

Abstract
Despite a growing interest in CHF2 in medicinal chemistry, there is a lack of efficient methods for the insertion of CHF18F into druglike compounds. Herein described is a photoredox flow reaction for 18F‐difluoromethylation of N‐heteroaromatics that are widely used in medicinal chemistry. Following the two‐step synthesis for a new 18F‐difluoromethylation reagent, the photoredox reaction is completed within two minutes and proceeds by C−H activation, circumventing the need for pre‐functionalization of the substrate. The method is operationally simple and affords straightforward access to radiolabeled N‐heteroaromatics with high molar activity suitable for biological in vivo studies and clinical application.
Keywords: C−H activation, flow chemistry, fluorine, photochemistry, radiochemistry
Make a trace: A new flow photoredox methodology allows the 18F‐difluoromethylation of heteroaromatics by C−H activation. The method is applicable to a wide range of heteroaromatics and can be used for the preparation of radiotracers with high molar activities that are suitable for positron emission tomography studies.
Introduction
PET (positron emission tomography)1 is a powerful non‐invasive imaging technology to evaluate disease states (diagnosis) and support the clinical development of new drug candidates. PET is the method of choice to measure the biodistribution of a drug and the receptor occupancy of a molecule in the central nervous system (CNS), enabling a more accurate assessment of the efficacy of a drug. The use of PET relies on the preparation of labeled compounds. Carbon‐112 or fluorine‐18 are the most commonly used isotopes for the synthesis and development of new radiotracers. Synthetic methodologies developed for the preparation of PET tracers should be compatible with the short half‐life of these radioisotopes. Because of a favorable half life of 110 minutes (vs. 20 min for carbon‐11) and the importance of fluorine and fluorinated groups such as CF3 and CHF2 in medicinal chemistry, fluorine‐18 is a very attractive isotope for PET studies. SNAr substitution reactions with [18F]fluoride is still the most prevalent method for the labeling of aromatics. This method is however limited to reactive electron‐deficient aromatics. Recently, a number of new synthetic methodologies have been developed for the incorporation of fluorine‐18 into electron‐neutral and electron‐rich aromatics to overcome this limitation, expanding significantly the chemical space accessible to radiolabeling.3a–3c Good progress has also been made for the introduction of other important fluorinated groups such as CF3.4a–4c Recently, there has been a growing interest in CHF2 in medicinal chemistry. The difluoromethyl group (CHF2) is less lipophilic than the trifluoromethyl one and has been successfully used to block oxidative metabolism because of aldehyde oxidase.5 In addition, CHF2 can serve as a hydrogen‐bond donor and thus can act as a lipophilic bioisoster of the hydroxy group.6a, 6b To the best of our knowledge, only three methodologies for the incorporation of CHF18F into druglike compounds have been reported (Figure 1). Two of them are applicable mainly to aromatics and require pre‐functionalization of the substrates.7a, 7b A limitation of these methods is the low to moderate molar activity obtained, and could make clinical application challenging. The third approach utilizes an oxidative benzylic fluorination of a pre‐labeled aromatic substrate and has not been exemplified on N‐heteroaromatics.7c In spite of these encouraging results, there is still a clear need to devise new methods to introduce CHF18F on drug‐like scaffolds typically found in medicinal chemistry programs. We reasoned that the use of a radical CHF18F could offer a straightforward way to label N‐heteroaromatics through C−H functionalization and overcome the tedious synthesis of pre‐functionalized substrates. Our aim was to produce the radical CHF18F through a photoredox reaction in flow to ensure optimal irradiation of the reaction mixture and a fast reaction time.
Figure 1.
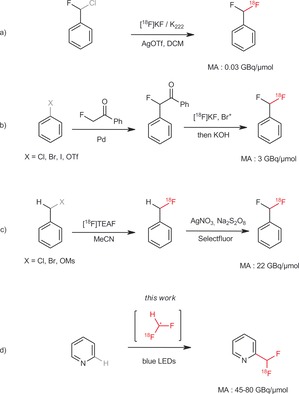
Existing methodologies for the introduction of CHF18F. a) Halex exchange.7a b) Prefunctionnalization and subsequent 18F‐fluorination.7b c) Prefunctionnalization and subsequent Halex and addition of a fluorine with selectfluor.7c d) Direct 18F difluoromethylation by C−H activation.
Several reagents have been described for the C−H functionalization of heteroaromatics with CHF2.8a–8c Based on the seminal contribution of Langlois8d, 8e on the radical trifluoromethylation of aromatics, Baran and co‐workers first disclosed the use of Zn(SO2CHF2)2 for C−H difluoromethylation of N‐heteroaromatics.8f More recently, the group of Maruoka reported that hypervalent iodonium reagents prepared from difluoroacetic acid are effective for the addition of CHF2 on heteroaromatics.8g The group of Nielsen disclosed a redox process utilizing difluoroacetic acid as a direct source of CHF2.8h A new and complementary method was published during the preparation of this manuscript describing an oxidative C−H difluoromethylation with copper complexes generated in situ from TMSCHF2.8i
However, all these described reagents are not available yet for fluorine‐18 application, either because they are not easy to label, or because they are difficult to purify and analyze (such as anionic compounds).
We envisioned that the benzothiazole sulfone reagent, described by Hu and co‐workers9a, 9b for the addition of CHF2 to alkenes, coupled to cyclization would serve our purpose well. The CHF2 radical can be produced under neutral conditions through a catalytic photoredox reaction. In addition, the opportunity to introduce fluorine‐18 through a halogen exchange (Halex) reaction and the excellent solubility of this reagent in organic solvent, as well as its easy purification, are attractive for PET chemistry. Below, we describe the labeling of Hu's reagent, as well as a novel methodology for the CHF18F labeling of N‐heteroaromatics under neutral conditions by a photoredox reaction in flow chemistry.
Results and Discussion
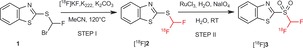
We first turned our attention to the preparation of the labeled benzothiazole sulfone [18F]3 (Table 1). The first step (the labeling step) was the introduction of no carrier added (nca) [18F]‐fluoride by a Halex reaction. Using K[18F]F, K222, and K2CO3 in MeCN at 120 °C, [18F]2 was obtained in 15.2±0.3 % radiochemical yield (RCY; entry 4). Unless otherwise specified, all the RCYs are based on HPLC and TLC analysis of the crude reaction mixture (see the Supporting Information). Lower temperature slightly decreased the RCY. Conducting the reaction in either DMSO or DCE either did not give the desired product or led to a lower RCY (entries 1 and 2). Increasing the quantity of 1, or replacement of the base, did not afford any improvement (entries 5 and 6). [18F]2 was directly engaged in the second step. Oxidation of [18F]2 to the corresponding sulfone [18F]3 using RuCl3, x H2O, and NaIO4 in water on a tC18 cartridge proceeded readily and quantitatively with a RCY of 13.4±0.4 % over the two steps (entry 7). No catalyst resulted in no reaction, and a lower amount of oxidant translated into a lower RCY (entries 8 and 9). After semi‐preparative HPLC purification, the molar activity of [18F]3 was 81.4±11.1 GBq μmol−1 [2.2±0.3 Ci μmol−1, decay corrected (dc)] at the end‐of‐bombardment (EOB), affording one of the best molar activities for CHF18F addition. The duration of the synthesis of [18F]3, including HPLC purification was about 45 minutes. With these encouraging results in hand, we started the exploration of the reaction of [18F]3 with N‐heteroaromatics in the presence of a photoredox catalyst.
Table 1.
Synthesis of [18F]3.
|
Entry |
Reaction Step |
Deviation from standard reaction conditions[a,b] |
RCY [%] (of the crude mixture) |
|---|---|---|---|
|
1 |
I |
85 °C, DCE |
7.2±0.5 |
|
2 |
I |
85 °C, DMSO |
0 |
|
3 |
I |
85 °C |
12.7±0.2 |
|
4 |
I |
– |
15.2±0.3 |
|
5 |
I |
1 (80 μmol) |
5.9±2.4 |
|
6 |
I |
Et4N+HCO3 ‐ |
12.8±1.1 |
|
7 |
II |
– |
13.4±0.4 |
|
8 |
II |
NaIO4 (0.12 mmol) |
9.6±0.9 |
|
9 |
II |
No RuCl3, x H2O |
0.2 |
[a] Standard reaction conditions for I: K[18F]F (100–150 MBq), 1 (40 μmol), K222 (10 μmol), K2CO3 (20 μmol), MeCN, 120 °C, 5 min, n=3. [b] Standard reaction conditions for II : NaIO4 (240 μmol), RuCl3⋅x H2O (80 μmol), H2O, RT, 5 min, n=3.
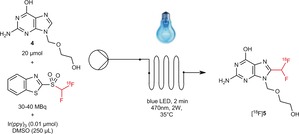
The 18F‐difluoromethylation reaction was conducted using flow chemistry to ensure a better irradiation of the solution and accelerate the reaction rate, since the reaction time is a crucial parameter for fluorine‐18 chemistry. The optimization of the photochemical reaction was performed on Acyclovir10 (4), an anti‐herpetic drug (Table 2). All reactions were done in triplicate with [18F]3, previously purified by semi preparative HPLC (PREP HPLC). Performing the reaction in DMSO with Ir(ppy)3 as a catalyst (0.01 μmol), under irradiation with blue LEDs (2 W, 470 nm) at 35 °C turned out to be optimal (entry 3). Under these reaction conditions, the desired difluoromethylated acyclovir [18F]5 was formed in only 2 minutes with an excellent RCY of 70±7 % (n=7). A slightly higher temperature (55 °C vs. 35 °C, entry 5), or a lower reaction time (30 s, entry 6) decreased the RCY. Changing the solvent and switching from DMSO to DMF also decreased the RCY from 70 to 44 %. The low solubility of acyclovir in MeCN and DCE prevented the use of these solvents. Interestingly, running the reaction with 0.001 μmol of Ir catalyst still afforded [18F]5 but in a lower RCY. In contrast, Ru(bpy)3 did not lead to any product at all, whereas benzophenone was less effective than the Ir(ppy)3 catalyst. It is worth mentioning that the presence of water (50 μL, entry 9) had a negative impact on the RCY. The use of dried DMSO and minimizing the presence of water within the reagent [18F]3 is recommended (see the Supporting Information). After HPLC purification, [18F]5 was obtained with a very satisfactory RCY of 42±4 %. Gratifyingly, the molar activity of [18F]5 was 44.4±11.1 GBq μmol−1 (1.2±0.3 Ci μmol−1 dc) at the EOB (around 90 min for the three steps). The estimated RCY for the three‐step synthesis is 3–4 % (dc). The desired labeled product [18F]5 was not formed without either a catalyst or light supporting the photoredox mechanism (entries 10 and 11). In addition, no reaction was observed with TEMPO (entry 12), suggesting that the reaction takes place through a radical mechanism. The mechanism of the reaction is likely to be similar to the one put forward by the group of MacMillan11 for C−H aromatic trifluoromethylation, and involves a CHF18F radical (see the Supporting Information).
Table 2.
Optimization of the 18F‐difluoromethylation reaction.
|
Entry |
Deviation from standard reaction conditions |
RCY [18F]5 [%] (of the crude reaction mixture) |
|---|---|---|
|
1 |
benzophenone[a] |
47±5 |
|
2 |
Ru(bpy)3 (0.01 μmol) |
0 |
|
3 |
– |
70±7 |
|
4 |
After HPLC purification of [18F]5 |
42±4[b] |
|
5 |
55 °C |
51±10 |
|
6 |
30 s |
60±8 |
|
7 |
Ir(ppy)3 (0.001 μmol) |
42±2 |
|
8 |
DMF |
44±1 |
|
9 |
DMSO/H2O (200/50 μL) |
45±10 |
|
10 |
no catalyst |
0 |
|
11 |
no light |
0 |
|
12 |
TEMPO |
0 |
[a] Benzophenone (10 μmol), 365 nm, [b] Radiochemical yield (n=4) of isolated product.
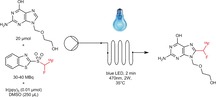
With the optimal reaction conditions in hand, the scope of the photochemical reaction was investigated (Figure 2). Pleasingly, C−H 18F‐difluoromethylation can be carried out on a broad range of heteroaromatics such as indole, benzimidazole, azaindole, pyridine, pyrimidine. 18F‐difluoromethylated compounds were obtained in low to excellent RCYs (from 18 to 75 % RCY). A higher amount of catalyst and a longer residence time were sometimes needed to complete the reaction ([18F]16–[18F]18). It should be noted that more than one isomer was usually observed when several C−H bonds were available for functionalization. However, isomers were readily separated and isolated by PREP‐HPLC (see [18F]31). We also observed that some substrates in Figure 2 proved to be less reactive or even unreactive under stoichiometric batch conditions using fluorine‐19, but could be readily labeled using our flow chemistry conditions (see Section 3 in the Supporting Information). The methodology was used to label more complex scaffolds of medical interest. Xanthines such as caffeine, theophylline, and pentoxifylline turned out to be good substrates and were labelled with RCYs ranging from 30 to 65 % ([18F]20–[18F]22). The methodology could also be used to label nucleic acid bases such as uracil, cytosine, and adenine ([18F]23–[18F]25). Guanine could not be used in flow because of its limited solubility in DMSO. However, guanosine and other nucleosides such as uridine, cytidine, and adenosine ([18F]26–[18F]29) were readily labeled under standard reaction conditions.
Figure 2.
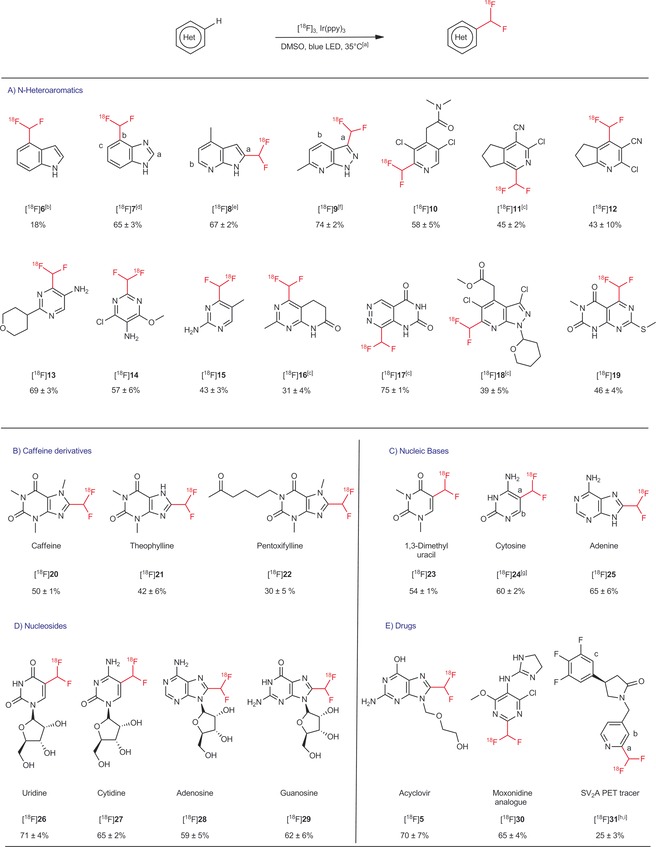
Scope of the 18F‐difluoromethylation reaction. [a] Standard reaction conditions: Substrate (20 μmol), [18F]3 (30–40 MBq), Ir(ppy)3 (0.01 μmol), residence time (2 min), 35 °C, DMSO, 470 nm, n=3. [b] 0.05 μmol Ir(ppy)3. [c] 0.05 μmol Ir(ppy)3 and residence time of 4 min. [d] Ratio a/b/c=3:6:1. [e] Ratio a/b=90:10. [f] Ratio a/b=70:30. [g] Ratio not determined. [h] Ratio a/b/c=4:1:2. [i] RCYs of isolated a: 4.2±0.3 % and b: 1.5±0.1 %.
To further demonstrate the benefit of the new methodology, the new procedure was applied to the preparation of the CHF18F analogue of moxonidine ([18F]30; Figure 2), and to the labeling of a new SV2A ligand ([18F]31). Moxonidine,12 a drug used for the treatment of hypertension is reported to exert its pharmacological effect through the imidazoline receptor. Starting from the precursor 36(see Section 1d in the Supporting Information), CHF18F was readily introduced in place of the methyl group under the optimal reaction conditions described above. [18F]30was obtained with 65±4 % RCY.
SV2A is the molecular target of the antiepileptic drugs Keppra (Levetiracetam13a) and Briviact (Brivaracetam13b). SV2A is widely expressed in the brain and the potential of SV2A PET tracers such as UCB‐J14 (Figure 3) as biomarkers of synaptic density are currently being explored for CNS disorders including neurodegenerative diseases. The University of Liège in collaboration with UCB reported the synthesis and evaluation of the first 18F‐SV2A radiotracer UCB‐H.15a, 15b However, introduction of a fluorine in place of the methyl group on the pyridine slightly reduced the affinity for SV2A (Figure 3). Our objective was to investigate the direct introduction of CHF18F onto the pyridine in place of the methyl substituent known to be important to maintain high affinity for SV2A. Application of our procedure (unsubstituted pyridine, see the Supporting Information) gave a mixture of three isomers, [18F]31 a, [18F]31 b, and [18F]31 c, which were readily separated by HPLC. After separation, [18F]31 b was obtained with a RCY of 1.5 %.
Figure 3.
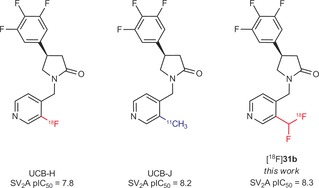
SV2A PET tracers.
In comparison to other reported procedures,16a, 16b our new methodology enables the direct labeling of the SV2A ligand (see Section 1d) and avoids the time and resource investment in chemistry for the preparation of a pre‐functionalized precursor. It provides access to a fluorine‐18 SV2A radiotracer with affinity similar to that of UCB‐J (Figure 3) and illustrates how the method can support rapid access to new PET tracers for biological evaluation.
Conclusion
We have disclosed the development of a new methodology for the direct C−H 18F‐difluoromethylation of a wide range of N‐heteroaromatics. The method utilizes a new CHF18F reagent and a photoredox reaction17a–17c for the labeling of druglike motifs by C−H activation using a three‐step synthesis. Molar activity is an important parameter to consider when assessing the value and utility of a new labeling methods for PET imaging. The methodology described herein gives access to 18F‐labeled products with molar activities (44.4±11.1 GBq μmol−1 as shown with acyclovir) suitable for biological in vivo studies and clinical application, affording a significant advantage over existing methods. The protocol is operationally simple, and the use of a flow process for the photoredox reaction enables very short reaction time for the labeling step (less than 2 min). The reaction proceeds under mild reaction conditions and displays high functional‐group tolerance. Most functional groups used in medicinal chemistry, such as free amine, alcohol, nitrile, chlorine, amide and ether, as well as aminals in sugars appear to be compatible with reaction conditions. This method does not require pre‐functionalization of substrates and can be realized at the very last step of the synthesis as demonstrated by the late‐stage labeling of four drugs (acyclovir, theophylline, pentoxifylline, and a moxonidine analogue) and a SV2A ligand. Furthermore, our flow method holds potential for automation. The development of a fully automated process is currently underway and will be reported in due course. We anticipate that this novel methodology will be very useful for late‐stage labeling of druglike compounds and the discovery of new PET tracers.
Experimental Section
General radiochemical procedure for 18F‐difluoromethylation. A solution of the substrate (20 μmol), Ir(ppy)3 (0.01 μmol) in DMSO (200 μL) was prepared. Then [18F]3 in solution in DMSO (around 37 MBq/1mCi, 50 μL) was added. The solution was injected in a 100 μL microchip, pumped with DMSO at a flow rate of 50 μL min−1 (residence time of 2 min) and irradiated under blue LED (470 nm, 2 W), at a temperature of 35 °C. The exited solution was analyzed by Radio‐TLC and Radio‐UPLC for radiochemical yield (RCY) determination.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska‐Curie grant agreement N°675071. The authors would like to thank Christelle Derwa, Fabien Chaix, and Jehan Claessens for their help in purification and characterization of the cold references, and Pr. Jean‐Christophe Monbaliu for the access to the flow equipment.
L. Trump, A. Lemos, B. Lallemand, P. Pasau, J. Mercier, C. Lemaire, A. Luxen, C. Genicot, Angew. Chem. Int. Ed. 2019, 58, 13149.
Contributor Information
Prof. André Luxen, Email: aluxen@uliege.be.
Dr. Christophe Genicot, Email: Christophe.genicot@ucb.com.
References
- 1. Mercier J., Provins L., Hannestad J. in Comprehensive Medicinal Chemistry III, Vol. 7.02 (Eds.: S. Chackalamannil, D. Rotella, E. W. Ward), Elsevier, Amsterdam, 2017, pp. 20–64. [Google Scholar]
- 2. Deng X., Rong J., Wang L., Vasdev N., Zhang L., Josephson L., Liang S. H., Angew. Chem. Int. Ed. 2019, 58, 2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Preshlock S., Tredwell M., Gouverneur V., Chem. Rev. 2016, 116, 719; [DOI] [PubMed] [Google Scholar]
- 3b. van der Born D., Pees A., Poot A. J., Orru R. V. A., Windhorst A. D., Vugts D. J., Chem. Soc. Rev. 2017, 46, 4709; [DOI] [PubMed] [Google Scholar]
- 3c. Coenen H. H., Ermert J., J. Clin. Trans. Imaging. 2018, 6, 169. [Google Scholar]
- 4.
- 4a. Huiban M., Tredwell M., Mizuta S., Wan A., Zhang X., Collier L. T., Gouverneur V., Passchier J., Nat. Chem. 2013, 5, 941; [DOI] [PubMed] [Google Scholar]
- 4b. van der Born D., Sewing C., Herscheid J. D. M., Windhorst A. D., Orru R. V. A., Vugts D. J., Angew. Chem. Int. Ed. 2014, 53, 11046; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11226; [Google Scholar]
- 4c. Verhoog S., Kee C. W., Wang Y., Khotavivattana T., Wilson T. C., Kersemans V., Smart S., Tredwell M., Davis B. G., Gouverneur V., J. Am. Chem. Soc. 2018, 140, 1572. [DOI] [PubMed] [Google Scholar]
- 5. O'Hara F., Burns A. C., Collins M. R., Dalvie D., Ornelas M. A., Vaz A. D. N., Fujiwara Y., Baran P. S., J. Med. Chem. 2014, 57, 1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Zafrani Y., Yeffet D., Sod-Moriah G., Berliner A., Amir D., Gershonov E., Saphier S., J. Med. Chem. 2017, 60, 797; [DOI] [PubMed] [Google Scholar]
- 6b. Sessler C. D., Rahm M., Becker S., Goldberg J. M., Wang F., Lippard S. J., J. Am. Chem. Soc. 2017, 139, 9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Verhoog S., Pfeifer L., Khotavivattana T., Calderwood S., Collier T. L., Wheelhouse K., Tredwell M., Gouverneur V., Synlett 2016, 27, 25; [DOI] [PubMed] [Google Scholar]
- 7b. Shi H., Braun A., Wang L., Liang S. H., Vasdev N., Ritter T., Angew. Chem. Int. Ed. 2016, 55, 10786; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10944; [Google Scholar]
- 7c. Yuan G., Wang F., Stephenson N. A., Wang L., Rotstein B. H., Vasdev N., Tang P., Liang S. H., Chem. Commun. 2017, 53, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews on difluoromethylation, see:
- 8a. Rong J., Ni C., Hu J., Asian J. Chem. 2017, 6, 139; [Google Scholar]
- 8b. Yerien D. E., Barata-Vallejo S., Postigo A., Chem. Eur. J. 2017, 23, 14676; [DOI] [PubMed] [Google Scholar]
- 8c. Lemos A., Lemaire C., Luxen A., Adv. Synth. Catal. 2019, 10.1002/adsc.201801121. For specific publications [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Ji Y., Bruecki T., Baxter R. D., Fujiwara Y., Seiple I. B., Su S., Blackmond D. G., Baran P. S., Proc. Natl. Acad. Sci. USA 2011, 108, 14411; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Langlois B. R. in Modern Synthesis Processes and Reactivity of Fluorinated Compounds (Eds.: H. Groult, F. R. Leroux, A. Tressaud), Elsevier, Amsterdam, 2017, chap. 5, pp. 125–140; [Google Scholar]
- 8f. Fujiwara Y., Dixon J. A., Rodriguez R. A., Baxter R. D., Dixon D. D., Collins M. R., Blackmond D. G., Baran P. S., J. Am. Chem. Soc. 2012, 134, 1494; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8g. Sakamoto A., Kashiwagi H., Maruoka K., Org. Lett. 2017, 19, 5126; [DOI] [PubMed] [Google Scholar]
- 8h. Tung T. T., Christensen S. B., Nielsen J., Chem. Eur. J. 2017, 23, 18125; [DOI] [PubMed] [Google Scholar]
- 8i. Zhu S., Liu Y., Li H., Xu X., Qing F., J. Am. Chem. Soc. 2018, 140, 11613. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Rong J., Deng L., Tan P., Ni C., Gu Y., Hu J., Angew. Chem. Int. Ed. 2016, 55, 2743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2793; [Google Scholar]
- 9b. Fu W., Han X., Zhu M., Xu C., Wang Z., Ji B., Hao X., Song M., Chem. Commun. 2016, 52, 13413. [DOI] [PubMed] [Google Scholar]
- 10. Dobson A. T., Little B. B., Scottie L. L., Am. J. Obstet. Gynecol. 1998, 179, 527. [DOI] [PubMed] [Google Scholar]
- 11. Nagib D. A., MacMillan D. W. C., Nature 2011, 480, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fenton C., Keating M. K., Lyseng-Williamson K. A., Drugs 2006, 66, 477. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Klitgaard H., Verdru P., Expert Opin. Drug Discovery 2007, 2, 1537; [DOI] [PubMed] [Google Scholar]
- 13b. Gillard M., Fuks B., Leclercq K., Matagne A., Eur. J. Pharmacol. 2011, 664, 36. [DOI] [PubMed] [Google Scholar]
- 14. Nabulsi N. B., Mercier J., Holden D., Carré S., Najafzadeh S., Vandergeten M.-C., Lin S., Deo A., Price N., Wood M., Lara-Jaime T., Montel F., Laruelle M., Carson R. E., Hannestad J., Huang Y., J. Nucl. Med. 2016, 57, 777. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Mercier J., Provins L., Valade A., Drug Discovery Today Technol. 2017, 25, 45; [DOI] [PubMed] [Google Scholar]
- 15b. Warnier C., Lemaire C., Becker G., Zaragoza G., Giacomelli F., Aerts J., Otabashi M., Bahri M. A., Mercier J., Plenevaux A., Luxen A., J. Med. Chem. 2016, 59, 8955. [DOI] [PubMed] [Google Scholar]
- 16.During the development of our methodology, another 18F-labeled SV2A radiotracer with affinity comparable to that of UCB-H was disclosed by two different groups. In both cases, the tracer was labeled with fluorine-18 through aromatic nucleophilic substitution and required the preparation of a pre-functionalized precursors.
- 16a. Constantinescu C. C., Tresse C., Zheng M., Gouasmat A., Caroll V. M., Mistico L., Alagille D., Sandiego C. M., Papin C., Marek K., Seibyl J. P., Tamagnan G. D., Barret O., Mol. Imaging Biol. 2019, 10.1007/s11307-018-1260-5, [DOI] [PubMed] [Google Scholar]
- 16b. Li S., Cai Z., Wu X., Holden D., Pracitto R., Kapinos M., Gao H., Labaree D., Nabulsi N., Carson R. E., Huang Y., ACS Chem. Neurosci. 2018, 10.1021/acschemneuro.8b00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For recent publications on fluorine-18 photochemistry, see:
- 17a. Nodwell M. B., Yang H., Colovic M., Yuan Z., Merkens H., Martin R. E., Bénard F., Schaffer P., Britton R., J. Am. Chem. Soc. 2017, 139, 3595; [DOI] [PubMed] [Google Scholar]
- 17b. Yuan Z., Nodwell M. B., Yang H., Malik N., Merkens H., Bénard F., Martin R. E., Schaffer P., Britton R., Angew. Chem. Int. Ed. 2018, 57, 12733; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12915; [Google Scholar]
- 17c. Chen W., Huang Z., Tay N. E. S., Giglio B., Wang M., Wang H., Wu Z., Nicewicz D. A., Li Z., Science 2019, 364, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary