

Abstract
Objective
Inflamed tissue is characterized by low availability of oxygen and nutrients. Yet CD4+ T helper lymphocytes persist over time in such tissue and probably contribute to the chronicity of inflammation. This study was undertaken to analyze the metabolic adaptation of these cells to the inflamed environment.
Methods
Synovial and blood CD4+ T cells isolated ex vivo from patients with juvenile idiopathic arthritis (JIA) and murine CD4+ T cells were either stimulated once or stimulated repeatedly. Their dependency on particular metabolic pathways for survival was then analyzed using pharmacologic inhibitors. The role of the transcription factor Twist 1 was investigated by determining lactate production and oxygen consumption in Twist1‐sufficient and Twist1‐deficient murine T cells. The dependency of these murine cells on particular metabolic pathways was analyzed using pharmacologic inhibitors.
Results
Programmed death 1 (PD‐1)+ T helper cells in synovial fluid samples from patients with JIA survived via fatty acid oxidation (mean ± SEM survival of 3.4 ± 2.85% in the presence of etomoxir versus 60 ± 7.08% in the absence of etomoxir on day 4 of culture) (P < 0.0002; n = 6) and expressed the E‐box–binding transcription factor TWIST1 (2–14‐fold increased expression) (P = 0.0156 versus PD‐1− T helper cells; n = 6). Repeatedly restimulated murine T helper cells, which expressed Twist1 as well, needed Twist1 to survive via fatty acid oxidation. In addition, Twist1 protected the cells against reactive oxygen species.
Conclusion
Our findings indicate that TWIST1 is a master regulator of metabolic adaptation of T helper cells to chronic inflammation and a target for their selective therapeutic elimination.
Introduction
CD4+ T lymphocytes are considered a driving force and relevant therapeutic target in chronic inflammatory rheumatic diseases. In the inflamed synovial tissue of patients, CD4+ T lymphocytes persist despite low levels of oxygen and nutrients, and they are refractory to conventional immunosuppressive therapies 1, 2. With respect to nutrients qualifying as a metabolic energy source, there is little glucose 3 and glutamine 4 in inflamed tissue, while fatty acids are readily available 5, 6. Herein, we describe the metabolic adaptation of CD4+ T lymphocytes to this inflamed environment.
Among the CD4+ T lymphocytes present in inflamed tissues, CD4+ T cells expressing programmed death 1 (PD‐1) protein 7 are a subpopulation of potential relevance for pathogenesis 8. In this study, we show that PD‐1+ Th1 cells isolated from the synovial fluid of patients with juvenile idiopathic arthritis (JIA) are dependent on fatty acid oxidation for survival. Their survival is blocked by the carnitine palmitoyltransferase 1 inhibitor etomoxir 9, which inhibits the transport of fatty acids from the cytoplasm into the mitochondria. We show that CD4+ PD‐1+ T cells in inflamed synovia express the E‐box–binding transcription factor TWIST1, a hallmark of T lymphocytes persisting in chronically inflamed human tissue 2.
Twist1 expression is selective for repeatedly activated murine Th1 cells, as compared to other types of T helper cells and Th1 cells activated only once. Twist1 expression by murine Th1 cells has been shown to dampen immunopathology in an autoregulatory, cell‐intrinsic manner 2. At the same time, Twist1 supports the persistence of repeatedly activated Th1 cells by inducing expression of microRNA‐148a (miR‐148a), which in turn regulates expression of the proapoptotic protein Bim 10. We previously demonstrated that selective depletion of Twist1‐expressing Th1 cells through blockade of miR‐148a, a Twist1‐induced miRNA, in vivo with antagomirs ameliorates inflammation, identifying Twist1‐expressing T helper cells as those driving inflammation 11. Conditional inactivation of Twist1 in repeatedly activated Th1 lymphocytes relieves their dependency on fatty acid oxidation and allows them to survive alternatively on glycolysis, demonstrating that Twist1 forces T helper cells into fatty acid oxidation, and thus regulates their proinflammatory activity 12, 13. Twist1 thus qualifies as an essential regulator of the metabolism of T helper lymphocytes in chronic inflammation, inhibiting glycolysis, and thus limiting immunopathology, while at the same time stimulating fatty acid oxidation, allowing the cells to persist in and contribute to the chronification of inflammation.
Materials and methods
Mice
C57BL/6J mice were purchased from Charles River. OT‐II × Twist1fl/fl × CD4Cre+/− and OT‐II × Twist1wt/wt × CD4Cre+/−, Twist1fl/fl × CD4Cre+/−, and Twist1wt/wt × CD4Cre+/− mice were bred in the Deutsches Rheuma‐Forschungszentrum animal facility under specific pathogen–free conditions in individually ventilated cages. Mice were handled in accordance with good animal practice as defined by the German animal welfare bodies, and killed by cervical dislocation. All experiments were approved by the State Office for Health and Social Affairs (Berlin, Germany).
Human patient samples
Peripheral blood and synovial fluid samples were collected at the Department of Pediatrics, Pediatric Rheumatology Section of Charité–Universitätsmedizin Berlin as approved by the ethics committee of Charité–Universitätsmedizin Berlin (approval no. EA2/069/15).
Human T cell phenotyping and cultivation
Mononuclear cells from peripheral blood were isolated by Ficoll density‐gradient centrifugation. Synovial fluid cells were depleted of CD14+ granulocytes by magnetic cell sorting using CD14 microbeads (Miltenyi Biotec). CD4+ T helper lymphocytes were isolated using CD4 microbeads (Miltenyi Biotec). For analysis of cytokine expression, synovial CD4+ T cells were stimulated with phorbol 12‐myristate 13‐acetate (PMA) (10 ng/ml) and ionomycin (1 μg/ml) (both from Sigma‐Aldrich) for a total of 5–6 hours in medium. After 1 hour, 5 μg/ml brefeldin A (BioLegend) was added to block the secretion of cytokines. Cells were fixed with Cytofix/Cytoperm (BD Biosciences) for 20 minutes at 4°C and stained intracellularly with anti–interferon‐γ (anti‐IFNγ) (4SB3; BioLegend), anti–interleukin‐17a (anti–IL‐17a) (BL168; BioLegend), anti–tumor necrosis factor (anti‐TNF) (cA2; Miltenyi Biotec), anti–IL‐2 (MQ1‐17H12; BioLegend), anti–IL‐10 (JES3‐9D7; Miltenyi Biotec), anti–IL‐4 (8D48; BioLegend), and anti–IL‐21 (7H20‐I19‐M3; BioLegend) according to published guidelines 14. For intracellular T‐bet staining (4B10; BioLegend), cells were additionally permeabilized with 0.01% Triton X‐100 for 10 minutes on ice. PD‐1+ and PD‐1− CD3+CD4+CD14−CD45RO+ T lymphocytes were sorted by fluorescence‐activated cell sorting (FACS) using a FACSAria cell sorter (BD Biosciences) and plated in RPMI medium containing human AB serum (Sigma), 100 units/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Etomoxir was added to the final concentrations as indicated. The numbers of viable CD4+ T cells were monitored over time by flow cytometry, using DAPI to exclude dead cells.
In vitro cultivation and differentiation of murine T helper cells
Murine CD4+CD62L+ (naive) T helper cells were isolated as previously described 15 and cultured at a concentration of 2.5 × 106 cells/5 ml in 6‐well plates in RPMI medium supplemented with 10% fetal calf serum (Merck), 300 μg/ml glutamine (Invitrogen), 100 units/ml penicillin, 100 μg/ml streptomycin (Life Technologies), and 50 μM β‐mercaptoethanol (Sigma‐Aldrich) at 37°C in 5% CO2 and 4.2% oxygen. The T helper cells were stimulated polyclonally with plate‐bound anti‐CD3 antibody (145‐2C11; 3 μg/ml) and soluble anti‐CD28 antibody (37.51; 1.5 μg/ml). For Th1 polarization, 5 ng/ml recombinant IL‐12, 10 ng/ml recombinant IL‐2, and 10 μg/ml anti–IL‐4 antibody (11B11) was added. To induce Th17 polarization, 10 μg/ml anti‐IFNγ (AN18.17.24), 10 μg/ml anti–IL‐4 (11B11), 20 ng/ml recombinant IL‐6, 20 ng/ml recombinant IL‐23, and 1 ng/ml recombinant transforming growth factor β (all from R&D Systems) were added. After 48 hours of stimulation, the cells were removed from the antibody‐coated culture dishes and cultured for an additional 3–4 days. T cell receptor–transgenic OT‐II lymphocytes were activated with 1 μg/ml of ovalbumin 327–339 peptide in the presence of irradiated (30 Gy) CD90‐depleted splenocytes from C57BL/6 mice. For repeated activation, viable T helper cells were isolated using Ficoll density‐gradient centrifugation and stimulated again under the original conditions.
Quantitative RNA expression analysis
Total RNA was isolated using an RNeasy kit (Qiagen) or a Direct‐zol RNA kit (Zymo Research) according to the manufacturer's instructions. RNA concentration and quality were determined by NanoDrop spectrometry. RNA (200 ng to 1 μg) was reverse transcribed using a TaqMan Reverse Transcription kit (ThermoFisher Scientific) according to the manufacturer's recommendations. Real‐time quantitative polymerase chain reaction was used to quantify the messenger RNA of interest with the following primer sets (TIB Molbiol Berlin): for murine hypoxanthine guanine phosphoribosyltransferase (Hprt), forward 5′‐TCCTCCTCAGACCGCTTTT‐3′ and reverse 5′‐CATAACCTGGTTCATCATCGC‐3′; for human HPRT, forward 5′‐ACCCTTTCCAAATCCTCAGC‐3′ and reverse 5′‐GTTATGGCGACCCGCAG‐3′; for murine Twist1, forward 5′‐CGCACGCAGTCGCTGAACG‐3′ and reverse 5′‐GACGCGGACATGGACCAGG‐3′; for human TWIST1, forward 5′‐GGCACCCAGTCGCTGAACG‐3′ and reverse 5′‐GACGCGGACATGGACCAGG‐3′; for murine Pdcd1, forward 5′‐CGTCCCTCAGTCAAGAGGAG‐3′ and reverse 5′‐GTCCCTAGAAGTGCCCAACA‐3′. Prior to Twist1 analysis, cultured cells were restimulated with PMA (10 ng/ml) and ionomycin (1 μg/ml) for 5 hours.
Glucose uptake assay
Murine naive CD4+ T helper cells were stimulated with 3 μg/ml of plate‐bound anti‐CD3 and 1.5 μg/ml of soluble anti‐CD28 antibodies at 3 × 105 cells per well under Th1‐inducing conditions for 5 hours. Medium was then aspirated, and 100 μl of glucose‐free medium with 1.5 μg/ml of soluble anti‐CD28 antibody was added to the cells. Following incubation for 60 minutes, 100 μl of glucose‐free RPMI with 300 μM 2‐([7‐nitro‐2,1,3‐benzoxadiazol‐4‐yl]amino)‐2 deoxyglucose (2‐NBDG) was added to the well, to reach a final concentration of 150 μM 2‐NBDG. Cultures with no 2‐NBDG added and cultures with 150 μM 2‐NBDG and 30 μM cytochalasin B were used as controls. Cells were incubated for 30 minutes at 37°C. After incubation, cells were washed twice in cold phosphate buffered saline (PBS) and maintained at 4°C. Uptake of 2‐NBDG was determined by flow cytometry using a MACSQuant analyzer. Dead cells were excluded by propidium iodide staining.
Determination of lactate production and oxygen consumption
Glycolysis, as determined by extracellular acidification, and oxidative phosphorylation, as determined by oxygen consumption, were measured using a Seahorse XP analyzer (Agilent). Prior to the assay, T lymphocytes were starved in glucose‐free RPMI assay medium and equilibrated in 5% CO2 at 37°C, under normoxic conditions.
T cell survival assay
CD4+ T helper cells were seeded in a 96‐well plate coated with 3 μg/ml anti‐CD3 antibodies and 1.5 μg/ml soluble anti‐CD28 antibodies under Th1‐ or Th17‐polarizing conditions. The inhibitors 2‐deoxy‐d‐glucose (2‐DG; 2 mM), 6‐diazo‐5‐oxo‐l‐norleucine (50 μM), oligomycin (2 μM), or etomoxir (150 μM) were added. Dead cells were excluded by staining with 100 ng/ml propidium iodide or 100 ng/ml DAPI. Numbers of viable cells were determined by flow cytometry using a MACSQuant analyzer.
Lipid peroxidation
BODIPY 581/591 C11 was added to CD4+ T lymphocytes in a 96‐well plate at a final concentration of 3 μM in RPMI without serum and 2‐mercaptoethanol. Cells were incubated for 40 minutes at 37°C and then washed with PBS/bovine serum albumin. Viable cells were quantified using a MACSQuant analyzer, and dead cells were excluded by DAPI staining.
Results
Survival of PD‐1+CD4+ T helper lymphocytes in inflamed synovia via fatty acid oxidation
CD4+ T helper lymphocytes were isolated from the synovial fluid of patients with JIA. More than 70% of the CD3+CD4+ cells were CD45RO+CD45RA− (Figure 1A). Upon stimulation with PMA and ionomycin ex vivo, ~50% of them expressed IFNγ, 70% expressed TNF, 50% expressed IL‐2, and 10% expressed IL‐10. IL‐4, IL‐17, and IL‐21 were each expressed by <10% of the cells (Figure 1B). Consistent with the cytokine expression pattern, almost all JIA synovial T helper cells expressed the T‐box–binding transcription factor T‐bet, identifying them as bona fide Th1 cells (Figure 1C). A significantly higher proportion of synovial T helper cells than peripheral blood T helper cells expressed PD‐1 (Figures 1D and E).
Figure 1.
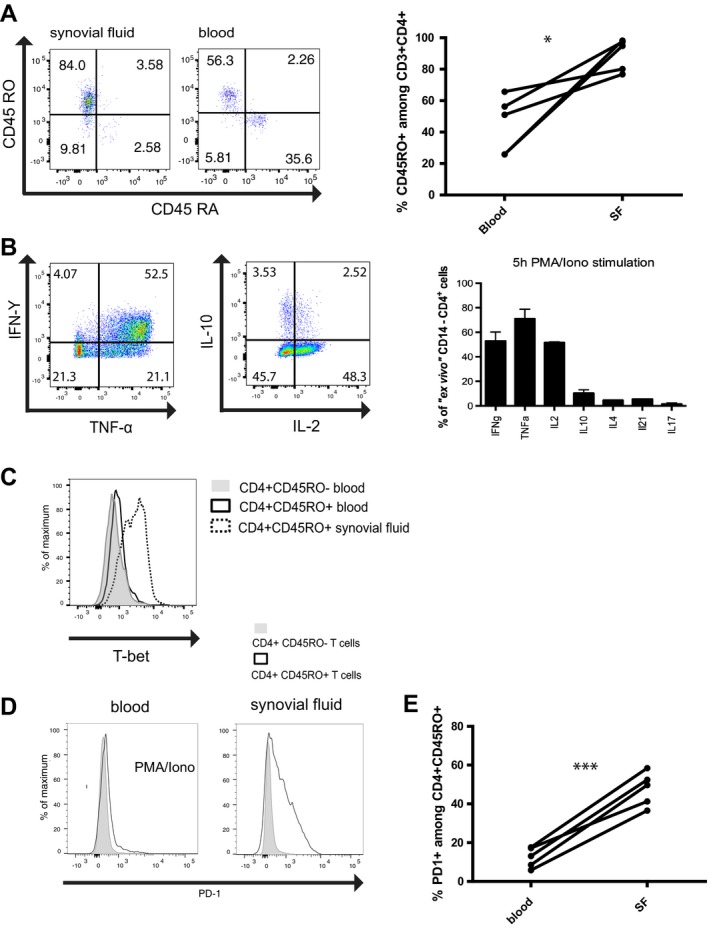
Synovial fluid (SF) T cells from patients with juvenile idiopathic arthritis (JIA) have a Th1 phenotype and express programmed death 1 (PD‐1). A, Left, Flow cytometric analysis indicating the frequencies of CD45RA+ and CD45RO+ cells among CD3+CD4+ T cells in synovial fluid and blood from patients with JIA. Results are representative of 5 experiments. Right, Percentage of CD3+CD4+ cells expressing CD45RO in blood and synovial fluid from patients with JIA (n = 5). * = P < 0.05 by 2‐tailed t‐test. B, Left, Expression of interferon‐γ (IFNγ), tumor necrosis factor (TNF), interleukin‐10 (IL‐10), and IL‐2 in ex vivo‐isolated synovial T cells after stimulation with phorbol 12‐myristate 13‐acetate (PMA) and ionomycin (iono), as analyzed by intracellular cytokine staining. Results are representative of 5 experiments. Right, Frequencies of synovial fluid CD4+ T cells expressing IFNγ, TNF, IL‐2, IL‐10, IL‐4, IL‐21, and IL‐17A after 5 hours of restimulation with PMA and ionomycin. Bars show the mean ± SEM (n = 5). C, T‐bet expression in CD4+CD45RO− and CD4+CD45RO+ T cells in blood and CD4+CD45RO+ T cells in synovial fluid from a patient with JIA. Results are representative of 3 experiments. D, PD‐1 expression in CD4+CD45RO− and CD4+CD45RO+ T cells isolated from blood stimulated with PMA and ionomycin and synovial fluid from a patient with JIA. Results are representative of 5 experiments. E, Percentage of CD4+CD45RO+ cells expressing PD‐1 in blood and synovial fluid from patients with JIA (n = 5). *** = P < 0.001 by 2‐tailed t‐test. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40939/abstract.
PD‐1+ and PD‐1− synovial T helper cells were separated by FACS (Figure 2A and Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40939/abstract), and their dependency on fatty acid oxidation was analyzed. CD4+CD45RO+ T cells isolated from the blood of healthy donors did not survive when cultured in vitro under hypoxic conditions (4.2% O2), with >90% of them dying within 2 days. Of the PD‐1− T helper cells isolated from synovial fluid, a mean ± SEM of 24.6 ± 2.29% survived until day 2 of culture and 19 ± 1.31% survived until day 4. Their survival was not affected by the addition of etomoxir, an inhibitor of fatty acid oxidation. In the presence of etomoxir, 20% of the cells survived until day 2 and 15.6 ± 4.5% survived until day 4 (Figure 2B). Of the PD‐1+ synovial T helper cells, 39.85 ± 7.35% survived until day 2, and these surviving cells expanded again to 60 ± 7.08% of the original number on day 4. Survival and expansion of these cells was entirely dependent on fatty acid oxidation, since they could be blocked with etomoxir, reducing cell numbers to a mean ± SEM of 3.4 ± 2.85% on day 4 (Figure 2B).
Figure 2.
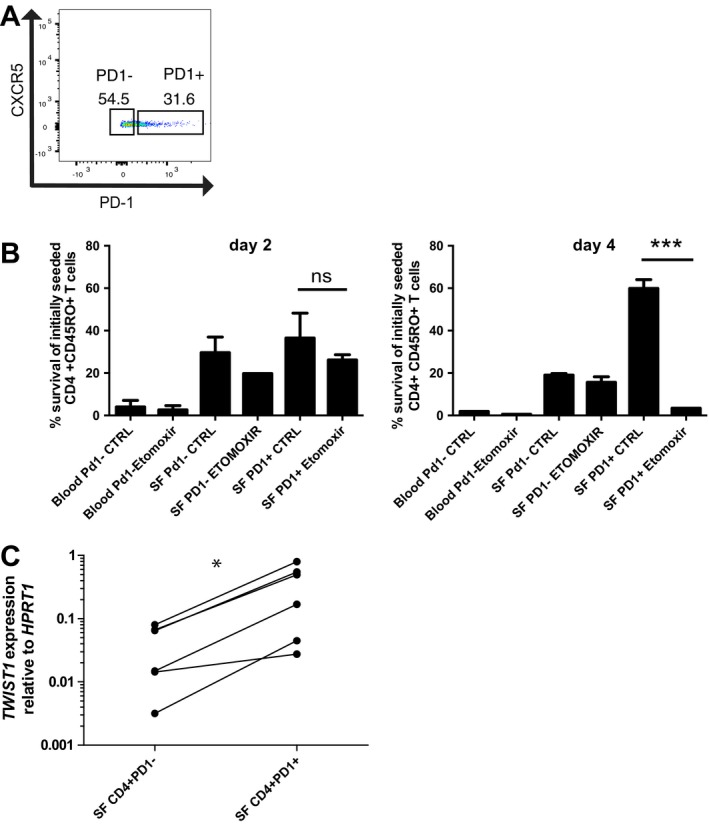
Survival of synovial CD4+CD45RO+PD‐1+ T cells in patients with JIA is dependent on fatty acid oxidation. A, Representative gating strategy for sorting PD‐1+ and PD‐1− CD4+CD45RO+ T cells from synovial fluid and blood. B, Frequency of viable cells relative to the number of initially seeded cells determined on day 2 and day 4 after stimulation with anti‐CD3 and anti‐CD28 antibodies in the presence or absence (control) of 200 μM etomoxir. Bars show the mean ± SEM. Data were pooled from 2 experiments (n = 3 blood and synovial fluid samples per experiment). *** = P < 0.0002 by unpaired 2‐tailed t‐test. NS = not significant. C, TWIST1 mRNA expression relative to HPRT in PD‐1+ and PD‐1− CD4+CD45RO+ T cells directly isolated from synovial fluid from patients with JIA (n = 6). * = P = 0.016 by Wilcoxon's 1‐tailed paired rank test. See Figure 1 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.40939/abstract.
We have previously shown that the E‐box–binding transcription factor TWIST1 is a hallmark of T helper lymphocytes isolated from the inflamed joints of patients with rheumatic diseases 2. In this study, we found that PD‐1+ synovial T cells isolated from the inflamed joints of patients with JIA expressed significantly higher levels of TWIST1 than PD‐1− T helper cells isolated from the same synovia (Figure 2C).
Upon repeated activation, murine Th1 cells shift from glycolysis to fatty acid oxidation
To investigate the role of Twist1 in the regulation of the metabolism of T helper lymphocytes, we used a murine model of repeatedly activated Th1 lymphocytes, which was previously used as a model to study transcriptional adaptations of chronic activation and in which we had previously demonstrated up‐regulated Twist1 expression 2. Naive CD4+CD62L+ T cells were stimulated with anti‐CD3 and anti‐CD28 antibodies 4 times at 6‐day intervals. We then compared the capacity of these cells to take up glucose and perform glycolysis, the latter reflected by the production of lactate, as outlined in Figure 3A. Uptake of the fluorescent glucose analog 2‐NBDG was monitored by flow cytometry. Repeatedly activated mouse Th1 cells took up 2.3‐fold less 2‐NBDG than mouse Th1 cells that were activated one time (Figure 3B).
Figure 3.
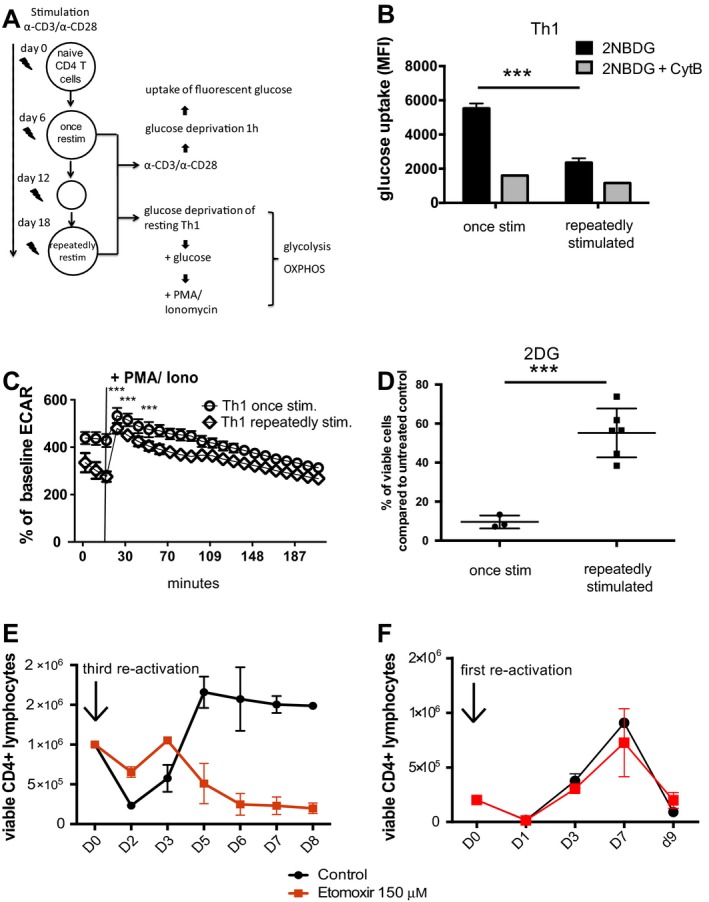
Repeatedly stimulated (stim) murine Th1 cells switch their metabolism from glycolysis to fatty acid oxidation. A, Experimental design. Naive CD4+CD62L+ T cells isolated from C57BL/6 mice were stimulated once or stimulated 2 additional times at 6‐day intervals with anti‐CD3/anti‐CD28 antibodies under Th1‐differentiating conditions. Cells were analyzed for energy metabolism. OXPHOS = oxidative phosphorylation. B, Glucose uptake in murine Th1 cells stimulated once and repeatedly stimulated murine Th1 cells, measured by flow cytometry using the fluorescent glucose analog 2‐([7‐nitro‐2,1,3‐benzoxadiazol‐4‐yl]amino)‐2 deoxyglucose (2‐NBDG). Th1 cells were restimulated for 6 hours with anti‐CD3 and anti‐CD28 antibodies prior to analysis. Cytochalasin B (CytB) was used to determine background fluorescence. Bars show the mean ± SEM. Results are representative of 3 independent experiments. *** = P < 0.0006 by Mann‐Whitney 2‐tailed test. C, Glycolytic activity, measured by extracellular acidification rate (ECAR), in murine Th1 cells stimulated once and repeatedly stimulated murine Th1 cells before and after restimulation with phorbol 12‐myristate 13‐acetate (PMA) and ionomycin (iono). Values are the mean ± SEM (n = 4 samples per group). *** = P < 0.0001 at 14 minutes, P = 0.0027 at 30 minutes, and P = 0.0027 at 50 minutes after glucose addition, by 2‐tailed t‐test. D, Frequency of viable murine CD4+ T cells stimulated once and murine CD4+ T cells stimulated repeatedly after restimulation with anti‐CD3/anti‐CD28 antibodies in the presence of the glycolysis inhibitor 2‐deoxy‐d‐glucose (2‐DG; 2 mM) for 72 hours, relative to cells cultivated without inhibitor (untreated control). Symbols represent individual samples; horizontal lines and error bars show the mean ± SEM. *** = P = 0.0005 by t‐test. E and F, Absolute number of repeatedly stimulated murine Th1 cells (E) and murine Th1 cells stimulated once (F) and restimulated with anti‐CD3/anti‐CD28 antibodies in the presence or absence of 150 μM etomoxir. Viability and cell count were determined by propidium iodide exclusion using flow cytometry. Values are the mean ± SEM. Results are representative of 3 experiments. MFI = mean fluorescence intensity.
Repeatedly activated murine Th1 cells were also less efficient in glycolysis, compared to Th1 cells activated once. Murine Th1 cells activated once produced 1.4‐fold more lactate than repeatedly activated murine Th1 cells in the presence of glucose, before and after stimulation with PMA and ionomycin (Figure 3C). Lactate production remained significantly lower in repeatedly activated murine Th1 cells thereafter. Of the Th1 cells that had been activated once, inhibition of glycolysis with 2‐DG killed 85% within 72 hours after activation, suggesting that such Th1 cells were dependent on glycolysis. In contrast, 50% of the repeatedly stimulated murine Th1 cells survived when glycolysis was inhibited by 2‐DG (Figure 3D), demonstrating that repeatedly stimulated T helper cells have alternative metabolic options, i.e., fatty acid oxidation. Blocking fatty acid oxidation with etomoxir for up to 8 days after a third reactivation showed that repeatedly stimulated murine Th1 cells use and are dependent on fatty acid oxidation, in particular in the late phase of expansion, between days 3 and 5 (Figure 3E). This was not the case for murine Th1 cells during the first week after the first reactivation (Figure 3F), which survived and proliferated even in the presence of etomoxir. The metabolism of repeatedly restimulated murine Th1 cells thus corresponds to that of PD‐1high human T helper cells isolated from inflamed synovia, in that they are dependent on fatty acid oxidation.
Inhibition of glycolysis of Th1 lymphocytes by Twist1
To analyze the role of Twist1 in regulating the metabolism of T helper cells functionally, we used mice with a conditional, cell type–specific knockout of Twist1 (CD4Cre × Twist1 fl/fl) 10 (Supplementary Figure 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40939/abstract). Both the Twist1‐deficient mouse Th1 cells that had been stimulated once and those that had been stimulated repeatedly showed an increased rate of glycolysis, as reflected by an increased production of lactate, when compared to their wild‐type counterparts (Figures 4A and B), indicating that Twist1 inhibits glycolysis. Twist1 selectively regulated the glycolysis of Th1, but not of repeatedly stimulated Th17 cells (Supplementary Figure 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40939/abstract).
Figure 4.
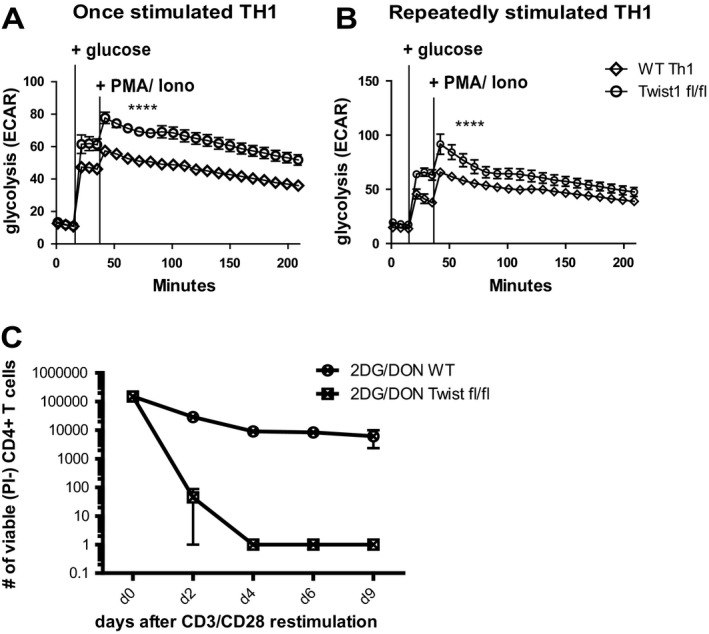
Twist 1 inhibits glycolysis in murine Th1 cells stimulated once and murine Th1 cells stimulated repeatedly. A and B, Glycolytic activity, measured by extracellular acidification rate (ECAR) of Twist1‐deficient (Twist1fl/fl) and Twist1‐sufficient (wild‐type [WT]) OT‐II TCR‐transgenic mouse Th1 cells stimulated once (A) or stimulated repeatedly (B) with ovalbumin peptide before and after the addition of glucose and restimulation with 12‐myristate 13‐acetate (PMA) and ionomycin (iono). **** = P < 0.0001 at all time points following glucose addition in A and at all time points from glucose addition to 120 minutes in B, by paired t‐test. C, Absolute number of viable repeatedly stimulated Twist1‐deficient and Twist1‐sufficient mouse Th1 cells treated with inhibitors of glycolysis (2‐deoxy‐d‐glucose [2‐DG]) and glutaminolysis (6‐diazo‐5‐oxo‐L‐norleucine [DON]). Viable cells were determined every 2 days by propidium iodide (PI) exclusion using flow cytometry. Values are the mean ± SEM. Results are representative of 2 experiments.
Requirement of Twist1 for the survival of Th1 lymphocytes via fatty acid oxidation
Twist1 is indispensable for the survival of repeatedly stimulated Th1 cells via fatty acid oxidation. When glycolysis and glutaminolysis were blocked by 2‐DG and 6‐diazo‐5‐oxo‐L‐norleucine, respectively, Twist1‐deficient mouse T helper cells did not survive (Figure 4C). In this situation, when fatty acid oxidation is the only metabolic pathway available, the competence to survive on this pathway depends on Twist1.
Twist1 protects Th1 cells against reactive oxygen species (ROS)
Consistent with fatty acid oxidation being the major energy source for repeatedly stimulated murine Th1 cells, these cells show a higher degree of oxidative phosphorylation than murine Th1 cells stimulated once, both in the resting phase and after reactivation ex vivo (Figure 5A). Oxidative phosphorylation is associated with the generation of ROS. ROS promote peroxidation of lipids in cell membranes, an effect that has been suggested to be involved in the pathogenesis of rheumatoid arthritis 16, 17. In T helper lymphocytes, lipid peroxides of the membrane, as detected by BODIPY C11 staining, were more abundant in T helper cells that had been stimulated 3 times, 8 days after reactivation, compared to T helper cells that had been stimulated once (Figure 5B). Compared to Twist1‐deficient mouse Th1 cells, Twist1‐sufficient (wild‐type) mouse Th1 cells, both those stimulated once and those stimulated repeatedly, showed significantly reduced lipid peroxidation (P = 0.002 for cells stimulated once and P = 0.0323 for cells stimulated repeatedly) (Figure 5B). These data suggest that in addition to promoting oxidative phosphorylation through fatty acid oxidation, Twist1 protects murine Th1 cells against harmful lipid peroxides, presumably by enhancing lipid peroxide scavenging.
Figure 5.
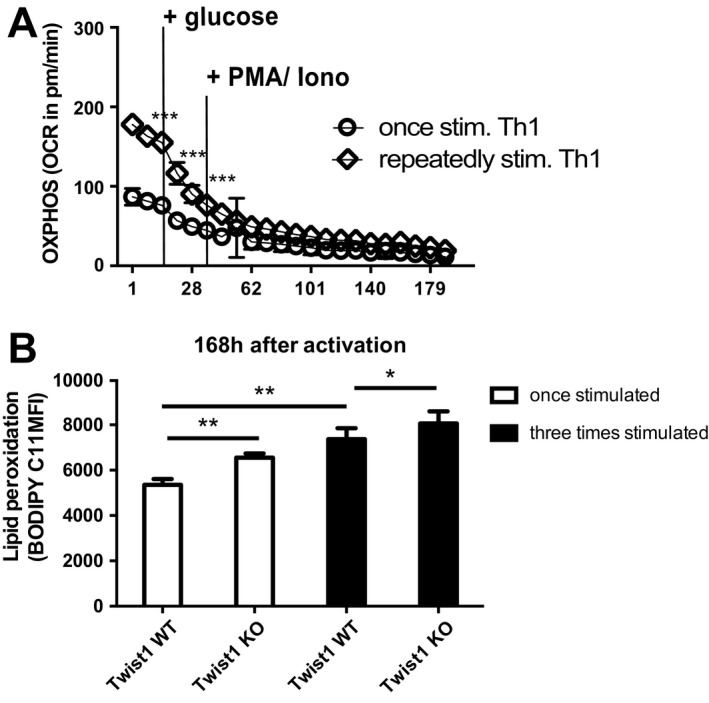
Repeatedly stimulated Th1 cells have increased oxidative phosphorylation. A, Oxidative phosphorylation (OXPHOS), measured by oxygen consumption rate (OCR) in Th1 cells stimulated once and Th1 cells stimulated repeatedly, before and after addition of glucose and restimulation with 12‐myristate 13‐acetate (PMA) and ionomycin (iono). Values are the mean ± SEM. *** = P < 0.0001 at 15 minutes; P = 0.0009 at 28 minutes; and P = 0.64 at 52 minutes, by paired t‐test. Results are representative of 3 independent experiments. B, Levels of lipid peroxides in wild‐type (WT) and Twist1‐deficient (knockout [KO]) mouse Th1 cells stimulated once or stimulated repeatedly. Levels were determined by BODIPY 581/591 C11 (2 μM) staining 7 days after the last reactivation with anti‐CD3/anti‐CD28 antibodies. Bars show the mean ± SEM. * = P = 0.0323; ** = P = 0.002, by paired 2‐tailed t‐test. MFI = mean fluorescence intensity.
Discussion
Acute, protective immune reactions are characterized by the apparent elimination of the antigen triggering them. The lymphocytes involved are either eliminated as well, or they develop into memory lymphocytes, resting in terms of activation and proliferation 18, 19, 20. Persistent antigens, whether they be pathogens, tumor antigens, or autoantigens, are a challenge to the immune system. Lymphocytes involved in chronic immune responses have to adapt to chronic inflammation and in doing so may become refractory to physiologic and conventional immunosuppression, resulting in chronic inflammatory diseases. CD4+ T helper lymphocytes in chronic inflammation express the E‐box–binding transcription factor TWIST1, which limits their ability to induce immunopathology 2 and promotes their resistance to apoptosis 10. In this study, we demonstrated that T helper lymphocytes in chronic inflammation also adapt their metabolism to become entirely dependent on fatty acid oxidation. These findings are consistent with the findings of 13C‐glucose tracing studies showing that chronically activated splenocytes in mice with lupus, and also chronically activated human T cells, have reduced lactate production, i.e., down‐regulate their glycolysis rate 21.
Reasoning that persistent antigen would lead to repeated restimulation of the T lymphocytes recognizing it, we previously compared the transcriptomes of murine T helper lymphocytes activated once with those activated 3 or 4 times. We identified transcription factors like Hopx 22 and Twist 1 2 and miRNAs like miR‐182 and miR‐148a 10, 23 as selectively expressed in repeatedly activated T helper cells. Of particular interest is the E‐box–binding transcription factor Twist1, originally identified as an anticachectic gene, with a strong dose dependency 24, since mice that are haploinsufficient for Twist1 and its isologue Twist2 are already prone to die young of cachexia 24. Expression of Twist1 is induced in activated T helper lymphocytes by STAT4 signaling, and thus a characteristic of Th1 lymphocytes, and its expression increases upon subsequent reactivations 2. The comparison of Twist1‐deficient and ‐sufficient, repeatedly activated T helper cells shows that Twist1 controls the ability of the cells to survive on fatty acid oxidation.
We previously showed that up‐regulation of the expression of Twist1, upon restimulation ex vivo, is a hallmark of CD4+ T lymphocytes isolated from the inflamed joints of patients with inflammatory rheumatic diseases, or from the inflamed intestinal mucosa of patients with inflammatory bowel diseases 2. Twist1 expression apparently is a hallmark of pathogenic CD4+ T cells, as selective targeting of such cells via the Twist1‐induced miR‐148a ameliorates inflammation without affecting memory CD4+ T cells induced by vaccination 11. In this study we confirm the observation that TWIST1 is highly expressed in CD4+ T lymphocytes isolated from the synovia of patients with JIA. We compared PD‐1high and PD‐1low synovial T helper cells, since PD‐1 expression by these cells has been invoked as a correlate of their involvement in chronic inflammation 7, 25, 26, 27, 28. PD‐1high cells from the synovial fluid of patients with JIA indeed showed a significant up‐regulation of expression of TWIST1, directly ex vivo. Like the repeatedly activated murine Th1 cells, the PD‐1high synovial human T helper cells were dependent on fatty acid oxidation for their survival, as evidenced by the fact that they could be killed by etomoxir, an inhibitor of fatty acid oxidation, unlike their PD‐1low synovial counterparts. This result points to an interesting therapeutic option to ablate PD‐1high CD4+ T lymphocytes selectively in chronic inflammatory diseases. Moreover, in preclinical models this would allow the determination of whether these cells indeed are the driving force of chronic inflammation, substantiating evidence that is thus far only correlative.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Drs. Chang and Radbruch had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Hradilkova, Chang, Radbruch.
Acquisition of data
Hradilkova, Maschmeyer, Westendorf, Schliemann, Husak, von Stuckrad, Kallinich, Minden.
Analysis and interpretation of data
Hradilkova, Durek, Grün, Chang, Radbruch.
Supporting information
Supplementary Figure Legends
Acknowledgments
We thank Toralf Kaiser, Jenny Kirsch, and Ana Teichmüller for support at the flow cytometry core facility, and Patrick Thiemann and Manuela Ohde for assistance with animal care.
Supported by the European Research Council Advanced Grant IMMEMO (project ERC‐2010‐AdG.20100317; grant 268987 to Dr. Radbruch), Deutsche Forschungsgemeinschaft (grant SFB 650 to Drs. Chang and Radbruch), the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement no. 777357, the Rheumastiftung (to Dr. Chang), and the Leibniz ScienceCampus Chronic Inflammation. Dr. Hradilkova's work was supported by the BSRT graduate school. Dr. Maschmeyer's work was supported by the state of Berlin and the European Regional Development Fund (ERDF 2014‐2020 and EFRE 1.8/11 to Deutsches Rheuma‐Forschungszentrum) and by EUTRAIN, a Seventh Framework Programme Marie Curie Initial Training Network for Early Stage Researchers funded by the European Union (FP7‐PEOPLE‐2011‐ITN‐289903).
Drs. Chang and Radbruch contributed equally to this work.
No potential conflicts of interest relevant to this article were reported.
References
- 1. Maciolek JA, Pasternak JA, Wilson HL. Metabolism of activated T lymphocytes. Curr Opin Immunol 2014;27:60–74. [DOI] [PubMed] [Google Scholar]
- 2. Niesner U, Albrecht I, Janke M, Doebis C, Loddenkemper C, Lexberg MH, et al. Autoregulation of Th1‐mediated inflammation by Twist1. J Exp Med 2008;205:1889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ciurtin C, Cojocaru VM, Miron IM, Preda F, Milicescu M, Bojincă M, et al. Correlation between different components of synovial fluid and pathogenesis of rheumatic diseases. Rom J Intern Med 2006;44:171–81. [PubMed] [Google Scholar]
- 4. Sido B, Seel C, Hochlehnert A, Breitkreutz R, Dröge W. Low intestinal glutamine level and low glutaminase activity in Crohn's disease: a rational for glutamine supplementation? Dig Dis Sci 2006;51:2170–9. [DOI] [PubMed] [Google Scholar]
- 5. Bole GG, Peltier DF. Synovial fluid lipids in normal individuals and patients with rheumatoid arthritis. Arthritis Rheum 1962;5:589–601. [DOI] [PubMed] [Google Scholar]
- 6. Leimer EM, Pappan KL, Nettles DL, Bell RD, Easley ME, Olson SA, et al. Lipid profile of human synovial fluid following intra‐articular ankle fracture. J Orthop Res 2017;35:657–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hatachi S, Iwai Y, Kawano S, Morinobu S, Kobayashi M, Koshiba M, et al. CD4+ PD‐1+ T cells accumulate as unique anergic cells in rheumatoid arthritis synovial fluid. J Rheumatol 2003;30:1410–9. [PubMed] [Google Scholar]
- 8. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017;542:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horn CC, Ji H, Friedman MI. Etomoxir, a fatty acid oxidation inhibitor, increases food intake and reduces hepatic energy status in rats. Physiol Behav 2004;81:157–62. [DOI] [PubMed] [Google Scholar]
- 10. Haftmann C, Stittrich AB, Zimmermann J, Fang Z, Hradilkova K, Bardua M, et al. MiR‐148a is upregulated by Twist1 and T‐bet and promotes Th1‐cell survival by regulating the proapoptotic gene Bim. Eur J Immunol 2015;45:1192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maschmeyer P, Petkau G, Siracusa F, Zimmermann J, Zügel F, Kühl AA, et al. Selective targeting of pro‐inflammatory Th1 cells by microRNA‐148a‐specific antagomirs in vivo. J Autoimmun 2018;89:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013;153:1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Millet P, Vachharajani V, McPhail L, Yoza B, McCall CE. GAPDH binding to TNF‐α mRNA contributes to posttranscriptional repression in monocytes: a novel mechanism of communication between inflammation and metabolism. J Immunol 2016;196:2541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cossarizza A, Chang HD, Radbruch A, Akdis M, Andrä I, Annunziato F, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur J Immunol 2017;47:1584–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lexberg MH, Taubner A, Förster A, Albrecht I, Richter A, Kamradt T, et al. Th memory for interleukin‐17 expression is stable in vivo. Eur J Immunol 2008;38:2654–64. [DOI] [PubMed] [Google Scholar]
- 16. Seven A, Güzel S, Aslan M, Hamuryudan V. Lipid, protein, DNA oxidation and antioxidant status in rheumatoid arthritis. Clin Biochem 2008;41:538–43. [DOI] [PubMed] [Google Scholar]
- 17. Dingjan I, Verboogen DR, Paardekooper LM, Revelo NH, Sittig SP, Visser LJ, et al. Lipid peroxidation causes endosomal antigen release for cross‐presentation. Sci Rep 2016;6:22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature 1997;388:133–4. [DOI] [PubMed] [Google Scholar]
- 19. Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, Grün JR, Löhning M, et al. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity 2009;30:721–30. [DOI] [PubMed] [Google Scholar]
- 20. Siracusa F, Alp ÖS, Maschmeyer P, McGrath M, Mashreghi MF, Hojyo S, et al. Maintenance of CD8+ memory T lymphocytes in the spleen but not in the bone marrow is dependent on proliferation. Eur J Immunol 2017;47:1900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus 2010;19:1492–501. [DOI] [PubMed] [Google Scholar]
- 22. Albrecht I, Niesner U, Janke M, Menning A, Loddenkemper C, Kühl AA, et al. Persistence of effector memory Th1 cells is regulated by Hopx. Eur J Immunol 2010;40:2993–3006. [DOI] [PubMed] [Google Scholar]
- 23. Stittrich AB, Haftmann C, Sgouroudis E, Kühl AA, Hegazy AN, Panse I, et al. The microRNA miR‐182 is induced by IL‐2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol 2010;11:1057–62. [DOI] [PubMed] [Google Scholar]
- 24. Šošić D, Richardson JA, Yu K, Ornitz DM, Olson EN. Twist regulates cytokine gene expression through a negative feedback loop that represses NF‐kB activity. Cell 2003;112:169–80. [DOI] [PubMed] [Google Scholar]
- 25. De Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross‐sectional study. Ann Rheum Dis 2007;66:589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murray KJ, Luyrink L, Grom AA, Passo MH, Emery H, Witte D, et al. Immunohistological characteristics of T cell infiltrates in different forms of childhood onset chronic arthritis. J Rheumatol 1996;23:2116–24. [PubMed] [Google Scholar]
- 27. Li S, Liao W, Chen M, Shan S, Song Y, Zhang S, et al. Expression of programmed death‐1 (PD‐1) on CD4+ and CD8+ T cells in rheumatoid arthritis. Inflammation 2014;37:116–21. [DOI] [PubMed] [Google Scholar]
- 28. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD‐1 alters T‐cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 2015;6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends