

Abstract
The direct reduction of arenes and heteroarenes by visible‐light irradiation remains challenging, as the energy of a single photon is not sufficient for breaking aromatic stabilization. Shown herein is that the energy accumulation of two visible‐light photons allows the dearomatization of arenes and heteroarenes. Mechanistic investigations confirm that the combination of energy‐transfer and electron‐transfer processes generates an arene radical anion, which is subsequently trapped by hydrogen‐atom transfer and finally protonated to form the dearomatized product. The photoreduction converts planar aromatic feedstock compounds into molecular skeletons that are of use in organic synthesis.
Keywords: dearomatization, electron transfer, energy transfer, photocatalysis, reduction
Splitting the work: Energy transfer (EnT) and photoinduced single‐electron transfer (SET) allow the reduction of arenes. Subsequent hydrogen‐atom transfer (HAT) yields the reduced dearomatized products.

Introduction
The reduction of arenes and heteroarenes is a valuable chemical transformation to access complex structures containing sp3‐hybridized carbon centers, structures that are typically present in bioactive molecules, natural products, flavours, and industrial materials.1, 2, 3, 4 The Birch reduction is the best‐known example of such transformations, whereby aromatic rings undergo a 1,4‐reduction that yields unconjugated cyclohexadienes. However, the reaction requires liquid ammonia as a solvent and pyrophoric alkali metals at cryogenic temperatures to generate solvated electrons.5 Mechanistically, a one‐electron reduction of the arene breaks the aromatic stabilization.6 The resulting radical anion is protonated, allowing a second reduction and protonation. Modifications of the Birch reduction employ several ammonia‐free variants that rely on single‐electron transfer (SET) reductants, namely alkali metal–silica gel adducts,7 alkali metal dispersions combined with crown ether additives,8 lithium di‐tertbutylbiphenyl (LiDBB)/bis(methoxyethyl)amine (BMEA) systems,9 inorganic electride [Ca2N]+⋅e−,10 and SmI2 complexes.11 Other arene reductions utilize catalytic amounts of transition metals at high hydrogen pressures.12 Recently, photocatalytic dearomatizing cycloadditions were reported, as well as an electrochemical Birch reduction.13, 14, 15
Photochemical arene reductions are well known, however, because of the absorption properties and high stabilities of arenes, UV light‐initiated reactions in the presence of super‐stoichiometric amounts of strong reducing agents are required.16, 17, 18, 19 Despite many applications of visible‐light photocatalysis in organic synthesis,20, 21 a direct reduction of aromatic compounds has not been achieved using visible‐light photoredox catalysis. One reason for this is that the energy of blue (455 nm) and green photons (530 nm) of 2.72 eV and 2.34 eV, respectively, are insufficient for most arenes to overcome the inherent aromatic stabilization that allows dearomatization. Alternatively, energy accumulation from more than one photon may open a path for visible‐light‐mediated dearomatization of stable arenes.
The energy accumulation of two photons by different strategies for the use in synthetic transformations has been demonstrated in visible‐light photoredox catalysis. Goez and co‐workers reported the consecutive use of two photons for the reduction of a ruthenium complex and excitation of the reduced complex to produce hydrated electrons (−2.5 V vs. SCE), however, high light intensities were required.22 We have previously reported a consecutive photoinduced electron transfer (conPET) using organic dye molecules, allowing the reduction of substrate molecules up to −2.4 V vs. SCE.23 Another approach for light energy accumulation is sensitized triplet–triplet annihilation (TTA), transforming the energy of two photons into radiation of higher energy.24, 25 A recent application in photocatalysis was reported by Campos, Rovis, and co‐workers, and involved the upconversion of near‐infrared light to visible light.26
We envisioned combining an energy‐transfer process27 with an electron‐transfer process28 to perform the arene dearomatization. In this approach, the sensitizer absorbs a photon and transfers energy to the arene. In a parallel process the excited sensitizer is reduced by a sacrificial electron donor. This sequence leads to an electron‐transfer process from the reduced sensitizer to the excited arene, reducing the arene to form a radical anion species (Figure 1). Most arenes have comparatively long excited‐state life times, the immediate reduction of which generates the corresponding radical anion. This step should be exergonic (ΔG<0),29 and is followed by a fast hydrogen‐atom transfer (HAT)30 that should trap the radical anion and transform it into a stable anion. Subsequent protonation would result in the dearomatized product.
Figure 1.
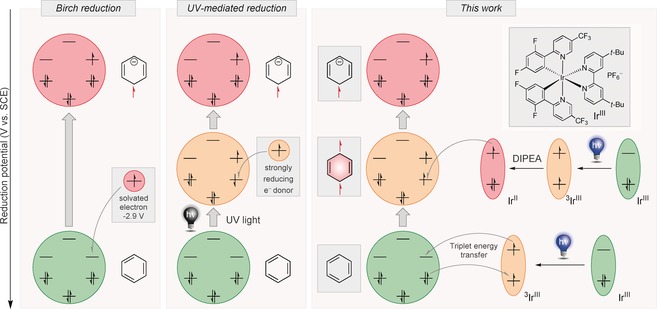
Overview of different strategies to generate radical anions of arenes by a simplified molecular orbital depiction. DIPEA=diisopropylethylamine.
Results and Discussion
We began our investigations with anthracene as the simplest model substrate because of its low aromatic stabilization energy and low triplet energy (E T=42.6 kcal mol−1), which is readily accessible by common photocatalysts. Control experiments (i.e., omitting each individual component) confirmed that the photocatalyst, DIPEA, and light irradiation were necessary for the photoreduction to occur (Table 1, entry 1; see Table S7 in the Supporting Information). Next, different organic and metal‐based photocatalysts were investigated in DMF under blue‐light irradiation, with DIPEA as the sacrificial electron donor. Ir[dF(CF3)ppy]2(dtbpy)PF6 (1; dF(CF3)ppy=2‐(2,4‐difluorophenyl)‐5‐(trifluoromethyl)pyridine and dtbpy=4,4′‐di‐tert‐butyl‐2,2′‐bipyridine) was the most efficient among them. Notably, the photocatalyst 2, which has a high reduction potential [E 0 red(*IrIII/IrIV)=−1.73 V), was not able to reduce anthracene (E 0 red=−1.98 V) efficiently (Table 1, entry 3), whereas the much weaker reductant 1 [E 0 red(*IrIII/IrIV)=−0.89 V] was a very efficient catalyst (entry 2). This observation can be rationally explained by the fact that 1 has a much higher triplet energy (E T=61.8 kcal mol−1) compared to that of 2 (E T=58.1 kcal mol−1). These results suggested that a photoinduced energy transfer from the excited photocatalyst to anthracene occurs, supporting our hypothesis. Surprisingly, no organic dye (entries 4 and 6) was able to reduce anthracene (E 0 red=−1.98 V vs. SCE), even though some of those dyes have reduction potentials of more than −2 V vs. SCE. A fast back‐electron transfer from anthracene to the catalyst may prevent this conversion.
Table 1.
Optimization of reaction conditions.[a]

|
Entry |
Photocatalyst[b] |
(e− + H+) Donor + HAT Reagent |
Yield [%][c] |
|---|---|---|---|
|
1 |
– |
DIPEA |
<5 |
|
2 |
1 |
DIPEA |
88 |
|
3 |
2 |
DIPEA |
47 |
|
4 |
Riboflavin tetraacetate |
DIPEA |
– |
|
5 |
3 |
DIPEA |
6 |
|
6 |
Rhodamine 6G |
DIPEA |
<5 |
|
7 |
1 |
NEt3 |
83 |
|
8 |
1 |
tributylamine |
85 |
|
9 |
1 |
NPh3 |
– |
|
10 |
1 |
pyridine |
– |
|
11 |
1 |
DIPEA + Hantzsch ester |
87 |
|
12 |
1 |
DIPEA + MeNH3Cl |
89[d] |
[a] The reactions were performed using 0.1 mmol 4 a in 1 mL DMF under air. [b] The triplet state energy decreases in the order 1>2>Riboflavin tetraacetate>3>Rhodamine 6G. [c] Yields were determined by GC analysis with naphthalene as an internal standard. [d] Reaction time=0.5 h.
Out of all the suitable HAT reagents able to compete with the back‐electron transfer, DIPEA was the best candidate. After electron donation, the oxidized form of DIPEA (DIPEA.+) can participate in a HAT. Additional HAT reagents, such as the Hantzsch ester (Table 1, entry 2 vs. 11), were found not to be essential for the reaction outcome, as the reaction yield did not improve. Other amines with α‐hydrogen atoms, including NEt3 and tributylamine, gave comparable yields to that of DIPEA (entries 7 and 8). However, amines without α‐hydrogen atoms did not yield the reduced arene (entries 9 and 10), confirming that DIPEA is the source of hydrogen atoms. The iminium ion generated after the HAT acts as a proton donor as a result of residual water present in the solvent. Indeed, the reduced product was obtained without any additional proton source, albeit longer reaction times (2 h). The presence of a catalytic amount (10 mol %) of MeNH3Cl was found to be beneficial in reducing the reaction time (from 2 to 0.5 h, entry 12). As a result, MeNH3Cl was chosen as an additive in further studies. Notably, the catalyst loading could be decreased to 0.25 mol % without compromising the yield of the product (see Table S4, entry 3) and a turnover (TON) of about 700 was achieved with 0.06 mol % catalyst loading (Table S4, entry 5).
Using the optimized reaction conditions, we investigated the scope and limitations for arenes and heteroarenes (Figure 2). Two factors determine the success of the photoreduction: the aromatic stabilization energy (ASE) and the triplet energy. High aromatic stabilization energy and triplet energy result in lower reactivity and yield of the isolated product of the corresponding reduction products: benzene<phenanthrene≈naphthalene<anthracene. ASE per benzene ring in kcal mol−1: benzene (36)>phenanthrene (30.3)≈naphthalene (30.5)>anthracene (27.7); triplet energy (E T in kcal mol−1): benzene (84.4)>phenanthrene (61.4)≈naphthalene (60.9)>anthracene (42.6).31
Figure 2.

Substrate scope of photoredox dearomatization. Yields of isolated products are shown. [a] Yield determined by NMR spectroscopy. DMF=N,N‐dimethylformamide.
Anthracene derivatives were readily reduced to their respective dihydro products (4 a′–e′; Figure 2) in good to excellent yields (80–90 % yield). There were no dimerizations and no over‐reduced byproducts were observed. Electron‐donating (e.g., ‐Me) and electron‐withdrawing (e.g., ‐Ph and ‐CONEt2) groups attached to the 9‐position of anthracene were equally compatible. However, halide and cyano substituents were cleaved during the reduction (see Figure S20). Phenanthrene and naphthalene were successfully reduced to 4 f′ and 4 j′,j′′, respectively, with corresponding yields of 88 and 75 %. The phenanthrene reduction was performed on a 10 mmol scale, yielding 83 % of the product (see the Supporting Information). The photoreduction of naphthalene derivatives gave mixtures of dihydro‐ and tetrahydronaphthalenes (4 j′–o′), where the ratios were dependant on reaction times and substituents. Tetrahydronaphthalenes resulted from a subsequent photoreduction of dihydronaphthalenes. In the case of aryl ethers, the reduction occurred on the electron‐deficient ring (4 m′–n′). In the presence of electron‐withdrawing groups para to the alkoxy group, a partial loss of the alkoxy group was observed that led to 4 l′′. A similar reaction to this has been reported using the classic Birch‐type reduction.32 Surprisingly, 4 o′ was obtained instead of a reduction of the substituted ring. DFT calculations indicated that 4 o′ is thermodynamically more stable than 4 l′′, and explains the observed selectivity. The triplet state of benzene (E T=84.4 kcal mol−1) cannot be sensitized by an energy transfer from photoexcited 1 (E T=61.8 kcal mol−1), therefore, no reactivity could be observed for the arenes like 1,4‐dimethoxybenzene and anisole.33
The nitrogen heterocycles phenazine (4 q), acridine (4 r), benzo‐cinnoline (4 s), 1,1,2‐trimethyl‐1H‐benzo[e]indole (4 t), quinoline (4 v), and isoquinoline (4 w) were reduced in moderate yields (22–60 %; Figure 2). The presence of electron‐withdrawing groups, such as an ester at the 4‐position, increased the reactivity of the quinoline 4 u by stabilizing the radical anion intermediate and decreasing the triplet energy (56 % yield for 4 u′; 25 % yield for 4 v′). Alkenes and alkynes that were conjugated to arenes were selectively reduced to 4 x′, y′ (>90 % yield) without any reduction of the arene. The ester of Neproxen, an anti‐inflammatory drug, was reduced to 4 z′ and 4 z′′ in 34 and 31 % yield, respectively.
Based on literature reports,29, 34 we propose that the reaction involves a triplet–triplet energy transfer (EnT), followed by SET, HAT, and protonation. Upon visible‐light photoexcitation of 1, the lowest triplet excited state, *IrIII, sensitizes the formation of the triplet state of anthracene, Anth*. As a result, one blue photon is utilized for an energy‐transfer process (EnT, Dexter energy transfer). In parallel, a second blue photon excited *IrIII is quenched by DIPEA to give an IrII complex and the radical cation of DIPEA (DIPEA.+). The excited Anth* (lifetime=3300 μs in polar solvents in inert atmosphere)31 is reductively quenched by the IrII complex to Anth.−, which extracts a hydrogen atom from DIPEA.+, yielding the Anth− carbanion. Finally, protonation from MeNH3Cl or the iminium ion of DIPEA results in the reduced product 9,10‐dihydroanthracene 4 a′.
Although a detailed spectroscopic study of transient intermediates would be necessary to prove the mechanistic hypothesis fully, several experimental observations support our mechanistic proposal already. UV‐visible spectroscopy confirmed that only 1 absorbs at λ=455 nm (see Figure S4). Therefore, a direct excitation of either anthracene or naphthalene is unlikely.35 Furthermore, Stern–Volmer quenching studies supported both the energy transfer from the excited photocatalyst to anthracene and the electron transfer from DIPEA to the excited photocatalyst (Figure 3 C). The energy transfer to anthracene and the electron transfer are similar in rate, but not identical. An excess of DIPEA does not interfere with the reaction (see Table S6), which indicates that the better overall performance with a ratio of 1:10 may have its reason in other chemical steps involving DIPEA. Online UV‐visible and EPR experiments provided evidence for the formation of the IrII complex (Figure 3 D). The excitation mechanism by an overall of two photons is supported by a quadratic dependency of the product yield (determined by GC analysis) on the irradiation intensity (Figure 3 E). Furthermore, substrate addition to an independently generated IrII complex while stirring in the dark resulted in no product formation (see the Supporting Information). This observation confirmed that an energy‐transfer step using another photon is crucial in generating the radical anion of anthracene. To prove the presence of a carbanion intermediate, a photochemical E1cB reaction was designed (Figure 3 B). A photosensitized electron transfer, which was followed by HAT, generated a carbanion intermediate of 5 b′. Subsequent leaving‐group elimination led to the alkene 5 b′′, which was then photoreduced to the corresponding alkane 4 b′.
Figure 3.

Mechanistic investigations. A) Mechanistic proposal. B) E1cB reaction proves carbanion intermediate. C) Stern–Volmer Quenching Study proves energy transfer from *IrIII to arene and electron transfer from DIPEA to *IrIII. D) EPR study proves generation of IrII species in the presence of DIPEA. E) Quadratic dependency of the product yield on irradiation density indicates the two‐photon process.
Finally, experiments with deuterated DMF excluded HAT and protonation processes that could involve the solvent. Protonation occurs from the iminium ion of DIPEA, however, MeNH3Cl also acts as an additional proton source as protonation from MeNH3Cl to 5 b′ is thermodynamically favorable (pK a values of benzylic C−H and MeNH3Cl are 30.1 and 11.1, respectively, in DMSO).36, 37 In addition, electrochemical data suggested that added MeNH3Cl lowers the reduction potential of the substrate by 0.10 V (see Figure S15), facilitating the electron transfer and thereby accelerating the reaction (Table 1, entry 14). The possibility of reducing the singlet–triplet gap (E T) of the substrate through a possible cation–π interaction38 in the presence of the ammonium salt was excluded, as no bathochromic shift in UV‐vis and phosphorescence spectra could be observed. Mechanisms involving either triplet–triplet annihilation or a conPET process of IrII are unlikely under the reaction conditions. For triplet–triplet annihilation, either laser irradiation or very high intensity light is typically required.25 Our LED set up is not able to produce singlet arenes via a triplet–triplet annihilation process.29 From time‐dependent UV measurements (see Figure S8) we can conclude that an IrII species is not stable for long in DMF under the reaction conditions. A further excitation of IrII is therefore unlikely, and excludes a conPET mechanism.
Conclusion
In conclusion, we have achieved the direct reduction of aromatic compounds using visible‐light photoredox catalysis. The method allowed the successful photoreduction of napthalenes, larger aromatic hydrocarbons, and heterocycles by using the energy of two visible light photons. Such photoreductions are valuable in synthesis. Triplet sensitizers that provide higher energies are currently being investigated to expand the scope of the reaction.
Experimental Section
General procedure for the reduction. A 5 mL crimp cap vial was equipped with the substrate (0.2 mmol, 1 equiv), DIPEA (0.2 mmol, 1 equiv), MeNH3Cl (10 mol %), the photocatalyst Ir[dF(CF3)ppy]2(dtbpy)PF6 (1 mol %) and a stirring bar. After adding the solvent (2 mL DMF) via syringe, the vial was capped under air. The reaction mixture was stirred and irradiated using a 455 nm (±10 nm) LED for 15–18 h at 25 °C. The progress was monitored by TLC and GC analysis. The reaction mixture was diluted with water (10 mL), extracted with ethyl acetate (3×20 mL), washed with brine (1×20 mL), and dried over anhydrous Na2SO4. The crude reaction mixture was obtained by removing the solvents under reduced pressure. Purification was performed by automated flash column chromatography (silica, 0–100 % EtOAc/PE).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank German Science Foundation (DFG, KO 1537/18‐1) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 741623) for financial support. We thank Dr. Rudolf Vasold, Regina Hoheisel, Julia Zach, Marsel Shafikov, and Jenny Phan for assistance with analytical measurements. We thank Prof. Jimmie Weaver, Matthias Schmalzbauer, Dr. Indrajit Ghosh, Dr. Stefano Crespi, and Sascha Grotjahn for helpful discussions and Ranit Lahmy for proof reading the manuscript.
A. Chatterjee, B. König, Angew. Chem. Int. Ed. 2019, 58, 14289.
References
- 1. Roche S. P., Porco J. A., Angew. Chem. Int. Ed. 2011, 50, 4068–4093; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4154–4179. [Google Scholar]
- 2. Przydacz A., Skrzyńska A., Albrecht Ł., Angew. Chem. Int. Ed. 2019, 58, 63–73; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 64–75. [Google Scholar]
- 3. Wertjes W. C., Southgate E. H., Sarlah D., Chem. Soc. Rev. 2018, 47, 7996–8017. [DOI] [PubMed] [Google Scholar]
- 4. Liu J., Krajangsri S., Singh T., De Seriis G., Chumnanvej N., Wu H., Andersson P. G., J. Am. Chem. Soc. 2017, 139, 14470–14475. [DOI] [PubMed] [Google Scholar]
- 5. Birch A. J. Q., Chem. Soc. Rev. 1950, 4, 69. [Google Scholar]
- 6. Holy N. L., Chem. Rev. 1974, 74, 243–277. [Google Scholar]
- 7. Dye J. L., Cram K. D., Urbin S. A., Redko M. Y., Jackson J. E., Lefenfeld M., J. Am. Chem. Soc. 2005, 127, 9338–9339. [DOI] [PubMed] [Google Scholar]
- 8. Lei P., Ding Y., Zhang X., Adijiang A., Li H., Ling Y., An J., Org. Lett. 2018, 20, 3439–3442. [DOI] [PubMed] [Google Scholar]
- 9. Donohoe T. J., Thomas R. E., Nat. Protoc. 2007, 2, 1888–1895. [DOI] [PubMed] [Google Scholar]
- 10. Yoo B. I., Kim Y. J., You Y., Yang J. W., Kim S. W., J. Org. Chem. 2018, 83, 13847–13853. [DOI] [PubMed] [Google Scholar]
- 11. Szostak M., Spain M., Procter D. J., J. Org. Chem. 2014, 79, 2522–2537. [DOI] [PubMed] [Google Scholar]
- 12. Giustra Z. X., Ishibashi J. S. A., Liu S.-Y., Coord. Chem. Rev. 2016, 314, 134–181. [Google Scholar]
- 13. James M. J., Schwarz J. L., Strieth-Kalthoff F., Wibbeling B., Glorius F., J. Am. Chem. Soc. 2018, 140, 8624–8628. [DOI] [PubMed] [Google Scholar]
- 14. Zhu M., Zheng C., Zhang X., You S.-L., J. Am. Chem. Soc. 2019, 141, 2636–2644. [DOI] [PubMed] [Google Scholar]
- 15. Peters B. K., Rodriguez K. X., Reisberg S. H., Beil S. B., Hickey D. P., Kawamata Y., Collins M., Starr J., Chen L., Udyavara S., Klunder K., Gorey T. J., Anderson S. L., Neurock M., Minteer S. D., Baran P. S. Science 2019, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mizuno K., Okamoto H., Pac C., Sakurai H., J. Chem. Soc. Chem. Commun. 1975, 839–840. [Google Scholar]
- 17. Yasuda M., Pac C., Sakurai H., J. Org. Chem. 1981, 46, 788–792. [Google Scholar]
- 18. Yoshimi Y., Ishise A., Oda H., Moriguchi Y., Kanezaki H., Nakaya Y., Katsuno K., Itou T., Inagaki S., Morita T., Hatanaka M., Tetrahedron Lett. 2008, 49, 3400–3404. [Google Scholar]
- 19. McCallum T., Pitre S. P., Morin M., Scaiano J. C., Barriault L., Chem. Sci. 2017, 8, 7412–7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marzo L., Pagire S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188–10228. [Google Scholar]
- 21. Qiao B., Jiang Z., ChemPhotoChem 2018, 2, 703–714. [Google Scholar]
- 22. Goez M., Kerzig C., Naumann R., Angew. Chem. Int. Ed. 2014, 53, 9914–9916; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10072–10074. [Google Scholar]
- 23. Ghosh I., Ghosh T., Bardagi J. I., König B., Science 2014, 346, 725–728. [DOI] [PubMed] [Google Scholar]
- 24. Kerzig C., Wenger O. S., Chem. Sci. 2018, 9, 6670–6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Islangulov R. R., Castellano F. N., Angew. Chem. Int. Ed. 2006, 45, 5957–5959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6103–6105. [Google Scholar]
- 26. Ravetz B. D., Pun A. B., Churchill E. M., Congreve D. N., Rovis T., Campos L. M., Nature 2019, 565, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strieth-Kalthoff F., James M. J., Teders M., Pitzer L., Glorius F., Chem. Soc. Rev. 2018, 47, 7190. [DOI] [PubMed] [Google Scholar]
- 28. Singh A., Fennell C. J., Weaver J. D., Chem. Sci. 2016, 7, 6796–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghosh I., Bardagi J. I., König B., Angew. Chem. Int. Ed. 2017, 56, 12822–12824; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12998–13000. [Google Scholar]
- 30. Guo X., Wenger O. S., Angew. Chem. Int. Ed. 2018, 57, 2469–2473; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2494–2498. [Google Scholar]
- 31. Montalti M., Murov S. L., Handbook of Photochemistry , 3rd ed., CRC/Taylor & Francis, Boca Raton, 2006. [Google Scholar]
- 32. Murthy A. R., Sundar N. S., Rao G. S. R. S., Tetrahedron 1982, 38, 2831–2836. [Google Scholar]
- 33.Notably, methyl terephthalate gave trace amounts of dimethyl-1,4-cyclohexandicarboxylate (10 % yield upon isolation), and may result from endergonic activation of the substrate by the photocatalyst. Although about 5 kcal mol−1 uphill energy trasfer seems to be feasible with such an iridium complex as reported: Glorious F. et al., Nat. Chem. 2018, 10, 981–988. Here we observe about 10 kcal mol−1 uphill energy transfer, and the reason for this is not clear to us at present.30082884 [Google Scholar]
- 34. Pal A. K., Li C., Hanan G. S., Zysman-Colman E., Angew. Chem. Int. Ed. 2018, 57, 8027–8031; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8159–8163. [Google Scholar]
- 35. Ghosh I., Shaikh R. S., König B., Angew. Chem. Int. Ed. 2017, 56, 8544–8549; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8664–8669. [Google Scholar]
- 36. Bordwell F. G., Bares J. E., Bartmess J. E., McCollum G. J., Van der Puy M., Vanier N. R., Matthews W. S., J. Org. Chem. 1977, 42, 321–325. [Google Scholar]
- 37. Crampton M. R., Robotham I. A., J. Chem. Res. Syn. 1997, 22–23. [Google Scholar]
- 38. Yamada S., Chem. Rev. 2018, 118, 11353–11432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary