kinetic parameters were calculated using at least three biological replicates and adjusted by nonlinear regression. Data were fitted with Prism 6 (GraphPad Software).
Circular Dichroism
All recombinant versions of ZmC4-NADP-ME were subjected to CD analysis in 20 mM NaPi, pH 8.0 or pH 7.0, and 5 mM MgCl2. Protein concentration was determined at 280 nm using a Jasco V-650 spectrophotometer. The protein concentration 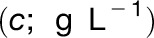 was calculated by the equation
was calculated by the equation 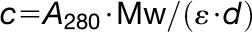 , where ε represents the molar extinction coefficient at 280 nm (76,250 L mol−1 cm−1), d is the cell path (1 cm), and Mw is molecular weight (80.7 kD for all three proteins).
, where ε represents the molar extinction coefficient at 280 nm (76,250 L mol−1 cm−1), d is the cell path (1 cm), and Mw is molecular weight (80.7 kD for all three proteins).
CD spectra between 240 and 190 nm were obtained by averaging 10 repetitive scans in a Jasco J-810 spectropolarimeter. Mean residue ellipticity ([θ], deg cm2 dmol−1) was obtained by the equation 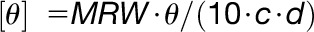 , where MRW is the mean amino acid residue weight calculated as the ratio of the protein’s Mw to the number of peptide bonds (N – 1, where N is the number of amino acids in the chain), d is the optical path length (0.1 cm), c is the protein concentration, and θ is the observed ellipticity in millidegrees (machine units). The protein secondary structure contents were determined using the CONTIN- LL method (van Stokkum et al., 1990) and the reference data set 4 (Sreerama and Woody, 2000) at the Dichroweb online server (http://dichroweb.cryst.bbk.ac.uk; Whitmore and Wallace, 2004). Prism software (http://www.graphpad.com/scientific-software/prism/) was used to visualize the deconvolution by Dichroweb.
, where MRW is the mean amino acid residue weight calculated as the ratio of the protein’s Mw to the number of peptide bonds (N – 1, where N is the number of amino acids in the chain), d is the optical path length (0.1 cm), c is the protein concentration, and θ is the observed ellipticity in millidegrees (machine units). The protein secondary structure contents were determined using the CONTIN- LL method (van Stokkum et al., 1990) and the reference data set 4 (Sreerama and Woody, 2000) at the Dichroweb online server (http://dichroweb.cryst.bbk.ac.uk; Whitmore and Wallace, 2004). Prism software (http://www.graphpad.com/scientific-software/prism/) was used to visualize the deconvolution by Dichroweb.
Analytical Ultracentrifugation
Sedimentation velocity experiments were performed using a Beckman Optima XL-A analytical ultracentrifuge. All recombinant versions of ZmC4-NADP-ME were assayed in 10 mM Tris-HCl buffer, pH 7.0 or pH 8.0, with addition of 5 mM MgCl2. Buffer exchange was performed using Amicon Ultra 0.5-mL centrifugal filters with a nominal molecular mass limit of 50 kD (Merck). The final protein concentration was adjusted to 0.6 mg mL−1. Samples (230 µg) and corresponding buffer solutions (400 µL) were loaded into aluminum double sector centerpieces separately and built up in a Beckman An-50 Ti rotor. Experiments were performed at 20°C and a rotor speed of 35,000 rpm. Protein samples were monitored by UV absorbance at 280 nm in a continuous mode with a radial resolution of 0.003 cm. In time intervals of ∼2 min, scans of the radial concentration profile were collected until the protein was fully sedimented. Data were analyzed using the c(s) model in the software package SEDFIT (Schuck and Rossmanith, 2000). For data analysis, a resolution of 0.1 S with a confidence level (F-ratio) of 0.95 was chosen for the appropriate s-value range within 0 to 30.0 S. The density and viscosity of the solvent had been calculated with the software Sednterp from tabulated values; ρ = 0.99885 g cm−3 and η = 0.01006 g cm−1 s−1. The protein partial specific volume applied for the wild type and mutants was 0.7387 cm3 g−1. Sedimentation coefficients are reported as s20,w values, that is, normalized to 20°C and water as a solvent. Graphic output was generated by Gussi 1.2.1 (Brautigam, 2015), and the final sedimentation coefficient distribution was normalized based on the maximum peak height.
NADP-ME Activity Measurements in Maize Leaf Extracts
Maize leaves were collected at ZT0.5, ZT2, ZT4, ZT8, ZT15.5, ZT16.5, ZT20, and ZT23.5, where ZT0 represents lights on and ZT16 represents lights off. One-third (from leaf tip) of maize leaves (third and fourth leaf) from 12-d-old seedlings were harvested, immediately frozen in liquid nitrogen, and stored at −80°C until further analysis. Soluble protein extracts were prepared by homogenizing 0.25 g of frozen maize leaves in 500 μL of ice-cold extraction buffer containing 100 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 0.1 mM EDTA, 10% (v/v) glycerol, 2 mM DTT, and 1 mM phenylmethylsulfonyl fluoride. The homogenate was clarified by centrifugation at 20,000g for 20 min at 4°C. Protein concentration of the extract was determined using the amido black assay (Schaffner and Weissmann, 1973). NADP-ME activity was measured in medium containing 50 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 0.5 mM NADP, 10 mM L-malate, and 1.5 to 2.2 µg of protein extract in a final volume of 0.2 mL. The reaction was started by addition of NADP+. Measurements were conducted using three independent replicates.
GC-MS Analysis
Frozen maize leaves were ground in liquid nitrogen until a fine powder was obtained. Ground material (∼50 mg) was extracted in methanol:chloroform:water (5:2:2) with ribitol for internal standardization as described previously by Lee and Fiehn (2008).
An aliquot of the supernatant (150 µL) was dried under vacuum, and the residue was derivatized for 120 min at 37°C (in 60 μL of 30 mg mL−1 methoxyamine hydrochloride in pyridine) followed by a 30-min treatment at 37°C with 120 μL of N-methyl-N-(trimethylsilyl)trifluoroacetamide. The gas chromatography (GC)-MS system used was a gas chromatograph coupled to a time-of-flight mass spectrometer (Leco Pegasus HT TOFMS system). An auto sampler Gerstel multi-purpose system injected the samples. Helium was used as carrier gas at a constant flow rate of 2 mL s−1, and GC was performed on a 30-m DB-35 column. The injection temperature was 230°C, and the transfer line and ion source were set to 250°C. The initial temperature of the oven (85°C) increased at a rate of 15°C/min up to a final temperature of 360°C. After a solvent delay of 180 s, mass spectra were recorded at 20 scans s−1 with m/z 70 to 600 scanning range. Chromatograms and mass spectra were evaluated using Chroma TOF 4.5 (Leco) and TagFinder 4.2 software (Ishizaki et al., 2005; Lisec et al., 2006).
Generation of the Complex Structures
For the complex preparation, we used the monomeric apo structure of ZmC4-NADP-ME (amino acids 132 to 628, PDB ID code: 5OU5). Based on the structural alignment with ME from pigeon liver (PDB ID code: 1GQ2) and SbC4-NADP-ME (PDB ID code: 6C7N), a Mg2+ ion and NADP were placed into the active site of ZmC4-NADP-ME. Subsequently, the complex was protonated using the Structure Preparation Tool and Protonate3D implemented in MOE (edition 2019.01, Chemical Computing Group ULC) at pH 8.0. The structures of the enzyme carrying a substitution or the phosphorylated residue were generated by replacing Ser419 either with glutamate or phospho-Ser in LEaP (Case et al., 2005).
Molecular Dynamics Simulations of NADP/ZmME Complexes
System Setup
The following procedures were performed using the Amber18 software package (Case et al., 2018). We used tLEaP to solvate each of the complexes in a periodic box of transferable intermolecular potential with 3 points water (Jorgensen et al., 1983), in such a way that the distance between the edges of the box and a solute atom is at least 11 Å. The system was charge neutralized by replacing random water molecules with sodium ions. The ff14SB force field (Hornak et al., 2006) was used for parameterizing the protein, and Mg2+ parameters were taken from the 12-6 LJ set of ion parameters as described by Li et al. (2013). The phospho-Ser parameters were taken from phosaa10 (Homeyer et al., 2006; Steinbrecher et al., 2012) and modified to be compatible with ff14SB. Partial charges according to the Restrained ElectroStatic Potential procedure (Bayly et al., 1993) for NADP were derived from the R.E.DD.B. (Dupradeau et al., 2008) project F-91, while for bond parameters the general AMBER force field (Wang et al., 2004) was used. To remove initial clashes due to the introduction of the larger side chains replacing Ser419, for the modified systems an energy minimization of this residue was performed with pmemd (Le Grand et al., 2013; Case et al., 2018) prior to the thermalization.
Thermalization
All simulations were performed using the graphics processing unit version of pmemd. The Langevin thermostat (Pastor et al., 1988) was used for temperature control with a collision frequency of γ = 2.0 ps−1. For treatment of long-range electrostatic interactions, the particle mesh Ewald method (Berendsen et al., 1984) was used with a cutoff of 8.0 Å. The SHAKE algorithm (Ryckaert et al., 1977) was used to constrain bond lengths involving hydrogen atoms, and hydrogen mass repartitioning (Hopkins et al., 2015) was used to allow for simulation steps of 4 fs.
Initially, an energy minimization was performed independently on solvent and solute atoms for 3000 steps each with the steepest decent algorithm, followed by 2000 steps with the conjugate gradient algorithm. Thereafter, all atoms were energy minimized in the same way. Next, the systems were heated to 300 K at a constant heating rate over 20 ps of constant temperature (NVT)-MD and simulated for a further 5 ps at 300 K. For density adaptation, 75 ps of isothermal-isobaric-MD were performed with a pressure relaxation time of τp = 1.0 ps, followed by 1700 ps of isothermal-isobaric-MD with the same restraints and τp = 2.0 ps. For all these steps, positional restraints of 2.0 kcalmol−1Å−2 were applied to the enzyme’s Cα-atoms and NADP. Ten further iterations of minimization were performed on the system with iteratively decreasing restraints on the solute atoms (force constants ranging from 2 to 0 kcal mol−1 Å−2), with 500 steps of steepest descent minimization and 500 steps of conjugate gradient minimization each. Finally, the systems were heated again to 300 K over 100 ps of NVT-MD, followed by 3100 ps NVT-MD. Again, positional restraints with a force constant of 2.0 kcal mol−1 Å−2 were applied to the enzyme’s Cα-atoms and NADP.
Production
For each of the three systems, 10 individual NVT-MD simulations of 100-ns length were performed. Since especially in the mutated systems the binding pose of NADP were not stable, and large conformational rearrangements would compromise the effective binding energy calculations, we restrained NADP within the binding site in all three systems by a distance restraint between the Cα of S435 and the nicotinamide of NADP using a one-sided harmonic potential with a force constant of 2.0 kcal mol−1 Å−2. Coordinates for post-processing and further analysis were extracted every 100 ps. For geometric analyses, CPPTRAJ (Roe and Cheatham, 2013) was used.
Calculation of Effective Binding Energies
The calculation was performed using MMPBSA.py from AmberTools18 (Miller et al., 2012; Case et al., 2018). For all systems the change of the effective energy due to binding of NADP was calculated using the single-trajectory MM-PBSA approach on the last 50 ns of trajectory (Gohlke and Case, 2004; Wang et al., 2016). All counter ions and water molecules were stripped from the trajectory, and the sum of electrostatic and van der Waals energies was calculated for each snapshot using force fields (internal energies cancel in this approach). The polar part of the solvation free energy was calculated at the level of Poisson-Boltzmann theory, with the dielectric constants set to 80 for the solvent and 4 for the solute and an ionic strength of 300 mM. The nonpolar part of the solvation free energy was calculated by separating the attractive and repulsive terms, as shown by Tan et al. (2007), using as a surface tension of 0.0378 kcal−1 Å−2. Previous studies have shown that inclusion of configurational entropy is crucial for calculating absolute binding free energies (Hou et al., 2011; Genheden and Ryde, 2015; Ben-Shalom et al., 2017). In this study, however, we were interested in relative binding free energies and thus decided to neglect contributions due to changes in the configurational entropy of the ligand or the receptor upon complex formation in order to avoid introducing additional uncertainty in the computations (Gohlke and Case, 2004; Weis et al., 2006; Hou et al., 2011).
Accession Numbers
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers GRMZM2G085019_T01 (C4-NADP-ME).
Supplemental Data
Supplemental Figure 1. Alignment of plastidic C4- and nonC4-NADP-ME sequences in Poaceae showing conservation of S419 and the BARK substrate motif (DSKGL).
Supplemental Figure 2. SDS-PAGE stained with Coomassie-Blue (lines 1-4) and analyzed by immunoblot (line 5) of protein fractions during the isolation of recombinant ZmC4-NADP-ME variants.
Supplemental Figure 3. Comparison of the CD spectra of ZmC4-NADP-ME WT obtained at pH 7.0 and 8.0.
Supplemental Figure 4. Representation of the bound cofactor NADP in SbC4-NADP-ME chain B.
Supplemental Figure 5. Root mean square deviations of ZmC4-NADP-ME’s Cα atoms determined by molecular dynamics (MD) simulations.
Supplemental Figure 6. Root mean square deviations of NADP in the binding site of ZmC4-NADP-ME determined by molecular dynamics (MD) simulations.
Supplemental Figure 7. Root mean square deviations of NADP in the binding site of ZmC4-NADP-ME after restraining NADP to the respective binding sites determined by molecular dynamics (MD) simulations.
Supplemental Table 1. Secondary structure contents of ZmC4-NADP-ME WT, S419A and S419E estimated from the CD-spectra obtained at pH of 7.0 and 8.0.
Supplemental Table 2. Polar interactions/hydrogen bonds between monomer A of SbC4-NADP-ME and the cofactor NADP with their distances.
Supplemental Table 3. P-values for Figure 4.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
Acknowledgments
This work was funded by the European Commission Directorate-General for Research and Innovation (3to4; 289582 to V.G.M. and I.A.A.) and by the Deutsche Forschungsgemeinschaft under Germany’s Excellence Strategy (EXC 2048/1; Project ID: 390686111 and EXC 1028 to V.G.M.). Support from the iGrad Plant International Graduate Program of the Heinrich Heine University was provided to A.B. The Center for Structural Studies is funded by the Deutsche Forschungsgemeinschaft (grant 417919780; INST 208/740-1 FUGG). Support was provided by the FCT Investigator programme and GREEN-it (UID/Multi/04551/2013 to I.A.A.); by the German Federal Ministry of Research and Education (Full Throttle grant 031B0205A to S.A. and A.R.F.); and by FCT postdoctorate fellowships (SFRH/BPD/98619/2013 to B.M.A. and PD/BD/113982/2015 within MolBioS PD/00133/2012 to I.M.L.). Liquid chromatography-tandem MS analyses were performed at the Mass Spectrometry Unit facility of Instituto de Biologia Experimental e Tecnológica/Instituto de Tecnologia Química e Biológica-Universidade Nova de Lisboa, Oeiras, Portugal. We are grateful for computational support by the Zentrum für Informations und Medientechnologie at the Heinrich Heine University and the computing time provided by the John von Neumann Institute for Computing to H.G. on the supercomputer JURECA at Jülich Supercomputing Centre (user ID: HKF7).
AUTHOR CONTRIBUTIONS
V.G.M. conceptualized the study and worked with I.A.A. to prepare the methodology. A.B., B.M.A, A.H., C.E.A., I.M.L., C.D., S.A., D.B., and H.G. performed the research. V.G.M., I.A.A., A.B., A.H., C.E.A., M.F.D., L.N.-S., D.B., and H.G. wrote the first draft of the article. Funding was acquired by V.G.M. and I.A.A. Resources were provided by V.G.M., I.A.A., M.F.D., and A.F.; V.G.M., I.A.A., M.F.D. supervised the study.
References
- Agarie S., Kai M., Takatsuji H., Ueno O. (1997). Expression of C3 and C4 photosynthetic characteristics in the amphibious plant Eleocharis vivipara: Structure and analysis of the expression of isogenes for pyruvate, orthophosphate dikinase. Plant Mol. Biol. 34: 363–369. [DOI] [PubMed] [Google Scholar]
- Alvarez C.E., Detarsio E., Moreno S., Andreo C.S., Drincovich M.F. (2012). Functional characterization of residues involved in redox modulation of maize photosynthetic NADP-malic enzyme activity. Plant Cell Physiol. 53: 1144–1153. [DOI] [PubMed] [Google Scholar]
- Alvarez C.E., Saigo M., Margarit E., Andreo C.S., Drincovich M.F. (2013). Kinetics and functional diversity among the five members of the NADP-malic enzyme family from Zea mays, a C4 species. Photosynth. Res. 115: 65–80. [DOI] [PubMed] [Google Scholar]
- Alvarez C.E., Bovdilova A., Höppner A., Wolff C.-C., Saigo M., Trajtenberg F., Zhang T., Buschiazzo A., Nagel-Steger L., Drincovich M.F., Lercher M.J., Maurino V.G. (2019). Molecular adaptations of NADP-malic enzyme for its function in C4 photosynthesis in grasses. Nat. Plants 5: 755–765. [DOI] [PubMed] [Google Scholar]
- Amanchy R., Periaswamy B., Mathivanan S., Reddy R., Tattikota S.G., Pandey A. (2007). A curated compendium of phosphorylation motifs. Nat. Biotechnol. 25: 285–286. [DOI] [PubMed] [Google Scholar]
- Arrivault S., Obata T., Szecówka M., Mengin V., Guenther M., Hoehne M., Fernie A.R., Stitt M. (2017). Metabolite pools and carbon flow during C4 photosynthesis in maize: 13CO2 labeling kinetics and cell type fractionation. J. Exp. Bot. 68: 283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astley H.M., Parsley K., Aubry S., Chastain C.J., Burnell J.N., Webb M.E., Hibberd J.M. (2011). The pyruvate, orthophosphate dikinase regulatory proteins of Arabidopsis are both bifunctional and interact with the catalytic and nucleotide-binding domains of pyruvate, orthophosphate dikinase. Plant J. 68: 1070–1080. [DOI] [PubMed] [Google Scholar]
- Bailey K.J., Gray J.E., Walker R.P., Leegood R.C. (2007). Coordinate regulation of phosphoenolpyruvate carboxylase and phosphoenolpyruvate carboxykinase by light and CO2 during C4 photosynthesis. Plant Physiol. 144: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly C.I., Cieplak P., Cornell W., Kollman P.A. (1993). A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97: 10269–10280. [Google Scholar]
- Bellasio C., Griffiths H. (2014a). Acclimation to low light by C4 maize: Implications for bundle sheath leakiness. Plant Cell Environ. 37: 1046–1058. [DOI] [PubMed] [Google Scholar]
- Bellasio C., Griffiths H. (2014b). The operation of two decarboxylases, transamination, and partitioning of C4 metabolic processes between mesophyll and bundle sheath cells allows light capture to be balanced for the maize C4 pathway. Plant Physiol. 164: 466–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom I.Y., Pfeiffer-Marek S., Baringhaus K.H., Gohlke H. (2017). Efficient approximation of ligand rotational and translational entropy changes upon binding for use in MM-PBSA calculations. J. Chem. Inf. Model. 57: 170–189. [DOI] [PubMed] [Google Scholar]
- Berendsen H.J.C., Postma J.P.M., Van Gunsteren W.F., Dinola A., Haak J.R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81: 3684–3690. [Google Scholar]
- Boxall S.F., Dever L.V., Kneřová J., Gould P.D., Hartwell J. (2017). Phosphorylation of phosphoenolpyruvate carboxylase is essential for maximal and sustained dark CO2 fixation and core circadian clock operation in the obligate crassulacean acid metabolism species Kalanchoë fedtschenkoi. Plant Cell 29: 2519–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam C.A. (2015). Calculations and publication-quality illustrations for analytical ultracentrifugation data. Methods Enzymol. 562: 109–133. [DOI] [PubMed] [Google Scholar]
- Burnell J.N. (2010). Cloning and characterization of Escherichia coli DUF299: A bifunctional ADP-dependent kinase–Pi-dependent pyrophosphorylase from bacteria. BMC Biochem. 11: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnell J.N., Chastain C.J. (2006). Cloning and expression of maize-leaf pyruvate, Pi dikinase regulatory protein gene. Biochem. Biophys. Res. Commun. 345: 675–680. [DOI] [PubMed] [Google Scholar]
- Burnell J.N., Hatch M.D. (1985). Regulation of C4 photosynthesis: Purification and properties of the protein catalyzing ADP-mediated inactivation and Pi-mediated activation of pyruvate,Pi dikinase. Arch. Biochem. Biophys. 237: 490–503. [DOI] [PubMed] [Google Scholar]
- Case D.A., et al. (2018). AMBER 2018. (San Francisco: University of California; ). [Google Scholar]
- Case D.A., Cheatham T.E. III, Darden T., Gohlke H., Luo R., Merz K.M. Jr., Onufriev A., Simmerling C., Wang B., Woods R.J. (2005). The Amber biomolecular simulation programs. J. Comput. Chem. 26: 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao Q., Liu X.Y., Mei Y.C., Gao Z.F., Chen Y.B., Qian C.R., Hao Y.B., Wang B.C. (2014). Light-regulated phosphorylation of maize phosphoenolpyruvate carboxykinase plays a vital role in its activity. Plant Mol. Biol. 85: 95–105. [DOI] [PubMed] [Google Scholar]
- Chastain C.J., Botschner M., Harrington G.E., Thompson B.J., Mills S.E., Sarath G., Chollet R. (2000). Further analysis of maize C(4) pyruvate,orthophosphate dikinase phosphorylation by its bifunctional regulatory protein using selective substitutions of the regulatory Thr-456 and catalytic His-458 residues. Arch. Biochem. Biophys. 375: 165–170. [DOI] [PubMed] [Google Scholar]
- Chastain C.J., Xu W., Parsley K., Sarath G., Hibberd J.M., Chollet R. (2008). The pyruvate, orthophosphate dikinase regulatory proteins of Arabidopsis possess a novel, unprecedented Ser/Thr protein kinase primary structure. Plant J. 53: 854–863. [DOI] [PubMed] [Google Scholar]
- Chen Y.B., Lu T.C., Wang H.X., Shen J., Bu T.T., Chao Q., Gao Z.F., Zhu X.G., Wang Y.F., Wang B.C. (2014). Posttranslational modification of maize chloroplast pyruvate orthophosphate dikinase reveals the precise regulatory mechanism of its enzymatic activity. Plant Physiol. 165: 534–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins B.C., Gillet L.C., Rosenberger G., Röst H.L., Vichalkovski A., Gstaiger M., Aebersold R. (2013). Quantifying protein interaction dynamics by SWATH mass spectrometry: Application to the 14-3-3 system. Nat. Methods 10: 1246–1253. [DOI] [PubMed] [Google Scholar]
- Detarsio E., Wheeler M.C., Campos Bermúdez V.A., Andreo C.S., Drincovich M.F. (2003). Maize C4 NADP-malic enzyme. Expression in Escherichia coli and characterization of site-directed mutants at the putative nucleoside-binding sites. J. Biol. Chem. 278: 13757–13764. [DOI] [PubMed] [Google Scholar]
- Detarsio E., Maurino V.G., Alvarez C.E., Müller G.L., Andreo C.S., Drincovich M.F. (2008). Maize cytosolic NADP-malic enzyme (ZmCytNADP-ME): A phylogenetically distant isoform specifically expressed in embryo and emerging roots. Plant Mol. Biol. 68: 355–367. [DOI] [PubMed] [Google Scholar]
- Dissmeyer N., Schnittger A. (2011). Use of phospho-site substitutions to analyze the biological relevance of phosphorylation events in regulatory networks. Methods Mol. Biol. 779: 93–138. [DOI] [PubMed] [Google Scholar]
- Dupradeau F.Y., Cézard C., Lelong R., Stanislawiak E., Pêcher J., Delepine J.C., Cieplak P. (2008). R.E.DD.B.: A database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 36: D360–D367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnenstich H., Saigo M., Niessen M., Zanor M.I., Andreo C.S., Fernie A.R., Drincovich M.F., Flügge U.I., Maurino V.G. (2007). Alteration of organic acid metabolism in Arabidopsis overexpressing the maize C4 NADP-malic enzyme causes accelerated senescence during extended darkness. Plant Physiol. 145: 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furbank R.T. (2011). Evolution of the C(4) photosynthetic mechanism: are there really three C(4) acid decarboxylation types? J. Exp. Bot. 62: 3103–3108. [DOI] [PubMed] [Google Scholar]
- Furbank R.T., Hatch M.D. (1987). Mechanism of c(4) photosynthesis: The size and composition of the inorganic carbon pool in bundle sheath cells. Plant Physiol. 85: 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furbank R.T., Jenkins C.L.D., Hatch M.D. (1990). C4 photosynthesis—quantum requirement, C4 acid overcycling and Q-cycle involvement. Aust. J. Plant Physiol. 17: 1–7. [Google Scholar]
- Furumoto T., Izui K., Quinn V., Furbank R.T., von Caemmerer S. (2007). Phosphorylation of phosphoenolpyruvate carboxylase is not essential for high photosynthetic rates in the C4 species Flaveria bidentis. Plant Physiol. 144: 1936–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S., Ryde U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10: 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohlke H., Case D.A. (2004). Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 25: 238–250. [DOI] [PubMed] [Google Scholar]
- Hartwell J., Gill A., Nimmo G.A., Wilkins M.B., Jenkins G.I., Nimmo H.G. (1999). Phosphoenolpyruvate carboxylase kinase is a novel protein kinase regulated at the level of expression. Plant J. 20: 333–342. [DOI] [PubMed] [Google Scholar]
- Hatch M.D. (1987). C4 photosynthesis: A unique blend of modified biochemistry, anatomy and ultrastructure. Biochim. Biophys. Acta 895: 81–106. [Google Scholar]
- Homeyer N., Horn A.H., Lanig H., Sticht H. (2006). AMBER force-field parameters for phosphorylated amino acids in different protonation states: phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J. Mol. Model. 12: 281–289. [DOI] [PubMed] [Google Scholar]
- Homeyer N., Stoll F., Hillisch A., Gohlke H. (2014). Binding free energy calculations for lead optimization: Assessment of their accuracy in an industrial drug design context. J. Chem. Theory Comput. 10: 3331–3344. [DOI] [PubMed] [Google Scholar]
- Hopkins C.W., Le Grand S., Walker R.C., Roitberg A.E. (2015). Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11: 1864–1874. [DOI] [PubMed] [Google Scholar]
- Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006). Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65: 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T., Wang J., Li Y., Wang W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias A.A., Andreo C.S. (1990). Kinetic and structural properties of NADP-malic enzyme from sugarcane leaves. Plant Physiol. 92: 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi N., Ku M.S., Ishihara K., Samejima M., Kaneko S., Matsuoka M. (1997). Characterization of the gene for pyruvate,orthophosphate dikinase from rice, a C3 plant, and a comparison of structure and expression between C3 and C4 genes for this protein. Plant Mol. Biol. 34: 701–716. [DOI] [PubMed] [Google Scholar]
- Ishizaki K., Larson T.R., Schauer N., Fernie A.R., Graham I.A., Leaver C.J. (2005). The critical role of Arabidopsis electron-transfer flavoprotein:ubiquinone oxidoreductase during dark-induced starvation. Plant Cell 17: 2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J.A., Chollet R. (1988). Light/dark regulation of maize leaf phosphoenolpyruvate carboxylase by in vivo phosphorylation. Arch. Biochem. Biophys. 261: 409–417. [DOI] [PubMed] [Google Scholar]
- Jiao J.A., Chollet R. (1989). Regulatory seryl-phosphorylation of C4 phosphoenolpyruvate carboxylase by a soluble protein kinase from maize leaves. Arch. Biochem. Biophys. 269: 526–535. [DOI] [PubMed] [Google Scholar]
- Jiao J.A., Chollet R. (1990). Regulatory phosphorylation of serine-15 in maize phosphoenolpyruvate carboxylase by a C4-leaf protein-serine kinase. Arch. Biochem. Biophys. 283: 300–305. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79: 926–935. [Google Scholar]
- Kromdijk J., Ubierna N., Cousins A.B., Griffiths H. (2014). Bundle-sheath leakiness in C4 photosynthesis: A careful balancing act between CO2 concentration and assimilation. J. Exp. Bot. 65: 3443–3457. [DOI] [PubMed] [Google Scholar]
- Laemmli U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Lambert J.P., et al. (2013). Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 10: 1239–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.Y., Fiehn O. (2008). High quality metabolomic data for Chlamydomonas reinhardtii. Plant Methods 4: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S., Gotz A.W., Walker R.C. (2013). SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 184: 374–380. [Google Scholar]
- Li P., Roberts B.P., Chakravorty D.K., Merz K.M. Jr. (2013). Rational design of particle mesh ewald compatible Lennard-Jones parameters for +2 metal cations in explicit solvent. J. Chem. Theory Comput. 9: 2733–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisec J., Schauer N., Kopka J., Willmitzer L., Fernie A.R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1: 387–396. [DOI] [PubMed] [Google Scholar]
- Luís I.M., Alexandre B.M., Oliveira M.M., Abreu I.A. (2016). Selection of an appropriate protein extraction method to study the phosphoproteome of maize photosynthetic tissue. PLoS One 11: e0164387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B.R. III, McGee T.D. Jr., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. (2012). MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8: 3314–3321. [DOI] [PubMed] [Google Scholar]
- Nimmo H.G. (2000). The regulation of phosphoenolpyruvate carboxylase in CAM plants. Trends Plant Sci. 5: 75–80. [DOI] [PubMed] [Google Scholar]
- Nimmo H.G. (2003). Control of the phosphorylation of phosphoenolpyruvate carboxylase in higher plants. Arch. Biochem. Biophys. 414: 189–196. [DOI] [PubMed] [Google Scholar]
- Pastor R.W., Brooks B.R., Szabo A. (1988). An analysis of the accuracy of langevin and molecular- dynamics algorithms. Mol. Phys. 65: 1409–1419. [Google Scholar]
- Roe D.R., Cheatham T.E. III (2013). PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9: 3084–3095. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.P., Ciccotti G., Berendsen H.J.C. (1977). Numerical-integration of cartesian equations of motion of a system with constraints - molecular-dynamics of n-alkanes. J. Comput. Phys. 23: 327–341. [Google Scholar]
- Sage R.F., Sage T.L., Kocacinar F. (2012). Photorespiration and the evolution of C4 photosynthesis. Annu. Rev. Plant Biol. 63: 19–47. [DOI] [PubMed] [Google Scholar]
- Saigo M., Bologna F.P., Maurino V.G., Detarsio E., Andreo C.S., Drincovich M.F. (2004). Maize recombinant non-C4 NADP-malic enzyme: A novel dimeric malic enzyme with high specific activity. Plant Mol. Biol. 55: 97–107. [DOI] [PubMed] [Google Scholar]
- Saigo M., Alvarez C.E., Andreo C.S., Drincovich M.F. (2013). Plastidial NADP-malic enzymes from grasses: unraveling the way to the C4 specific isoforms. Plant Physiol. Biochem. 63: 39–48. [DOI] [PubMed] [Google Scholar]
- Schaffner W., Weissmann C. (1973). A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 56: 502–514. [DOI] [PubMed] [Google Scholar]
- Schuck P., Rossmanith P. (2000). Determination of the sedimentation coefficient distribution by least-squares boundary modeling. Biopolymers 54: 328–341. [DOI] [PubMed] [Google Scholar]
- Sharwood R.E., Sonawane B.V., Ghannoum O. (2014). Photosynthetic flexibility in maize exposed to salinity and shade. J. Exp. Bot. 65: 3715–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N., Woody R.W. (2000). Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287: 252–260. [DOI] [PubMed] [Google Scholar]
- Steinbrecher T., Latzer J., Case D.A. (2012). Revised AMBER parameters for bioorganic phosphates. J. Chem. Theory Comput. 8: 4405–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C., Tan Y.-H., Luo R. (2007). Implicit nonpolar solvent models. J. Phys. Chem. B 111: 12263–12274. [DOI] [PubMed] [Google Scholar]
- van Stokkum I.H., Spoelder H.J., Bloemendal M., van Grondelle R., Groen F.C. (1990). Estimation of protein secondary structure and error analysis from circular dichroism spectra. Anal. Biochem. 191: 110–118. [DOI] [PubMed] [Google Scholar]
- Walker R.P., Leegood R.C. (1996). Phosphorylation of phosphoenolpyruvate carboxykinase in plants. Studies in plants with C4 photosynthesis and Crassulacean acid metabolism and in germinating seeds. Biochem. J. 317: 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R.P., Chen Z.H., Acheson R.M., Leegood R.C. (2002). Effects of phosphorylation on phosphoenolpyruvate carboxykinase from the C4 plant Guinea grass. Plant Physiol. 128: 165–172. [PMC free article] [PubMed] [Google Scholar]
- Wang C., Nguyen P.H., Pham K., Huynh D., Le T.B., Wang H., Ren P., Luo R. (2016). Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 37: 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25: 1157–1174. [DOI] [PubMed] [Google Scholar]
- Wei M., Li Z., Ye D., Herzberg O., Dunaway-Mariano D. (2000). Identification of domain-domain docking sites within Clostridium symbiosum pyruvate phosphate dikinase by amino acid replacement. J. Biol. Chem. 275: 41156–41165. [DOI] [PubMed] [Google Scholar]
- Weis A., Katebzadeh K., Söderhjelm P., Nilsson I., Ryde U. (2006). Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J. Med. Chem. 49: 6596–6606. [DOI] [PubMed] [Google Scholar]
- Whitmore L., Wallace B.A. (2004). DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32: W668-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell M.B., Fahnenstich H., Maier A., Saigo M., Voznesenskaya E.V., Edwards G.E., Andreo C., Schleifenbaum F., Zell C., Drincovich M.F., Maurino V.G. (2010). Analysis of Arabidopsis with highly reduced levels of malate and fumarate sheds light on the role of these organic acids as storage carbon molecules. Plant Physiol. 152: 1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]