

Abstract
A focused PROTAC library hijacking cancer therapeutic target CDK6 was developed. A design principle as “match/mismatch” was proposed for understanding the degradation profile differences in these PROTACs. Notably, potent PROTACs with specific and remarkable CDK6 degradation potential were generated by linking CDK6 inhibitor palbociclib and E3 ligase CRBN recruiter pomalidomide. The PROT AC strongly inhibited proliferation of haematopoietic cancer cells including multiple myeloma and robustly degraded copy-amplified/mutated forms of CDK6, indicating future potential clinical applications.
Keywords: Protein degradation, CDK6, match/mismatch, multiple myeloma
Graphical Abstract
INTRODUCTION
Cell cycle regulation is one of the vital and house-keeping activities in most types of the cells1. Cyclin-CDK dual complexes are the major driving machinery for cell cycle progression1. Among the CDKs, CDK6, plays a significant role in cell cycle entrance and is frequently overexpressed or hyper-activated in cancer samples2. Therefore, small molecule inhibitors of CDK6 have been officially approved or clinically tested to act against cancers including breast cancer, lymphoma and multiple myeloma3–5. However, overexpression of CDK6, instead of its close homolog CDK4, induced by gene amplification or loss of FATl gene functions, has been reported to be correlated to CDK4/6 inhibitor resistance in breast cancer cell lines and patient samples (Fig. 1A)6,7. Moreover, point mutation of CD K6 could possibly result in attenuation of drug binding affinity or hyper-activation of CDK6, though not yet reported in clinical samples (Fig. 1A)8,9. Thus, there is an urgent need to develop a practical strategy against CDK6-centered malignancy.
Figure 1.

Resistance to CDK6 inhibitor and PROTAC technique for overcoming resistance. (A) Mechanisms for CDK6-centered resistance to CDK6 inhibitors. CDK6i, CDK6 inhibitor. Ovcrexpression here indicate CDK6 overexpression resulting from CDK6 gene amplification or upstream FAT1 loss. Mutation here implies potential point mutation that hampers binding affinity of CDK6 inhibitor. (B) induced degradation of wild type (wt) and mutated (mt) CDK6 proteins by PROTAC. E3ligase is recruited to the proximity of target protein CDK6 and mediates poly-ubiquitination of CDK6, which is then processed into proteasome for degradation
PROTAC (PROteolysis TArgeting Chimera) is an emerging chemical biology approach for targeted protein depletion by exploiting the intracellular ubiquitin-proteasome system10. A typical PROTAC molecule is bifunctional by combining a target-selective ligand and a specific E3 ligase recruiting ligand via a linker11. The resulting PROTAC could thus recruit the E3 ligase onto target protein and induce the ubiquitination and protein degradation via the proteasome (Fig. 1B)11,12. The concept of targeted protein degradation was first proved by the groups of Crews and Deschaies in 2001 and was later successfully applied in multiple targets with different subcellular localizations, especially in hijacking cancer-related kinases13–26. Notably, overexpressed BRD4 and drug-resistant mutated form of BTK and AR, which have been implicated in several different cancers, could be efficiently degraded through the PROTAC technique15–17,27,28. These studies present the PROTAC technique as a promising alternative approach against cancer. Although this field has observed tremendous achievements in recent years, there is still a vast amount of challenges for PROTAC design to overcome. Various factors including ligand binding affinity, linker, spatial orientation, E3 ligase ligand and cell permeability play vital roles in the functional capacity of PROTAC. Therefore, further investigation and evaluation of how these factors cooperate and function is a significant scientific issue to resolve. We selected CDK6 as the primary target to discuss how these critical factors are involved in the functional readout of successful PROTACs. We also propose that the resulting PROTAC molecules hold potential advantages over parental inhibitors in certain contexts with clinical significance. For example, specific subtype of cancers and intrinsic or acquired resistance caused by target (here as CDK6) overexpression or mutation (Fig. 1B).
In this study, we report the design and synthesis of CDK6-targeting degraders using PROTAC strategy. These PROTACs could efficiently and specifically degrade CDK6 in low concentrations. More importantly, our newly designed PROTACs also significantly induced the degradation of mutant CDK6. Furthermore, our PROTAC molecules strongly inhibited proliferation of multiple myeloma, leukemia and mantle cell lymphoma cells. These data demonstrate the significance and potential for developing PROTAC-based therapeutic molecules.
RESULTS AND DISCUSSION
Based on literatures14,26,29 and our previous experiences17–19, we hypothesized that the degradation strength or efficiency of a certain PROTAC molecule was at least dictated by the following three major factors: the linker length, the spatial orientation of the target and the E3ligase upon PROTAC conjugation, and binding affinity between PROTAC and its target. The success or failure of PROTAC is decided by the “match” or “mismatch” between PROTAC, E3 ligase and target. Therefore, we applied various CDK6-targeting compounds, distinct E3 ligases and multiple types of linker in PROTAC design. Specifically, we chose the three FDA-approved CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) which possess strong binding affinity but different terminal orientation to target CDK630. The common terminal piperazine in the inhibitors from palbociclib, ribociclib and abemaciclib stretched into solvent via distinct angles31 (Fig. 2A).
Figure 2.

Design of CDK6-targeting PROTACs. (A) Superimposed complex of CDK6 with palbociclib (yellow, PDB: 2euf), abemaciclib (green, PDB: Sl2s), ribociclib (orange, PDB: 512t). (B) Representative structures of designed PROTACs library. Models were prepared with Pymol.
For palbociclib and abemaciclib, their piperazine moieties both stretch into the solvent horizontally, with slight deviation to N-terminal β-sheet of CD K6. In contrast, the terminal piperazine ring of ribociclib is farther from the solvent zone and folds toward the C-terminal α-helix. This may determine the overall rough spatial directions of deriving PROTACs. Nonetheless, the aminopyrimidine stretches into the pocket and forms pivotal hydrogen bonds with surrounding amino acids to stabilize the complexes31. The NH group in piperazine of palbociclib is exposed to the solvent, making it the most suitable site for attachment of linkers without disrupting critical binding interactions31,32. Meanwhile, linking groups at E3 ligase end can also profoundly affect the interacting angle between CDK6 and ligase, prompting us to introduce flexible and rigid groups, such as alkyl and alkyne, into the CRBN recruiting moiety pomalidomide. Additionally, nutlin-3b, VH032, and bestatin were also applied as recruiting moiety for E3 ligase MDM2, VHL and cIAP respectively, for the reason that we could not predict which ligase match or mismatch with CDK630,33. Integrating the above principles, we designed and constructed a small focused library containing different types of PROTACs (Fig. 2B and Table S1). For control, we also synthesized two negative control PROTAC molecules named CP-9 and CP-23 (corresponding to CP-2 and CP-10, respectively) by ethylation of pomalidomide end and thus blocking the binding between pomalidomide and CRBN, and abrogating C DK6 ubiquitination33.
We evaluated the degradation potential of all the designed CDK6-targeting PROTACs by in vitro assays (Summarized in Tables 1, 2, see also Fig. S2). Interestingly, we found that CDK6 proteins were degraded only when PROTAC recruited CRBN but not the other three E3 ligases (Fig. 3A, B). The degradation of CDK4 was less significant compared to CDK6 even though the target-binding molecules were believed to target both CDKs at similar potency, which will be discussed later. And again, CDK4 degradation was only observed in samples treated with CRBN-recruiting PROTACs. We ruled out the possibilities of E3 ligase deficiency and cell context specificity because the U251 glioblastoma cells express all these E3 ligases and the somehow CRBN-preference in CDK6 degradation was also observed in medullablastoma cell line DAOY (Fig. S1). In theory, updating E3 recruiting ligand potency can enhance the degradation potential of PROTAC, for example, introducing cIAP ligands with more potency than bestatin may facilitate CDK6 degradation. However, previous successful target degradation by PROTACs harboring the same ligands we used suggested that the CRBN preference we observed in this study could be more on the intrinsic/PROTAC-induced substrate selectivity of E3 ligases, which needs future investigation. Among the CRBN-recruiting PROTACs, palbociclib-derived degraders CPs were moderately superior to abemaciclib-derived CP-As, while ribociclib-derived CPRs could barely induce CDK6 degradation (Fig. S2 and Tables 2, S1). This may be explained by the distinct binding mode of ribociclib from the other two drugs (Fig. 2A). Herein, we discussed PROTAC molecules based on palbociclib and pomalidomide primarily.
Table 1.
Structures and DC50 values for PROTACs deriving from palbociclib and pomalidomide.
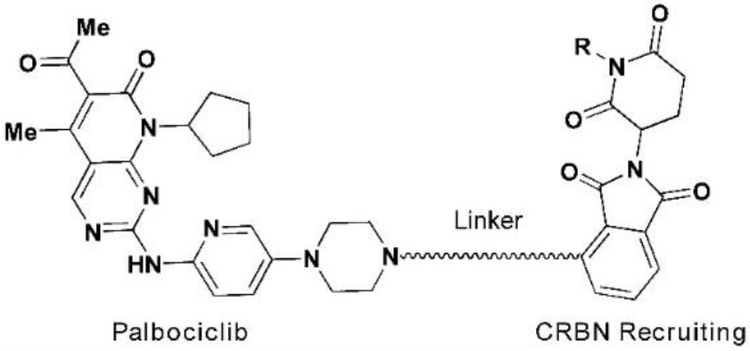 | ||||
|---|---|---|---|---|
| Compounds | Linker | R | DC50/nM CDK6 | DC50/nM CDK4 |
| CP-10 | H | 2.1 | >100 | |
| CP-5 | H | 1.1 | >100 | |
| CP-6 | H | 24.2 | >100 | |
| CP-7 | H | 14.3 | >100 | |
| CP-8 | H | 31.0 | >100 | |
| CP-13 | H | 5.3 | >100 | |
| CP-14 | H | 10.6 | >100 | |
| CP-15 | H | 1.6 | >50 | |
| CP-16 | H | 1.7 | >50 | |
| CP-21 | H | 81.6 | >100 | |
| CP-22 | H | 86 8 | >100 | |
| CP-9 | CH2CH3 | ND | ND | |
| CP-23 | CH2CH3 | ND | ND | |
Notes. ND indicates no degradation.
Table 2.
Structures and DC50 values for PROTACs deriving from abemaciclib and pomalidomide.
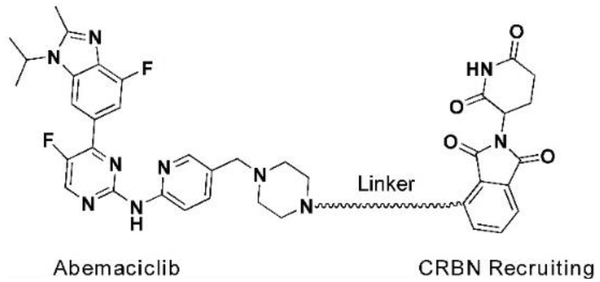 | |||
|---|---|---|---|
| Compounds | Linker | DC50/nM CDK6 | DC50/nM CDK4 |
| CP-A1 | 8.6 | >500 | |
| CP-A2 | 129.7 | >500 | |
| CP-A3 | 271.9 | >500 | |
| CP-A4 | 145.5 | >500 | |
Figure 3.

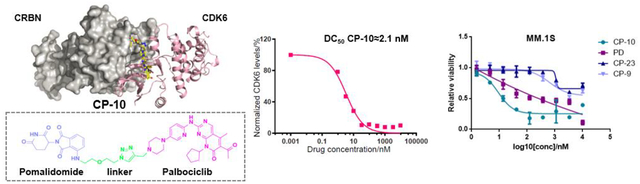
Screen and characterization of potent CDK6-degrading PROTACs. (A/B) CDK6 levels in U251 cells upon 24 h drug treatment. All drugs were administrated at 1 μM. PD (palbociclib) and PD.HCI (commercialized inhibitor, salt) were applied here as control. (C) CP-10 induced more significant degradation of CDK6 than CDK4. Relative expression levels of CDK4/6 normalized to β-Tubulin were labeled. Thus calculated DC50 of CDK6 was about 2.1 nM while DC50 of CDK4 was about 150~180 nM. (D) CP-10 induced specific degradation of CDK6. Immunoblots of representative proteins in samples from two groups of replicates. U2Sl cells were treated with 500 nM PD, CP-1 0 or vehicle control (DMSO) for 4 h. (E) Docking of CP-10 onto CDK4(PDB: 2w96) or CDK6(PDB: 2euf)via Maestro 11.3 (Schrödinger). Docking scores were presented.
To be noted, linker length dependence was observed in CDK6 degraders. PROTACs with shorter linker such as CP-10 and CP-5, possessed higher degradation capacity, which implied these shorter molecules held preferable spatial positions for CRBN recruitment towards CDK6. Moreover, exchanging linker-attaching end at palbociclib side among amide, triazole and methylene yielded PROTACs with similar degradation potential (DC50 of CP-5 ≈ 1.1 nM, DC50 of CP-10 ≈ 2.1 nM, DC50 of CP-15 ≈ 1.6 nM).
At pomalidomide side, however, degradation potency was decreased by nearly 8 fold when flexible imino group on linkers was replaced by rigid alkyne (DC50 of CP-14 ≈ 10.6 nM, DC50, of CP-21 ≈ 81.6 nM, DC50 of CP-22 ≈ 86.8 nM), but slightly enhanced when imino group was substituted by methylene (DC50, of CP-13 ≈ 5.3 nM, DC50, of CP-16 ≈ 1.7 nM). These data suggested that CDK6 protein was insensitive to the rotation of linkage moieties near its kinase active pocket, allowing the accession of CRBN from various obvious preference of flexible linker due to its narrow binding pocket. We also found out that, CP-13/14 possessing alkyl linker and CP-15 removing triazole group excelled CP-10 in CDK6 degradation strength, which may be resulted from enhancement of molecule flexibility, hydrophobic solubility and membrane permeability. Among these CRBN-recruiting PROTACs, CP-10 with the conjugation of palbociclib and pomalidomide demonstrated the best degradation efficacy with the simplest synthesis procedures (Tables 1,2, S1). CP-10 induced nearly 72% degradation of CDK6 at l0 nM and 89% at 100 nM inhuman glioblastoma U251 cells (Fig. 3C). The degradation of CDK4 induced by CP-10 was far weaker than that of CDK6 (DC50 50~80 fold), albeit similar palbociclib affinity and sequence homology of these two kinases (Fig. 3C)31. Time-lapse analysis showed that CDK6 degradation began at roughly 2 h, and was completed by 6 h (Fig. S3).
Control experiments dearly demonstrated that palbociclib, pomalidomide or unconjugated PROTAC arms could not induce CDK6 degradation (Fig. S3). In contrast, palbociclib or pomalidomide could competitively inhibit the degradation effect of CP-10. Additionally, carfilzomib, a proteasome inhibitor, could completely disable the PROTAC effect34. Strong inhibition was also observed with the neddylation inhibitor MLK-4924, which was in accordance with the requirement of neddylation for processive E3 ligase activity of CRllN35 (Fig. S3). The above observations confirmed that degradation of COK6 was mediated by the ubiquitin-proteasome system.
We also examined the responses of several other CDKs and kinases to CP-10 and found out that CP-10 was highly selective for CDK6 without significant off-target effect (Fig. 3D). Quantitative proteomic analysis also showed that CDK6 stood out as the most down-regulated protein in the cell upon 4-hour CP-10 treatment, further confirming the selectivity of CP-10 for CDK6 (Fig. S4A). We did observe several other down-regulated proteins, including ZFP91 and MAP2K7 (MKK7) (Fig. S4B). The modest degradation of COK4 was reasonable because of the high concentration of CP-10 applied. ZFP91 was previously reported to be IMiD-dependent substrate of the CRL4CRBN ubiquitin ligase36. However, ZFP91 remained unchanged after 24 h CP-10 treatment (Fig. S4), indicating that the ZFP91 decrease observed in proteomic analysis was a transient response to high-dose pomalidomide arm. MKK7 also stayed unchanged after 24 h CP-10 incubation, in contrast to the steady and significant degradation of CDK6 (Fig. S4).
In vitro kinase assay indicated that the kinase inhibitory activity of CP-10 for CDK4 or CDK6 was 10–25 fold weaker than that of PD, implying the compromised binding affinity between CP-10 for CDK4 or CDK6 (Fig. S4C). Difference in endogenous abundance for both could be ruled out since FLAG-tagged CDK6 was degraded with greater percentage than FLAG-tagged CDK4 when they were expressed at the same level (Fig. S4D). The selectivity could be partially explained by the more stable complex formed by CP-10 and CDK6 than CDK4, or more “match” as displayed by the docking simulation (Fig. 3E). The fact that CDK4 has less lysine residues (11 in CDK4 vs 18 in CDK6) for ubiquitination may also contribute to the selectivity, which awaits further exploration. Nonetheless, preferred degradation of CDK6 over CDK4 indicates that PROTAC CP-10 is in a better match for CDK6 than CDK4, via proper linker and favorable orientation. A stable and “matched” ternary complex of target, E3 ligase and PROTAC could be formed despite attenuated target-binding affinity.
As CDK6-dependency was claimed mostly in haematopoietic cells37, we selected several such cell lines to further assess the effect of CP-10 in vitro. Human leukemia cell lines THP-1 and HL-60, human mantle cell lymphoma cell line Mino and jeKo-1, human multiple myeloma cell lines MM.lS and RPMI8226 were then treated with increasing concentrations of CP-10 and the corresponding inhibition of cell proliferation was evaluated with CCK-8. For control, we also performed parallel experiments with parental drug PD and non-degrading control molecules CP-9 and CP-23. We found that CP-10 displayed a much better cell inhibition potential (IC50≈10 nM) than PD(IC50≈200 nM) in multiple myeloma cell MM.lS and mantle cell lymphoma cells (in Mino, CP-10 IC50≈8 nM, PD IC50≈45 nM) or comparable activities in leukemia cells (Fig. 4A and Fig. S5). However, CP-10 was far weaker than PD in another multiple myeloma cell RPMI8226 (Fig. S5). As expected, non-degrading PROTACs CP-9 and CP-23 barely inhibited the cell proliferation, or at very high concentrations (Fig. 4A and Fig. S5). Protein levels ofCDK4/6 and phospho-Rb (S780), as an indicator of their kinase activities, were in line with the proliferation assay results (Fig. S5). These data dearly demonstrated that the inhibition on proliferation by CP-10 was mainly achieved via degradation of target protein instead of residual kinase inhibitory activities. The differential response to CP-10 in multiple cell lines suggested cell context specificity or specific dependence on endogenous CDK4 or CDK6 levels or their ratios. For example, the residual CDK4 proteins upon CP-10 treatment in HL-60 cells probably attenuated the cellular sensitivity to CP-10 compared to PD, and also, indicating that HL-60 cells were not simply dependent on CDK6. We were also surprised to observe the superior potency of CP-1 0 in MM.1S than pomalidomide and the combination of PD and pomalidomide, as pomalidomide is an approved agent for multiple myeloma treatment (Fig. S5). Collectively, these data strongly supported the promising potential of CDK6-degraders in fighting against cancers of hematopoietic origin.
Figure 4.

CP-10 inhibited proliferation of cancer cells and induced degradation of amplified/mutated forms of CDK6. (A) A panel of different cancer cells were treated with CP-10/PD/CP-9/CP-23 for 84 h before cell viability measurement by CCK-8. Relative absorbance was plotted. Error bar, SD. (B) CP-10 induced robust degradation of WT, D163G and S178P forms of CDK6. WT, D163G and S178P forms of CDK6-FLAG were stably introduced into MCF-7 cells via lentivirus. Cells were then treated as indicated for 24h before harvest for immunoblotting. Data were representatives of independent replicate experiments for at least three times.
As mentioned in the beginning, the CDK6 overexpression could result in resistance to CDK6 inhibitors in reported preclinical and clinical cases. Therefore, we introduced additional copies of CDK6 into the Ewing’s sarcoma cell line A-673 which expressed scarce CDK6, by lentivirus infection. Indeed, CP-10 cut down the expression levels of over-expressed CDK6 (Fig. S6). This result indicates that the CDK6-targeting PROTAC degraders may represent a new strategy against CDK6-overexpression induced drug resistance. We tested CP-10 in two independent palbociclib-resistant breast cancer cell lines resulting from CDK6 copy amplification (MCF-7 CDK6N2) and FAT1 loss (MCF-7 FAT1 CR)6,7. We found CP-10 could induce degradation of CDK6 and CDK4 and inhibit the proliferation of these palbociclib resistant cell lines (Fig. S6).
Moreover, D163G mutation in CDK6 was previously predicted to attenuate palbociclib binding and S178P was predicted to min1ic CDK4 activation and result in CDK6 hyper-activation8,9. Both mutations may lead to clinical resistance for CDK6 inhibitors. As observed, CP-10 induced degradation of these mutants as efficiently as the wild type CDK6, indicating the robustness for CP-10 induced degradation (Fig. 4B). The degradation of CDK6 D 163G was a little bit compromised compared to wildtype but still significant, suggesting that target binding affinity of palbociclib arm of CP-10 was compromised due to binding hotspot mutation, but sufficient to facilitate CDK6 degradation. Again, successful degradation of palbociclib binding site mutant supported the “match” between CP-10 and mutated CDK6 in spite of compromised binding affinity in theory. These preliminary data supported the application potential of overcoming palbociclib resistance via approaches of CDK4/6 degradation.
CONCLUSION
In summary, a focused library of CDK6-degrader was developed and factors including linker length, spatial orientation and binding affinity were systematically evaluated to help understand the match/mismatch between PROTAC and target and deduce the best strategy for future design or optimization of CDK6 degradation. Remarkably and interestingly, we found out that i) pomalidomidebased PROTACs recruiting CRBN, instead of other tested E3 ligases, resulted in functional degraders; ii) the dual CDK4/CDK6 ligand palbociclib we applied surprisingly resulted in CDK6 selective PROTACs; iii) the representative palbociclib-derived PROTAC CP-10 could inhibit proliferation of several haemetopoietic cancer cells with impressive potency including multiple myeloma; iv) mutated and overexpressed CDK6 can be still degraded by CP-10. These data added to the growing trends of potential clinical benefits of PROTAC techniques and also suggested the specific application of CDK6 degradation in certain cancers.
EXPERIMENTAL SECTION
Chemistry.
All reactions were carried out under atmosphere or argon. Glassware was oven-dried prior to use. Unless otherwise indicated, common reagents or materials were obtained from commercial source and used without further purification. Flash column chromatography was performed using silica gel60 (200–300 mesh). Analytical thin layer chromatography (TLC) was carried out on Yinlong silica gel plates with QF-254 indicator and visualized by UV. The 1H and 13C NMR spectra were recorded at 400 MHz, respectively, on Bruker 400 MHz NMR spectrometer.
All NMR spectra were measured at 25 °C in CDCl3 or DMSO-d6. Chemical shifts (δ) are reported in parts per million, and coupling constants (J) are reported in hertz. The resonance multiplicities in the 1H NMR spectra are described as “s” (singlet), “d” (doublet), “t” (triplet), “quint” (quintet), and “m” (multiplet), and broad resonances are indicated by “br.” Residual protic solvent of CDCl3 (1H, δ 7.26 ppm; 13C, δ 77.16 ppm), DMSO-d6 (1H, δ 2.50 ppm; 13C 39.50 ppm) was used as the internal reference in the 1H- and 13C-NMR spectra. Low-resolution mass spectral analyses were performed with a Waters AQUITY UPLCTM/MS. Purities of the tested compounds, determined by HPLC, were > 95%. Preparative HPLC was carried out on 250 × 10 mm C-18 column using gradient conditions (l – 90% B, flow rate = 3.5 mL/min, 30min). The eluents used were: solvent A (H20 with 0.1% TFA) and solvent B (CH3CN with 0.1% TFA).
Synthetic route of target compound CP-10.
As shown in Scheme 1. Intermediate 12 was prepared according to the patent W02014128588A138. Firstly, compound 10 was synthesized via a substitution reaction between 8 and 9 in presence of isopropylmagnesium chloride. I-PrMgCl was chosen as the base. With this condition, the transformation in step 1, scheme 1 afforded target product in excellent yield.
Scheme 1.

Synthetic route of target compound CP-10
Subsequent intermolecular heck reaction coupled 10 with n-butyl vinyl ether followed by acidolysis led to compound 11. The Bocprotecting group of piperazine was then removed under acid condition to afford 12. An alkynyl group was introduced to 12 by reacting with propargyl bromide, generating intermediate 6. Finally, desired degrader CP-10 was obtained through a click reaction coupling the azide group in 13 with the alkynyl group in 639.
6-acetyl-8-cycolpentyl-5-methyl-2-((5-(4(prop-2-yn-1-yl)piperazin-1-yl)pyridine-2-yl)amino)pyrido[2,3-δ]pyrimidin-7(8H)-one (6).
To the solution of Palbociclib (500 mg, 1.12 mmol) in DMF were added 3-bromopropyne (106 μl, 1.23 mmol), K2CO3 (136 mg, 1.34 mmol) and TBAB (36 mg, 0.11 mmol). The reaction was stirred at 90 °C for 3 h, then washed with water and the organic layer was concentrated to dryness. Flash column chromatography (CH2Cl2/MeOH, 40:1, vol/vol) give compound 9 (438 mg, 81%) as yellow solid. Rf = 0.4, CH2Cl2/MeOH 20:1; 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 8.38 (s, 1H), 8.16 (d, J = 9.0 Hz, 1H), 8.07 (d, J = 2.8 Hz, 1H), 7.33 (dd, J1 = 9.1 Hz, J2 = 3.0 Hz, 1H), 5.92 to 5.83 (m, 1H), 3.39 (d, J = 2.4 Hz, 2H), 3.24 (t, J = 4.8 Hz, 4H), 2.77 (t, J = 4.8 Hz, 4H), 2.54 (s, 3H), 2.37 to 2.33 (m, 5H), 2.29 (t, J = 2.4 Hz, 1H), 2.07 to 2.04 (m, 2H), 1.92 to 1.85 (m, 2H), 1.70 to 1.66 (m, 2H); 13CNMR (400 MHz, CDCl3) δ 202.84, 161.59, 158.25, 157.40, 155.70, 145.09, 143.73, 14.95, 136.81, 130.87, 126.06, 113.76,107.88, 78.55, 73.70, 54.19, 51.78, 49.54, 47.07, 31.70, 28.23, 25.91, 14.12, 8.23; LC-MS (EST+): m/z calculated for C27H31N7O2: 486.25 [M+H]+; found 487.6194.
4-((2-(2-(4-((4-(6-((6-acetyl-8-cyclopentyl-5-methyl-7-oxo-7,8-dihydropyrido [2,3-d]pyrimidin-2-yl)amino)pyridin-3-yl)piperazin-1-yl)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (CP-10).
Compound 6 (15 mg, 0.03 mmol), compound 13 (0.04 mmol, 1.2 eq), sodium ascorbate (17.8 mg, 0.09 mmol) was dissolved in tBuOH/DCM: 1 ml/0.5 ml, then the solution of CuSC4 (9.6 mg, 0.06 mmol) in 0.5 ml water was added. The resulting mixture was stirred at room temperature for 10 min. After the reaction was completed, the solvent was removed and then dealt with 7 M ammonium hydroxide, organic layer was separated and concentrated to dryness. Flash column chromatography (CH2Cl2/MeOH, 20:1, vol/vol) give compound CP-10, (0.019 mmol, 63%) as solid. Rf = 0.4, CH2Cl2/MeOH 10:lH NMR (400MHz, CDC3) δ 10.34 (s, 1H), 8.79 (s, 1H), 8.22 (s, 1H), 8.12 (d, J = 9.1 Hz, 1H), 8.02 (d, J = 2.5 Hz, 1H), 7.75 (s, 1H), 7.51 (dd, j1 = 8.3 Hz, J2 = 7.3 Hz, 1H), 7.30 (dd, J1 = 9.1 Hz, J2 = 2.5 Hz, 1H), 7.14 (d, J = 7.3 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.63 (t, J = 5.3 Hz, 1H), 5.89 to 5.84 (m, 1H), 4.92 to 4.88 (m, 1H), 4.60 to 4.60 (m, 2H), 3.94 to 3.87 (m, 3H), 3.72 to 3.63 (m, 3H), 3.43 to 3.41 (m, 2H), 3.24 to 3.19 (m, 4H), 2.76 to 2.69 (m, 7H), 2.54 (s, 3H), 2.36 to 2.32 (m, 5H), 2.19 to 2.15 (m, 1H), 2.07 to 2.03 (m, 2H), 1.89 to 1.83 (m, 2H), 1.70 to 1.66 (m, 2H); 13C NMR (400 MHz, CDCI3) δ 202.87, 712.18, 169.99, 169.54, 167.62, 161.58, 158.25, 157.39, 155.68, 147.07, 141.99, 136.42, 132.69, 130.81, 126.09, 124.75, 117.17,112.30, 110.93, 107.79, 77.39, 69.91, 69.61, 68.12, 54.23, 52.84, 52.49, 50.67, 49.18, 49.06, 42.40, 31.69, 31.55, 29.84, 28.20, 27.06, 25.87, 25.76, 23.20, 14.10; LC-MS (ESI+): m/z calculated for C44H49N13O7: 437.195 [M+2H]2+; found 437.2669. Purities of CP-10 determined by HPLC, were > 95% (Shown in Supporting information).
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Jiahua Han and Dr. Wensheng Wei for sharing reagents. We also thank Dr. Haitao Li, Dr. Qiaoran Xi, Dr. Xiaonan Su, Dr. Xiuyun Sun, Ms. Ye Sun and Ms. Dan Wang for helpful discussions and suggestions and Dr. Libing Mu for cartoon illustration assistance.
S.C. has received research support (to institution) from Genentech, Novartis, Eli Lilly, Daiichi Sankyo, and Sanofi and ad hoc consulting fees (to SC) from Eli Lilly, Novartis, Sermonix, Revolution Medicines, Context Therapeutics, and BMS. All the other authors declare no conflicts of interest.
Funding Sources
Y.R’s research is funded by National Natural Science Foundation of China (# 81573277, 81622042, 81773567) and National Major Scientific and Technological Special Project for “Significant New Drugs Development” (# SQ2017ZX095003). W.W’s research was funded by National Natural Science Foundation of China (# 81672950). S.C’s research is funded by NIH Cancer Center Support Grant (CCSG P30 CA08748) and the Breast Cancer Research Foundation.
Footnotes
Supporting Information.
Details for Synthesis Routines, HPLC analysis, Predictive model construction and biological experiments, Supplementary Figures and Tables, Molecular Formula Strings (provided as separate CSV files) and Abbreviation used in this article could be found in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Vermeulen K; Van Bockstaele DR; Berneman ZN The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif 2003, 36, 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Otto T; Sicinski P Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tadesse S; Yu M; Kumarasiri M; Le BT; Wang S Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle 2015, 14, 3220–3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Niesvizky R; Badros AZ; Costa LJ; Ely SA; Singhal SB; Stadtmauer EA; Haideri NA; Yacoub A; Hess G; Lentzsch S; Spicka I; Chanan-Khan AA; Raab MS; Tarantolo S; Vij R; Zonder JA; Huang X; Jayabalan D; Di Liberto M; Huang X; Jiang Y; Kim ST; Randolph S; Chen-Kiang S Phase 1/2 study of cyclin-dependent kinase (CDK)4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leuk. Lymphoma 2015, 56, 3320–3328. [DOI] [PubMed] [Google Scholar]
- (5).National Cancer Institute. Clinical trials using palbociclib page. https://www.cancer.gov/about-cancer/treatment/clinicaltrials/intervetion/palbocilib (accessed Mar 29, 2019)
- (6).Yang C; Li Z; Bhatt T; Dickler M; Giri D; Scaltriti M; Baselga J; Rosen N; Chandarlapaty S Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Li Z; Razavi P; Li Q; Toy W; Liu B; Ping C; Hsieh W; Sanchez-Vega F; Brown DN; Da Cruz Paula AF; Morris L; Selenica P; Eichenberger E; Shen R; Schultz N; Rosen N; Scaltriti M; Brogi E; Baselga J; Reis-Filho JS; Chandarlapaty S Loss of the FAT1 tumor suppressor promotes resistance to CDK4/6 inhibitors via the Hippo pathway. Cancer Cell 2018, 34, 893–905 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bockstaele L; Bisteau X; Paternot S; Roger PP Differential regulation of cyclin-dependent kinase 4 (CDK4) and CDK6, evidence that CDK4 might not be activated by CDK7, and design of a CDK6 activating mutation. Mol Cell. Biol 2009, 29, 4188–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hernandez Maganhi S; Jensen P; Caracelli I; Zukerman Schpector J; Frohling S; Friedman R Palbociclib can overcome mutations in cyclin dependent kinase 6 that break hydrogen bonds between the drug and the protein. Protein Sci. 2017, 26, 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Neklesa TK; Winkler JD; Crews CM Targeted protein degradation by PROTACs. Pharmacol. Ther 2017, 174, 138–144. [DOI] [PubMed] [Google Scholar]
- (11).Ottis P; Toure M; Cromm PM; Ko E; Gustafson JL; Crews CM Assessing different E3 Ligases for small molecule induced protein ubiquitination and degradation. ACS Chemical Biology 2017, 12, 2570–2578 [DOI] [PubMed] [Google Scholar]
- (12).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 2017,16,101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skpl-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Burslem GM; Smith BE; Lai AC; Jaime-Figueroa S; McQuaid DC; Bondeson DP; Toure M; Dong H; Qian Y; Wang J; Crew AP; Hines J; Crews CM The advantages of targeted protein degradation over inhibition: An RTK Case Study. Cell Chem Biol 2018, 25, 67–77 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lu J; Qjan Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Bioi 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sun Y; Zhao X; Ding N; Gao H; Wu Y; Yang Y; Zhao M; Hwang J; Song Y; Lin W; Rao Y PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhao Q; Lan T; Su S; Rao Y Induction of apoptosis in MDAMB-231 breast cancer cells by a PARP1-targeting PROTAC small molecule. Chemical Communications 2019,55, 369–372. [DOI] [PubMed] [Google Scholar]
- (19).An Z; Lv W; Su S; Wu W; Rao Y Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein & Cell 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jiang B; Wang ES; Donovan KA; Liang Y; Fischer ES; Zhang T; Gray NS Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angewandte Chemie 2019, 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hatcher JM; Wang ES; Johannessen L; Kwiatkowski N; Sim T; Gray NS Development of highly potent and selective steroidal inhibitors and degraders of CDKS. ACS Med. Chem. Lett 2018, 9, 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Olson CM; Jiang B; Erb MA; Liang Y; Doctor ZM; Zhang Z; Zhang T; Kwiatkowski N; Boukhah M; Green JL; Haas W; Nomanhhoy T; Fischer ES; Young RA; Bradner JE; Winter GE; Gray NS Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Bioi 2018, 14, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faclth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher, l.; Crews, C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chcm. Biol 2015, 11, 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Crew AP; Raina K; Dong H; Qian Y; Wang J; Vigil D; Serebrenik YV; Hamman BD; Morgan A; Ferraro C; Sin K; Neklesa TK; Vlinkler JD; Coleman KG; Crews CM Identification and characterization of Von Hippel-Lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-Binding Kinase I. J. Med. Chem 2018, 61, 583–598. [DOI] [PubMed] [Google Scholar]
- (25).Cramm PM; Samarasinghe KTG; Hines J; Crews CM Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J.Am. Chem. Soc 2018, 140, 17019–17026. [DOI] [PubMed] [Google Scholar]
- (26).Smith BE; Wang SL; Jaime-Figueroa S; Harbin A; Wang J; Hamman BD; Crews CM Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nature Comnnmicatiom 2019, 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Buhimschi AD; Armstrong HA; Toure M; Jaime-Figueroa S; Chen TL; Lehman AM; Woyach JA; Johnson AJ; Byrd JC; Crews CM Targeting the C481S ibrutinib-resistance mutation in Bruton’s Tyrosine Kinase using PROTAC-mediated degradation. Biochemistry 2018, 57, 3564–3575. [DOI] [PubMed] [Google Scholar]
- (28).Salami J; Alabi S; Willard RR; Vitale NJ; Wang J; Dong H; Jin M; McDonnell DP; Crew AP; Neklesa TK; Crews CM Androgen receptor degradation by the proteolysis-targeting chimera ARCC-4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Communicatiom biology 2018, 1, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angewandte Chemie 2016, 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sherr CJ; Beach D; Shapiro GI Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov 2016, 6, 353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chen P; Lee NV; Hu W; Xu M; Ferre RA; Lam H; Bergqvist S; Solowiej J; Diehl W; He YA; Yu X; Nagata A; Van Arsdale T; Murray BW Spectrum and degree of CDK drug interactions predicts clinical performance. Mol. Cancer Ther. 2016, 15,2273–2281. [DOI] [PubMed] [Google Scholar]
- (32).Lu H; Schulze-Gahmen U Toward understanding the structural basis of cyclin-dependent kinase 6 specific inhibition. J. Med. Chem 2006, 49, 3826–3831. [DOI] [PubMed] [Google Scholar]
- (33).Toure M; Crews CM Small-Molecule PROTACS: New Approaches to Protein Degradation. Angewandte Chemie 2016, 55, 1966–1973. [DOI] [PubMed] [Google Scholar]
- (34).Kuhn DJ; Chen Q; Voorhees PM; Strader JS; Shenk KD; Sun CM; Demo SD; Bennett MK; van Leeuwen FW; Chanan-Khan AA; Orlowski RZ Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-protcasome pathway, against preclinical models of multiple myeloma. Blood 2007, 110, 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Nawrocki ST; Griffin P; Kelly KR; Carew JS MLN4924: a novel first-in-class inhibitor of NEDDS-activating enzyme for cancer therapy. Expert Opin Investig Dmgs 2012, 21, 1563–1573. [DOI] [PubMed] [Google Scholar]
- (36).An J; Ponthier CM; Sack R; Seebacher J; Stadler MB; Donovan KA; Fischer ES pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4CRBN ubiquitin ligase. Nature Communications 2017, 8, 15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kim S; Tiedt R; Loo A; Horn T; Delach S; Kovats S; Haas K; Engstler BS; Cao A; Pinzon-Ortiz M; Mulford I; Acker MG; Chopra R; Brain C; di Tomaso E; Sellers WR; Caponigro G The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 2018, 9, 35226–35240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chekal BP; IDE, N.D. Solid forms of a selective cdk4/6 inhibitor. W.O. Patent 2014128588, August 28,2014.
- (39).Wurz RP; Dellamaggiore K; Dou H; Javier N; Lo MC; McCarter JD; Mohl D; Sastri C; Lipford JR; Cee VJA “Click Chemistry Platform” for the Rapid Synthesis of Bispeciflc Molecules for Inducing Protein Degradation. J. Med. Chcm 2018, 61, 453–461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.