

Abstract
Humans and animals are repeatedly exposed to endocrine disruptors, many of which are ubiquitous in the environment. Endocrine disruptors interfere with hormone action; thus, causing non-monotonic dose responses that are atypical of standard toxicant exposures. The female reproductive system is particularly susceptible to the effects of endocrine disruptors. Likewise, exposures to endocrine disruptors during developmental periods are particularly concerning because programming during development can be adversely impacted by hormone level changes. Subsequently, developing reproductive tissues can be predisposed to diseases in adulthood and these diseases can be passed down to future generations. The mechanisms of action by which endocrine disruptors cause disease transmission to future generations are thought to include epigenetic modifications. This review highlights the effects of endocrine disruptors on the female reproductive system, with an emphasis on the multi- and transgenerational epigenetic effects of these exposures.
Keywords: developmental origins of health and disease, environmental contaminants and toxicants, endocrine disruptors, epigenetics, DNA methylation, female reproductive tract, histone modifications, chromatin
Endocrine disruptors alter reproductive tissues and functions across generations via epigenetic mechanisms.
Endocrine disruptors
Synthetic chemicals have become a part of people's everyday lives and some of these chemicals have been identified as endocrine disruptors. Endocrine disruptors are exogenous chemicals, mixtures of chemicals, or non-chemical exogenous factors that interfere with the body's normal endocrine system, leading to adverse effects on hormonally controlled functions [1]. Endocrine disruptors are heterogeneous and vary from synthetic to natural chemicals. Specifically, synthetic chemicals such as polychlorinated biphenyls, plasticizers, pesticides, fungicides, and pharmaceutical agents are known endocrine disruptors [1]. Natural chemicals such as phytoestrogens found in food products are also known endocrine disruptors. These chemicals serve various purposes and are ubiquitous in the environment [1]. Endocrine disruptors interfere with hormone actions by mimicking hormones, promoting inappropriate responses at improper times, or by blocking hormone action, leading to alterations in the hormonal and homeostatic systems and interfering with the ability of the body to communicate with and respond to the environment [1]. Endocrine disruptors tend to have a low binding affinity for hormone receptors and their ability to activate or block hormone receptors may vary. Although it is often difficult to define adverse effects, some researchers consider any biological response to an endocrine disruptor to be an adverse event [2].
Endocrine disruptors are found in food, consumer products, water, soil, and in wildlife and people who are exposed through ingestion, inhalation, dermal contact, or injection [1]. Examples of endocrine disruptors vary from chemical to non-chemical exogenous factors [1, 3]. Chemical endocrine disruptors can be categorized into three major groups: pesticides, chemicals in consumer products, and food contact materials [4]. Examples of pesticides that induce endocrine disruptive activities include glyphosate, dichlorodiphenyltrichloroethane (DDT), atrazine, chlorpyrifos, and methoxychlor [4–6]. Endocrine disruptors found in consumer products include, but are not limited to, brominated flame-retardants, phthalates, parabens, heavy metals, polychlorinated biphenyls, nonylphenols, diethylstilbestrol (DES), and perfluorochemicals [4, 7–9]. Additional endocrine disruptors described as food contact materials are bisphenol A (BPA), phthalates, and phenols. [4, 8].
Numerous endocrine disruptors exist, but this review will focus on eight well-documented endocrine disruptors and their epigenetic effects on female reproduction. The chosen chemicals range from chemicals in consumer products, food contact materials, and pesticides and collectively, are ubiquitously found in the environment. Therefore, it is critical to thoroughly investigate and analyze these chemicals across generations on female reproduction. Specifically, BPA is a well described endocrine disruptor [10]. BPA is a synthetic chemical used mostly in polycarbonate plastics, epoxy resin liners in aluminum cans, and thermal receipts. It can act through various sex steroid hormone receptors, including estrogen receptors (ESRs) 1 and 2, androgen receptors, and thyroid hormone receptors [11]. Phthalates are a class of chemicals that serve as plasticizers and act as endocrine disruptors [12, 13]. DES is an endocrine disruptor that was used as an anti-abortive drug until the 1970s, but it is no longer used due to its reproductive toxicity [12]. DDT and its metabolite dichlorodiphenyldichloroethylene (DDE) are organochlorine insecticides and are well known endocrine disruptors [12, 14, 15]. Methoxyclor (MXC) is another organochlorine pesticide and endocrine disruptor that replaced DDT, but methoxychlor is now banned in many countries due to its toxicity [15]. Vinclozolin is a dicarboximide fungicide used in agriculture, but more specifically in the viniculture industry, and it exhibits endocrine disrupting effects [16]. Further, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is a potent polychlorinated dibenzodioxin and endocrine disruptor. It is formed as a by-product of organic combustion and is a major component of the Agent Orange that was used during the Vietnam War. It also is a major environmental contaminant from an industrial explosion in Seveso, Italy [12, 17].
Endocrine disruptors cause non-monotonic dose responses such as sigmoid, U-shaped, or inverted-U-shaped curves [1, 18]. For example, endocrine disruptors that mimic estrogen stimulate MCF-7 human breast cancer cell proliferation at low doses, but saturate the cell growth response and do not increase proliferation at high doses [19]. Although the mechanisms behind such non-monotonic effects are not fully understood, they may be due to receptor type and abundance in specific cells or tissues [20], receptor down-regulation and desensitization [21, 22], and endocrine feedback loops [23, 24].
Endocrine disruptors also have been shown to act at low levels and in the range of normal human exposure [1]. This is not surprising because endocrine disruptors mimic endogenous hormones, which act at low concentrations. Therefore, studies that utilize environmentally relevant and low doses are important for understanding the effects of endocrine disruptors in the body.
Recently, several studies have linked exposure to endocrine disruptors to adverse reproductive outcomes. For example, the incidence and prevalence of diseases involving reproductive tissues such as breast cancer, prostate cancer, and polycystic ovarian syndrome have increased over time [25–27]. These diseases are influenced by both genetic and environmental factors, but dramatic increases in incidence and prevalence suggest that these diseases are largely due to environmental factors. An analysis of about 44,800 pairs of twins from Sweden, Denmark, and Finland shows that the environment, and not genetics, substantially influenced the rates of sporadic prostate and breast cancers [26]. Another study demonstrates that exposure to BPA plays a major role in polycystic ovarian syndrome pathogenesis [25].
The reproductive system is especially vulnerable to endocrine disruptors during development. The developmental origins of health and disease (DOHaD) is a paradigm in which environmental exposures during development can lead to health and disease risk later in childhood and adult life [28]. The concept is that environmental stressors including malnutrition and exposure to environmental endocrine disruptors during critical periods of development cause subtle changes in gene expression that lead to permanent alterations in an organ, tissue, or structure. The alteration will then lead to a health and disease risk later in life [28–30]. In addition, DOHaD disease risk can be transmitted across generations [28].
Endocrine disruptors act through multiple pathways to influence developmental programming. During early development, the fetus is protected from exogenous estrogens by a plasma protein, α-fetoprotein, which binds estrogens and protects the fetus. However, some endocrine disruptors bypass α-fetoprotein due to weak binding affinity, subsequently rendering the fetus vulnerable to toxicity [31]. Further, other hormone-binding proteins circulate through the blood and endocrine disruptors may bind to these proteins, disrupting the balance between hormone-binding proteins and endogenous hormones [32]. Further, endogenous hormones may be less bioavailable, whereas the endocrine disruptors are physiologically available, causing inappropriate hormone signaling [32, 33]. The ability of endocrine disruptors to interfere with hormone levels during development is of concern because cell differentiation and tissue development can be adversely impacted by hormone level changes. Subsequently, these tissues can be predisposed to diseases in adulthood and disease can be passed down to future generations [34].
Mechanisms of multigenerational and transgenerational epigenetic inheritance
The ability of endocrine disruptors to influence developmental programming and cause disease and infertility is a major concern, but even more troubling is that some of the effects of endocrine disruptors may be multigenerational or transgenerational in nature. To obtain a multigenerational effect, the phenotype or effect must occur in generations that were directly exposed to the endocrine disruptor during development. However, to observe a transgenerational effect, the phenotype or the effect must be inherited by the generation that was not directly exposed to the endocrine disruptor during development [35].
Multiple exposure paradigms produce multigenerational and transgenerational effects (Figure 1). The first paradigm is by adult life exposure. The F0 generation is exposed to an endocrine disruptor during adult life. During this window, the F1 generation experiences preconception exposure as the germ-line. Once the F0 generation produces the F1 generation, any effects observed in the F1 generation are due to multigenerational effects of endocrine disruptor exposure. To observe a transgenerational effect from adult exposure, the subsequent generation, the F2 generation, must be produced. This is the first generation that is not directly exposed to the endocrine disruptor and any effects observed in this generation are considered transgenerational [35–37].
Figure 1.
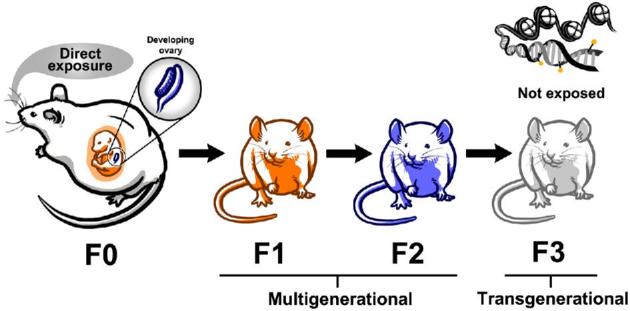
Exposure to endocrine disruptors during prenatal development causes multigenerational effects in the F1 and F2 generations and transgenerational effects in the F3 generation. The F1 and F2 generations are directly exposed to the endocrine disruptor as a fetus and germ cell, respectfully. The F3 generation is not directly exposed and the mechanisms governing the effects in the F3 generation are thought to be epigenetic in nature.
The second exposure paradigm is by prenatal exposure (Figure 1). The F0 generation is exposed to an endocrine disruptor during pregnancy. During this exposure window, the F1 generation is exposed as a developing fetus and the F2 generation is exposed as the developing germ cells inside the fetus. Both the F1 and F2 generations are directly exposed and effects observed in these generations are considered multigenerational effects. The subsequent generation, the F3 generation, is the first generation that is not directly exposed to the chemical and any effects observed are considered transgenerational effects [35–37]. The transgenerational phenomenon does not involve direct exposure and usually involves epigenetic changes induced in the germline [34, 38–40].
Epigenetics are mitotically and meiotically heritable changes in gene function without changing DNA sequences [41, 42]. Broadly, these heritable changes in the epigenome define and control cell and tissue development by controlling gene expression [43]. Multiple molecular mechanisms alter the epigenome, including changes DNA methylation, chromatin modification, and some noncoding RNAs (ncRNAs) (Figure 2). Epigenetic modifications must be transmitted through the germline to the unexposed generation to cause a transgenerational phenomenon [36].
Figure 2.

An overview of epigenetic mechanisms including chromatin modifications, DNA methylation, and ncRNA interactions. Chromatin is made of DNA, histone proteins, and nucleosomes and regulates gene expression by controlling the access of transcription factors to DNA. DNA methylation creates a physical barrier that generally impedes transcription factor binding. Noncoding RNA interacts with chromatin and modifies domains.
DNA methylation is a commonly studied epigenetic mechanism [44]. Specifically, methylation of DNA is a highly dynamic modification that occurs on the cytosine residue in “CpG” dinucleotides [45]. Cytosines in 5′ promoter region that are methylated hinder the transcription of the gene, thus causing gene silencing. This is because DNA methylation is associated with providing a physical barrier, which impedes transcription factor binding, resulting in downregulation of gene expression [46, 47].
In addition to epigenetic modifications occurring by DNA methylation, they can occur due to chromatin modifications. Chromatin modifications are epigenetic modifications that directly regulate the packaging of DNA. Over 3 billion base pairs are contained in chromosomes and need to fit inside the nucleus. Chromatin serves to compact the DNA while allowing transcription factor access to relevant DNA sequences. Chromatin is made of DNA, histone proteins, and nucleosomes. Histones are critical for condensing DNA and their functions are primarily controlled by modifying the N-terminus, also known as histone tail domains. These histone tails are modifiable by non-histone proteins and these modifications reflect DNA compaction. The type of modification on the specific histone and the position of the modification influences gene expression. Histone tail modifications include acetylation, methylation, proline isomerization, SUMOlyation, ubiquitination, phosphorylation, ADP ribosylation, and deamination [48]. Post-translational modifications interact with the histone tails to modify the transcriptional regulatory readout. Post-translational modifications may occur on any of these histones [48].
Another type of epigenetic modification involves ncRNA. Noncoding RNAs are forms of epigenetic modifications that serve housekeeping and regulatory functions and are involved in function. Long noncoding RNAs (lncRNAs) are longer than 200 nucleotides in length, do not encode protein, and mediate gene expression. The mechanisms by which lncRNAs exert their epigenetic effects are diverse. Long ncRNA have the ability to interact with genomic loci and chromatin by forming stable domains for protein binding and chromatin localization. Therefore, the lncRNA interactions allow sequence specific localization of chromatin-modifying complexes and the lncRNAs direct the chromatin-modifying complexes to target genes, thus regulating transcriptional activity [49, 50]. Further, lncRNAs can directly interact with DNMT1 and affect global methylation patterns [51]. Finally, lncRNAs have been shown to interact with other ncRNAs such as miRNA. Specifically, lncRNAs can bind and sequester miRNAs and prevent them from binding to their target mRNAs [52, 53]. Further detailed information about epigenetic mechanisms and modifications is described in several extensive epigenetic reviews [44, 46, 54–59].
Epigenetic effects of endocrine disruptors on female reproduction
Multiple organs are required to facilitate healthy reproduction and studies show that endocrine disrupting chemicals can interfere with the function of these organs. Organs within the hypothalamic-pituitary-ovary axis consist of the hypothalamus, anterior pituitary, and the ovary. A summary of studies demonstrating the multigenerational and transgenerational impacts of endocrine disrupting chemicals on each of these organs are described below.
Epigenetic effects of endocrine disruptors on the hypothalamus
Studies indicate that endocrine disrupting chemicals can target the hypothalamus in the brain, leading to epigenetic changes and transgenerational effects. For example, BPA has been shown to cause transgenerational inheritance in the hypothalamus [60]. Prenatal BPA (5 mg/kg) exposure disrupts the number of ERα-cells in brain regions (bed nucleus of the stria terminalis and anteroventral periventricular nucleus) important for reproductive function in female mice in a transgenerational manner [60]. However, the epigenetic mechanism governing the change in ERα cell numbers was not explored in the studies. Further, perinatal BPA exposure (5 mg/kg) increases the expression of Meg3, a maternally expressed lncRNA, in the female hypothalamus of the F3 generation [61]. The expression of Meg3 is important because it is correlated with the central control of precocious puberty [62] and increased levels of BPA are associated with precocious puberty in women [63] and in laboratory animals [64]. The increase in Meg3 is significant because it is an epigenetic modifier and mRNA expression is increased in the generation that was ancestrally exposed to BPA. These findings are significant because the dose of BPA used (5 mg/kg) is estimated to be what is found in human maternal blood (0.3–18.9 ng/mL maternal plasma) and is therefore environmentally relevant [60, 65, 66].
Epigenetic effects of endocrine disruptors on the ovary
Endocrine disruptors can affect several processes in ovary, including the formation of a healthy primordial follicle pool, maintenance of a constant stream of growing follicles, and normal steroidogenic capacity, all of which are required for normal female fertility [13, 67–70]. Any chemical that interferes with these processes can cause severe reproductive outcomes. Specifically, chemicals that target the formation of the primordial follicle pool cause infertility because they deplete the finite follicle reserve used for the growth of ovulatory follicles [67, 68]. Additionally, an increased loss of primordial follicles leads to an early onset of reproductive senescence [69]. This is of concern because early onset of reproductive senescence is associated with increased risk of chronic diseases [69, 71–74]. Chemicals that specifically target primary, preantral, and antral follicles may lead to temporary infertility or permanent infertility. Temporary infertility may occur when the toxicant only targets the mature population of follicles, but not immature follicles. Thus, when the toxicant is removed, the immature follicle types can grow and replenish the mature population of follicles, restoring fertility. Permanent infertility occurs when the toxicant is not removed and continuously targets the growth and function of ovarian follicles [70]. Permanent infertility is more likely to occur in humans because chemical exposure occurs on a daily basis and it is difficult to remove chemical exposure. Chemicals that target the production of sex steroid hormones from the ovary may lead to infertility and other non-reproductive disorders [70, 71, 75–77].
Exposure to DES has been associated with multigenerational effects on the ovaries in women. In particular, one case study describes small cell carcinoma of the ovary in a 15 year old girl whose maternal grandmother had taken DES during her pregnancy [78]. This study provides one example that prenatal DES exposure is associated with a multigenerational increase in ovarian cancer in the F2 generation in humans. Similarly, another study has shown that prenatal exposure to DES is associated with ovarian cancer in the F2 generation [79]. Unfortunately, the epigenetic mechanisms for these ovarian effects have not been fully investigated across generations. In contrast, some studies have shown that DES exposure does not lead to female genital tract anomalies. In particular, 28 daughters that were F2 generation descendants were evaluated for lower genital tract abnormalities. None of the daughters were found to have abnormalities usually associated with DES exposure [80]. Additionally, a retrospective cohort study of 2268 women exposed to DES in utero shows that their daughters, the F2 generation, did not experience a significant increase in female genital tract anomalies [81]. It is likely that because half of the F2 females were less than 18 years old, the females may not yet show signs of anomalies. Therefore, further evaluation is necessary to understand the effects of DES on female genital tract anomalies in women.
MXC is a banned insecticide once used as a replacement for DDT. It is an endocrine disruptor shown to directly affect ovarian functions. Studies have shown the exposure to MXC causes various ovarian-related diseases in both multigenerational and transgenerational manners. MXC exposure (200 mg/kg BW/day) causes ovarian disease in the F1 generation of rats and ancestral MXC (200 mg/kg BW/day) exposure increases polycystic ovarian-like syndrome in the F3 generation of rats [82]. Epigenetic analyses show that MXC (100 mg/kg) hypermethylates CpGs in the ERβ promoter of the ovary. Further, MXC (100 mg/kg) hypermethylates multiple loci critical for ovarian signaling pathways and concurrently decreases gene expression [83, 84]. MXC (20 μg and 100 mg/kg) exposure also increases expression of Dnmt3b in the ovaries, suggesting that Dnmt3b plays a critical role in DNA hypermethylation [83]. The adult general population exposure to MXC ranges between 0.1–0.3 ng/kg/day based on the Food and Drug Administration’s Total Diet Study for the period 1986–1991 [85]. Therefore, it is likely that the doses used in previous studies are too high to be relevant to human exposure. Thus, future studies are needed that assess the effects of environmentally relevant levels of methoxychlor on the ovary across generations. Although both multigenerational and transgenerational effects on ovarian functions are observed in experimental animals, it is not yet determined if the epigenetic changes observed in some experiments are specifically linked to an ovarian phenotype. This indicates a need for future studies on epigenetic mechanisms underlying MXC-induced toxicity.
Phthalates are a class of chemicals commonly used as plasticizers, but are also known endocrine disrupting chemicals. Previous studies show that prenatal exposure to a mixture of phthalates (21% di (2-ethyhexyl) phthalate (DEHP), 35% diethyl phthalate (DEP), 15% dibutyl phthalate (DBP), 8% diisobutyl phthalate (DiBP), 5% benzylbutyl phthalate (BzBP), and 15% diisononyl phthalate (DiNP)) that mimics human exposure causes multigenerational effects on mouse ovaries [86, 87]. Specifically, prenatal phthalate mixture exposure (20 μg/kg/day, 200 μg/kg/day, 200 mg/kg/day, and 500 mg/kg/day) induces cystic ovaries in the F1 and F2 generations [86, 87]. The phthalate mixture was designed from levels of phthalate metabolites measured in urine samples from pregnant women in Illinois [87, 88]. Additionally, the lowest dose used in the phthalate mixture studies (20 μg/kg/day) mimics human exposure levels and is within the high end of phthalate detection levels in humans [87]. Another mixture study shows exposure to mixtures of plastic derivatives (BPA 50 mg/kg BW/day, DEHP 750 mg/kg BW/day and DBP 66 mg/kg/BW/day and BPA 25 mg/kg BW/day, DEHP 375 mg/kg BW/day, DBP 33 mg/kg BW/day) causes polycystic ovaries in both the F1 and F3 generations [89]. Although the doses in these studies are high in relation to human exposures, the study was designed to examine pharmacological actions of the chemicals on epigenetic transgenerational inheritance [89].
Exposure to a single phthalate, DEHP, also causes adverse transgenerational effects on the ovary in mice. Specifically, prenatal DEHP exposure (20 μg/kg/day, 200 μg/kg/day, 500 mg/kg/day, 750 mg/kg/day) dysregulates folliculogenesis, alters sex steroid hormone levels, and increases the presence of ovarian cysts in a multigenerational manner [90, 91]. Further, ancestral exposure to DEHP (20 μg/kg/day, 200 μg/kg/day, 500 mg/kg/day, 750 mg/kg/day) accelerates early folliculogenesis in a transgenerational manner [91]. Although studies demonstrate that phthalate exposure causes transgenerational effects on the ovary, the mechanisms causing these effects are not well understood. A few studies suggest that DEHP exposure (80 mg/kg/day, 2 g/kg, and mixtures of benzo[a]pyrene + DEHP at 5 mg/kg and 300 mg/kg, respectively and at 10 mg/kg and 600 mg/kg, respectively) causes multigenerational effects through both the ESR1 and peroxisome proliferator-activated receptor alpha [92–95]. However, further studies at human relevant doses are necessary to investigate the effects of phthalate exposure on the epigenome of the ovary, especially because the body burden of DEHP is approximately 19.17 μg/kg as of 2015–2016 [96]. Although previous studies provide insight in to the mechanism of phthalate exposure on ovarian phenotypes, it is necessary to elucidate epigenetic mechanisms across generations.
BPA causes both multigenerational and transgenerational effects on the ovary. Prenatal exposure to BPA (20 μg/kg/day) decreases serum testosterone levels in the F2 generation and dysregulates steroidogenic enzymes in the F2 ovaries of mice [97]. Ancestral exposure to BPA (0.5, 20, and 50 μg/kg/day) dysregulates gene expression of ovarian apoptotic factors, oxidative stress factors, and autophagy factors in mice [98]. Interestingly, some studies have linked BPA exposure to DNA methylation across generations in females [89, 99]. Specifically perinatal exposure to BPA (10 mg/kg/BW/day) altered DNA methylation at a differentially methylated region that regulates expression of Igf2 gene in F1 and F2 generations; however, this was found only in male mice [99]. Another study demonstrated that prenatal exposure to a mixture of BPA and phthalates (BPA 50 mg/kg BW/day, DEHP 750 mg/kg BW/day, and DBP 66 mg/kg/BW/day) promotes epigenetic transgenerational inheritance of disease. However, this study only showed that the plasticizer mixture affected the differentially methylated regions in sperm. Epigenetic analyses were not performed on the females [89]. Thus, future studies should examine the epigenetic mechanism underlying the toxic effects of BPA on the ovary using exposure levels that mimic human daily exposure (about 46.8 ng/kg/day) [100].
Vinclozolin, a fungicide used on fruits, acts as an endocrine disruptor and causes transgenerational effects [54, 101]. Prenatal vinclozolin exposure (100 mg/kg BW/day) decreases primordial follicle counts in both the F1 and F3 generations of rats at 1 year of age [101]. In addition, ancestral exposure to vinclozolin (100 mg/kg BW/day) causes small and large cysts in the ovaries and increases circulating androstenedione levels in the F3 generation [101]. The observed phenotype in the F3 generation is similar to the clinical phenotype in women with polycystic ovarian syndrome [101]. Further, vinclozolin causes differential gene expression in the F3 ovaries. These genes are associated with ovarian diseases such as polycystic ovarian syndrome. Ancestral vinclozolin exposure (100 mg/kg BW/day) also alters DNA methylated regions in promoter regions of the granulosa cells; however, the DNA methylation changes do not overlap with the promoters of the differential gene expression in the F3 generation [101]. Instead, the DNA methylation changes may influence distal gene expression through ncRNA, which may regulate the differential gene expression in the granulosa cells of the F3 ovaries [101]. Further investigations reveal that purified rat granulosa cells from the vinclozolin (100 mg/kg BW/day) exposed F3 generation have differentially expressed lncRNA and sncRNAs and that these changes contribute to the vinclozolin-induced dysregulation of the ovary [102]. Overall, these studies suggest that vinclozolin exposure (100 mg/kg BW/day) induces transgenerational epigenetic effects via ncRNA in the ovary. These studies provide a mechanistic approach to understanding vinclozolin exposure. However, the dose used in these studies is high compared to the estimated daily intake of vinclozolin, which is between 2.0–11.5 ng/kg/day [85]. Therefore, it is necessary for future studies to expand these transgenerational studies with additional epigenetic analyses using environmentally relevant levels of vinclozolin.
Epigenetic effects of endocrine disruptors on the uterus
The uterus is critical for fertility in females; it acts as an endocrine sensitive organ that facilitates both embryo implantation and parturition. Studies indicate that endocrine disruptors can affect the uterus and that these changes may lead to epigenetic and transgenerational inheritance of diseases. The prescription of DES to pregnant women is one of the best examples of multi- and transgenerational impact because it is associated with fetal endocrine disruption and adverse reproductive health outcomes in subsequent generations in humans [103]. Women who were exposed to DES as a fetus, also known as “DES daughters”, have more frequent benign reproductive tract problems, including reproductive organ dysfunction, abnormal pregnancies, structural changes of the uterus, and reduced fertility [80]. These women have an increased risk of a rare clear-cell cervicovaginal adenocarcinoma and squamous-cell and cervicovaginal carcinoma [104]. Further, these “DES daughters” report that their in utero exposure led to cancer in their daughters, the F2 generation [105]. Effects seen in this F2 generation demonstrate a multigenerational effect of prenatal DES exposure on the uterus in humans [105]. Animal studies further demonstrate multigenerational effects of DES. Specifically, prenatal DES exposure (2.5, 5, 10 μg/kg/day) decreases fertility in the F1 generation of female mice and increases the incidence of malignant reproductive tract tumors such as adenocarcinomas in the F2 generation of female mice [106]. Additional studies show that perinatal exposure to DES (1 μg/kg BW) increases the susceptibility of uterine developmental abnormalities and cancer in both the F1 and F2 generations of female mice [107, 108].
Currently, the mechanisms explaining the multigenerational effects of DES exposure on the uterus are not fully understood. However, studies in mice suggest epigenetic alterations in DNA methylation involving hormone responsive families of genes including lactoferrin, homeobox, wnt signaling pathway, and epidermal growth factor genes are involved with the reproductive tract developmental changes in a multigenerational manner [79, 109]. Another study demonstrates that neonatal DES exposure (1 mg/kg) alters the expression of chromatin-modifying proteins, DNA methylation mediators, and DNA methylation in the adult mouse uterus, causing persistently altered epigenetic marks [110]. Further, neonatal DES exposure decreases Dnmt gene expression and alters DNA methylation in the mouse uterus [111]. Although these epigenetic markers are observed from neonatal exposure within the same generation, these epigenetic changes may help contribute to the multigenerational effects of DES exposure. Although some of the epigenetic mechanisms underlying the effects of DES exposure on the F3 uterus have been identified, the epigenetic mechanisms underlying DES toxicity in the F3 generation and beyond are not understood.
Another endocrine disruptor that targets the uterus is 2,3,7,8-TCDD. TCDD is a byproduct of incomplete combustion of a variety of products such as fossil fuels, wood, and industrial wastes. Studies show that TCDD exposure causes uterine dysfunction across generations [112–116]. For example, perinatal TCDD (10 μg/kg) exposure causes endometriosis-like reproductive phenotypes in F1–F4 generations of female mice, indicating both multi- and transgenerational effects [112, 113]. Further, TCDD exposure (10 μg/kg) reduces uterine progesterone responsiveness and causes subfertility [112, 114]. TCDD exposure (10 μg/kg) increases both stromal cell and epithelial cell ERS2 protein expression in the F1–F3 generations and causes adenomyosis in the F3 generation [115]. Interestingly, ancestral exposure to TCDD (10 μg/kg) causes hypermethylation of Pgr, which is associated with the development of the endometriosis-like histological and functional phenotypes [116]. The studies above demonstrate that a single dose of TCDD at 10 μg/kg causes transgenerational inheritance of uterine endometriosis-like phenotypes and epigenetic modification of Pgr, suggesting that this may be a mechanism of action. The dose 10 μg/kg was used because it is not teratogenic or abortigenic, is below the LD50 for the strain, and is rapidly cleared in mice [116, 117]. However, the NHANES study shows that between 1999–2010, TCDD levels ranged between 0.4–12.1 pg/g lipid in human serum [96]. Thus, it may be likely that the 10 μg/kg dose used in previous studies is relatively high compared to human exposure. Therefore, even though previous studies provide insight on the mechanism of action of TCDD exposure, future studies should be conducted using lower doses of TCDD.
Transgenerational and multigenerational effects of endocrine disruptors on female reproductive outcomes
Plasticizers that act as endocrine disruptors interfere with female reproductive outcomes. Some of these reproductive outcomes are due to impacts on the hypothalamus, pituitary, ovary, and uterus. For example, prenatal BPA exposure (0.5, 20, and 50 μg/kg/day) reduces the ability of mice to maintain pregnancies in a multigenerational manner and it delays the onset of puberty and compromises the ability of mice to become pregnant in a transgenerational manner [118]. BPA exposure in the diet (5 mg/kg) has been shown to interfere with brain and ovarian functions in a transgenerational manner, likely contributing to altered reproductive outcomes [61, 118]. A study on Chinese textile workers shows that in utero serum DDE levels (15 μg/L) were associated with an early menarche in the offspring [119]. Both prenatal and ancestral exposure to a mixture of phthalates (21% DEHP, 35% DEP, 15% DBP, 8% DiBP, 5% BBzP, and 15% DiNP at 20 μg, 200 mg, and 500 mg/kg/day) cause pregnancy complications in the F2 and F3 generations of mice [86]. Further, ancestral exposure to a phthalate mixture (21% DEHP, 35% DEP, 15% DBP, 8% DiBP, 5% BBzP, and 15% DiNP at 500 mg/kg/day) reduces total litter size and the percentage of dams that produce live litters in the F3 generation [86], whereas exposure to DEHP, a single phthalate, causes multi- and transgenerational effects on reproductive outcomes [91, 120]. Specifically, prenatal DEHP exposure (500 mg/kg/day) increases litter size and decreases the percentage of dams that give birth in the F2 generation [120]. Ancestral DEHP exposure (500 mg/kg/day) accelerates the onset of puberty and reproductive senescence in the F3 generation of female mice [90, 120]. The ability of phthalates to interfere with ovarian functions may contribute to these altered reproductive outcomes [86, 90, 91, 120, 121].
Recently, a study was published on the reproductive and hormone-related outcomes in women over the age of 18 that were third generation exposed to DES. This study shows that in the third generation, women had an increased risk of irregular menses, amenorrhea, pre-term delivery, and ectopic pregnancies. These changes were more apparent in women whose mother was affected by DES-associated vaginal epithelial changes [122].
Summary/Conclusion
Overall, the literature shows that several endocrine disrupting chemicals cause reproductive dysfunction in females in a multigenerational and transgenerational manner and that some of these effects are due to epigenetic changes. Epidemiological data show that exposure to endocrine disruptors is associated with adverse ovarian and uterine health outcomes in women across generations [78, 105, 119]. Experimental data demonstrate that endocrine disruptors cause female reproductive abnormalities in the hypothalamus, ovary, and uterus in multigenerational and transgenerational manners [61, 82, 86, 90, 91, 106, 120, 123]. Generally, the consensus is that epigenetic changes are induced by chemical exposures and are inherited through the germline, thus causing transgenerational phenotypes in reproductive functions in the generation that was not directly exposed to the endocrine disruptor. However, it is critical that future studies continue to investigate the epigenetic basis of transgenerational inheritance and demonstrate that the epigenetic changes are inherited through the germline. Specifically, it would be beneficial to map a time course of endocrine disruptor exposure in the developing gonad and how the exposure directly influences the epigenetic machinery for each generation. Such information could provide insight into why some studies, but not all studies, observe effects of endocrine disruption on reproduction and the epigenome. Additionally, determining whether histone modifications at a certain chromosomal location and/or DNA methylation profiles are conserved throughout generations would provide potential targets of interest for future mammalian studies on environmental endocrine disruptors. Finally, it is imperative that future studies use doses that are environmentally relevant to accurately assess the reproductive and epigenetic effects on the body. Endocrine disruptors do not follow a monotonic dose response, and therefore different doses will produce different effects on the body. It is crucial to fill the gap in knowledge about how endocrine disruptors affect the epigenome so that potential interventions can be developed and used to stop endocrine disruption of female reproductive health.
Acknowledgment
We thank Catheryne (Katie) Chiang for her exemplary illustrations in this review.
Notes
Edited by Dr. Lane K. Christenson, PhD, University of Kansas Medical Center
Author Biographical
 Saniya Rattan recently received her PhD from the University of Illinois at Urbana-Champaign. She completed her dissertation work on the transgenerational effects of phthalate exposure on female reproduction in Dr. Jodi Flaws' laboratory. Saniya has published several manuscripts in the field of reproduction and developmental toxicology. Some of her major publications include a manuscript titled "Prenatal exposure to di(2-ethylhexyl) phthalate (DEHP) disrupts ovarian function in a transgenerational manner" in Biology of Reproduction, a manuscript titled "Di(2-ethylhexyl) phthalate exposure during prenatal development causes adverse transgenerational effects on female fertility in mice" in Toxicological Sciences, and an article titled "Exposure to endocrine disruptors during adulthood: Consequences for female fertility" in Journal of Endocrinology. Thus summer, Saniya will begin postdoctoral training in the laboratory of Dr. Humphrey Yao at the National Institute of Environmental Health Sciences.
Saniya Rattan recently received her PhD from the University of Illinois at Urbana-Champaign. She completed her dissertation work on the transgenerational effects of phthalate exposure on female reproduction in Dr. Jodi Flaws' laboratory. Saniya has published several manuscripts in the field of reproduction and developmental toxicology. Some of her major publications include a manuscript titled "Prenatal exposure to di(2-ethylhexyl) phthalate (DEHP) disrupts ovarian function in a transgenerational manner" in Biology of Reproduction, a manuscript titled "Di(2-ethylhexyl) phthalate exposure during prenatal development causes adverse transgenerational effects on female fertility in mice" in Toxicological Sciences, and an article titled "Exposure to endocrine disruptors during adulthood: Consequences for female fertility" in Journal of Endocrinology. Thus summer, Saniya will begin postdoctoral training in the laboratory of Dr. Humphrey Yao at the National Institute of Environmental Health Sciences.
 Dr. Jodi Flaws is a Professor in the Department of Comparative Biosciences in the College of Veterinary Medicine at the University of Illinois, Urbana-Champaign. The focus of her laboratory is to determine the environmental factors that affect the female reproductive system. Specifically, the research in Dr. Flaws' laboratory is designed to determine the effects of endocrine disrupting chemicals (phthalates, bisphenols, water disinfection by-products, and pesticides) on the ovary and female fertility using animal models and epidemiological approaches. Dr. Flaws and her trainees have published over 200 manuscripts on the impact of environmental chemicals on female reproduction, including several manuscripts in Biology of Reproduction.
Dr. Jodi Flaws is a Professor in the Department of Comparative Biosciences in the College of Veterinary Medicine at the University of Illinois, Urbana-Champaign. The focus of her laboratory is to determine the environmental factors that affect the female reproductive system. Specifically, the research in Dr. Flaws' laboratory is designed to determine the effects of endocrine disrupting chemicals (phthalates, bisphenols, water disinfection by-products, and pesticides) on the ovary and female fertility using animal models and epidemiological approaches. Dr. Flaws and her trainees have published over 200 manuscripts on the impact of environmental chemicals on female reproduction, including several manuscripts in Biology of Reproduction.
Footnotes
Grant Support: This work was supported by the Billie A. Field Fellowship in Reproductive Biology (SR), NIH P01 ES022848 (JAF), EPA RD83 543401 (JAF), NIH F31 ES030467 (SR), and NIH T32 ES007326 (SR).
References
- 1. Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, Toppari J, Zoeller RT. EDC-2: the Endocrine Society's second scientific statement on endocrine-disrupting chemicals. Endocr Rev 2015; 36(6):E1–E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr, Lee DH, Shioda T, Soto AM, vom Saal FS, Welshons WV, Zoeller RT, Myers JP. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev 2012; 33(3):378–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Russart KLG, Nelson RJ. Light at night as an environmental endocrine disruptor. Physiol Behav 2018; 190:82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gore AC, Crews D, Doan LL, Merrill ML, Patisaul H, Zota A. Introduction to endocrine disrupting chemicals (EDCs) – a guide for public interest organizations and policy-makers. Endocrine Society 2014. [Google Scholar]
- 5. Mostafalou S, Abdollahi M. Pesticides: an update of human exposure and toxicity. Arch Toxicol 2017; 91(2):549–599. [DOI] [PubMed] [Google Scholar]
- 6. Patel S, Zhou C, Rattan S, Flaws JA. Effects of endocrine-disrupting chemicals on the ovary1. Biol Reprod 2015; 93(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burke RD, odd SW, Lumsden E, Mullins RJ, Mamczarz J, Fawcett WP, Gullapalli RP, Randall WR, Pereira EFR, Albuquerque EX. Developmental neurotoxicity of the organophosphorus insecticide chlorpyrifos: from clinical findings to preclinical models and potential mechanisms. J Neurochem 2017; 142(Suppl 2):162–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dodson RE, Nishioka M, Standley LJ, Perovich LJ, Brody JG, Rudel RA. Endocrine disruptors and asthma-associated chemicals in consumer products. Environ Health Perspect 2012; 120(7):935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Long NE, Holloway AC. Early-life chemical exposures and risk of metabolic syndrome. DMSO 2017; 10:101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, Padmanabhan V, Taylor HS, Swan SH, VandeVoort CA, Flaws JA. Bisphenol A and reproductive health: update of experimental and human evidence. Environ Health Perspect 2014; 122(8):775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richter CA, Birnbaum LS, Farabollini F, Newbold RR, Rubin BS, Talsness CE, Vandenbergh JG, Walser-Kuntz DR, vom Saal FS. In vivo effects of bisphenol A in laboratory rodent studies. Reprod Toxicol 2007; 24(2):199–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rattan S, Zhou C, Chiang C, Mahalingam S, Brehm E, Flaws JA. Exposure to endocrine disruptors during adulthood: consequences for female fertility. J Endocrinol 2017; 233(3):R109–R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hannon PR, Flaws JA. The effects of phthalates on the ovary. Front Endocrinol 2015; 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schafer KS, Kegley SE. Persistent toxic chemicals in the US food supply. J Epidemiol Community Health 2002; 56(11):813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Craig ZR, Wang W, Flaws JA. Endocrine-disrupting chemicals in ovarian function: effects on steroidogenesis, metabolism and nuclear receptor signaling. Reproduction 2011; 142(5):633–646. [DOI] [PubMed] [Google Scholar]
- 16. Walker DM, Gore AC. Transgenerational neuroendocrine disruption of reproduction. Nat Rev Endocrinol 2011; 7(4):197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tavakoly Sany SB, Hashim R, Salleh A, Rezayi M, Karlen DJ, Razavizadeh BB, Abouzari-Lotf E. Dioxin risk assessment: mechanisms of action and possible toxicity in human health. Environ Sci Pollut Res 2015; 22(24):19434–19450. [DOI] [PubMed] [Google Scholar]
- 18. Conolly RB, Lutz WK. Nonmonotonic dose-response relationships: mechanistic basis, kinetic modeling, and implications for risk assessment. Toxicol Sci 2004; 77(1):151–157. [DOI] [PubMed] [Google Scholar]
- 19. Welshons WV, Thayer KA, Judy BM, Taylor JA, Curran EM, vom Saal FS. Large effects from small exposures. I. Mechanisms for endocrine-disrupting chemicals with estrogenic activity. Environ Health Perspect 2003; 111(8):994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katzenellenbogen BS, Montano MM, Ediger TR, Sun J, Ekena K, Lazennec G, Martini PG, McInerney EM, Delage-Mourroux R, Weis K, Katzenellenbogen JA. Estrogen receptors: selective ligands, partners, and distinctive pharmacology. Recent Prog Horm Res 2000; 55:163–193; discussion 194–5. [PubMed] [Google Scholar]
- 21. Ismail A, Nawaz Z. Nuclear hormone receptor degradation and gene transcription: an update. IUBMB Life 2005; 57(7):483–490. [DOI] [PubMed] [Google Scholar]
- 22. Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog Horm Res 1996; 51:319–351; discussion 352–3. [PubMed] [Google Scholar]
- 23. McKenna NJ. Chapter 9 - Gonadal Steroid Action. In: Knobil and Neill's Physiology of Reproduction (Fourth Edition). San Diego: Academic Press; 2015:313–333. [Google Scholar]
- 24. Vandenberg LN. Non-monotonic dose responses in studies of endocrine disrupting chemicals: bisphenol a as a case study. Dose-Response 2014; 12(2):259–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hossein Rashidi B, Amanlou M, Behrouzi Lak T, Ghazizadeh M, Haghollahi F, Bagheri M, Eslami B. The association between bisphenol A and polycystic ovarian syndrome: a case-control study. Acta Med Iran 2017; 55(12):759–764. [PubMed] [Google Scholar]
- 26. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343(2):78–85. [DOI] [PubMed] [Google Scholar]
- 27. WHO Global status report on noncommunicable diseases - 2014. W.H. Organization (ed.), Geneva, Switzerland: WHO Press; 2014:1–298. [Google Scholar]
- 28. Heindel JJ, Vandenberg LN. Developmental origins of health and disease. Curr Opin Pediatr 2015; 27(2):248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gluckman PD, Buklijas T, Hanson MA. Chapter 1 - The Developmental Origins of Health and Disease (DOHaD) Concept: Past, Present, and Future. In: Rosenfeld CS. (ed.), The Epigenome and Developmental Origins of Health and Disease. Boston: Academic Press; 2016:1–15. [Google Scholar]
- 30. Uzumcu M, Zama AM. Chapter 9 - Developmental Effects of Endocrine-Disrupting Chemicals in the Ovary and on Female Fertility. In: Rosenfeld CS. (ed.), The Epigenome and Developmental Origins of Health and Disease. Boston: Academic Press; 2016:143–170. [Google Scholar]
- 31. Hong H, Branham WS, Dial SL, Moland CL, Fang H, Shen J, Perkins R, Sheehan D, Tong W. Rat alpha-Fetoprotein binding affinities of a large set of structurally diverse chemicals elucidated the relationships between structures and binding affinities. Chem Res Toxicol 2012; 25(11):2553–2566. [DOI] [PubMed] [Google Scholar]
- 32. Milligan SR, Khan O, Nash M. Competitive binding of xenobiotic oestrogens to rat alpha-fetoprotein and to sex steroid binding proteins in human and rainbow trout (Oncorhynchus mykiss) plasma. Gen Comp Endocrinol 1998; 112(1):89–95. [DOI] [PubMed] [Google Scholar]
- 33. Sheehan DM, Young M. Diethylstilbestrol and estradiol binding to serum albumin and pregnancy plasma of rat and human. Endocrinology 1979; 104(5):1442–1446. [DOI] [PubMed] [Google Scholar]
- 34. Hanson MA, Skinner MK. Developmental origins of epigenetic transgenerational inheritance. Environ Epigenet 2016; 2(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skinner MK. What is an epigenetic transgenerational phenotype? Reprod Toxicol 2008; 25(1):2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skinner MK. Epigenetic transgenerational inheritance. Nat Rev Endocrinol 2016; 12(2):68–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rattan S, Chiang C, Brehm E, Flaws JA. What are the Possible Consequences of Pre-conception Female Germ Cell Exposures on Fertility and Pregnancy Outcomes? In: Hales B, Scialli A, Tassinari M (eds.), Teratology Primer. The Teratology Society; 2018. [Google Scholar]
- 38. Nilsson EE, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of reproductive disease. Biol Reprod 2015; 93(6):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab 2010; 21(4):214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Skinner MK. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 2011; 6(7):838–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rubin H. Etymology of epigenetics. Science 2001; 294(5551):2477c–2478. [DOI] [PubMed] [Google Scholar]
- 42. Zama AM, Uzumcu M. Epigenetic effects of endocrine-disrupting chemicals on female reproduction: an ovarian perspective. Front Neuroendocrinol 2010; 31(4):420–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. WHO/UNEP State of the science of endocrine disrupting chemicals - 2012. W. H. Organization (ed.), Geneva, Swizterland: WHO Press; 2012:1–296. [Google Scholar]
- 44. McCarthy MM, Rissman EF. Chapter 52 - Epigenetics of Reproduction. In: Knobil and Neill's Physiology of Reproduction (Fourth Edition). San Diego: Academic Press; 2015:2439–2501. [Google Scholar]
- 45. Sales VM, Ferguson-Smith AC, Patti ME. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab 2017; 25(3):559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord 2007; 8(2):173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Long MD, Smiraglia DJ, Campbell MJ. The genomic impact of DNA CpG methylation on gene expression; relationships in prostate cancer. Biomolecules 2017; 7(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403(6765):41–45. [DOI] [PubMed] [Google Scholar]
- 49. Forrest ME, Khalil AM. Review: regulation of the cancer epigenome by long non-coding RNAs. Cancer Lett 2017; 407:106–112. [DOI] [PubMed] [Google Scholar]
- 50. Chaumeil J, Le Baccon P, Wutz A, Heard E. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 2006; 20(16):2223–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Merry CR, Forrest ME, Sabers JN, Beard L, Gao XH, Hatzoglou M, Jackson MW, Wang Z, Markowitz SD, Khalil AM. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum Mol Genet 2015; 24(21):6240–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014; 505(7483):344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kartha RV, Subramanian S. Competing endogenous RNAs (ceRNAs): new entrants to the intricacies of gene regulation. Front Genet 2014; 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology 2006; 147(6):s43–s49. [DOI] [PubMed] [Google Scholar]
- 55. Cabot B, Cabot RA. Chromatin remodeling in mammalian embryos. Reproduction 2018; 155(3):R147–R158. [DOI] [PubMed] [Google Scholar]
- 56. Meller VH, Joshi SS, Deshpande N. Modulation of chromatin by noncoding RNA. Annu Rev Genet 2015; 49(1):673–695. [DOI] [PubMed] [Google Scholar]
- 57. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology 2013; 38(1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ramassone A, Pagotto S, Veronese A, Visone R. Epigenetics and microRNAs in cancer. Int J Mol Sci 2018; 19(2):459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ross PJ, Canovas S. Mechanisms of epigenetic remodelling during preimplantation development. Reprod Fertil Dev 2016; 28(2):25–40. [DOI] [PubMed] [Google Scholar]
- 60. Goldsby JA, Wolstenholme JT, Rissman EF. Multi- and transgenerational consequences of bisphenol A on sexually dimorphic cell populations in mouse brain. Endocrinology 2017; 158(1):21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Drobna Z, Henriksen AD, Wolstenholme JT, Montiel C, Lambeth PS, Shang S, Harris EP, Zhou C, Flaws JA, Adli M, Rissman EF. Transgenerational effects of bisphenol A on gene expression and DNA methylation of imprinted genes in brain. Endocrinology 2018; 159(1):132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tao YH, Sharif N, Zeng BH, Cai YY, Guo YX. Lateral ventricle injection of orexin-A ameliorates central precocious puberty in rat via inhibiting the expression of MEG3. Int J Clin Exp Pathol 2015; 8(10):12564–12570. [PMC free article] [PubMed] [Google Scholar]
- 63. Supornsilchai V, Jantarat C, Nosoognoen W, Pornkunwilai S, Wacharasindhu S, Soder O. Increased levels of bisphenol A (BPA) in Thai girls with precocious puberty. J Pediatr Endocrinol Metab 2016; 29(11):1233–1239. [DOI] [PubMed] [Google Scholar]
- 64. Yang F, Chen LQ, Jin MF, Zhou WW, Wu HY. Impact of neonatal exposure to different doses of bisphenol A on puberty in female rats. Zhongguo Dang Dai Er Ke Za Zhi 2014; 16(7):754–758. [PubMed] [Google Scholar]
- 65. Wolstenholme JT, Taylor JA, Shetty SR, Edwards M, Connelly JJ, Rissman EF. Gestational exposure to low dose bisphenol A alters social behavior in juvenile mice. PLoS One 2011; 6(9):e25448–e25448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect 2002; 110(11):a703–a707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hannon PR, Brannick KE, Wang W, Flaws JA. Mono(2-ethylhexyl) phthalate accelerates early folliculogenesis and inhibits steroidogenesis in cultured mouse whole ovaries and antral follicles1. Biol Reprod 2015; 92(5):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hannon PR, Niermann S, Flaws JA. Acute exposure to di(2-ethylhexyl) phthalate in adulthood causes adverse reproductive outcomes later in life and accelerates reproductive aging in female mice. Toxicol Sci 2016; 150(1):97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gore AC, Hall JE, Hayes FJ. Chapter 37 - Aging and Reproduction. In: Knobil and Neill's Physiology of Reproduction (Fourth Edition). San Diego: Academic Press; 2015:1661–1693. [Google Scholar]
- 70. Pangas SA, Rajkovic A. Chapter 21 - Follicular Development: Mouse, Sheep, and Human Models. In: Knobil and Neill's Physiology of Reproduction (Fourth Edition). San Diego: Academic Press; 2015:947–995. [Google Scholar]
- 71. Cauley JA. Estrogen and bone health in men and women. Steroids 2015; 99(Pt A):11–15. [DOI] [PubMed] [Google Scholar]
- 72. Szamatowicz M. How can gynaecologists cope with the silent killer - osteoporosis? Prz Menopauzalny 2016; 15(4):189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cramer DW, Vitonis AF. Signatures of reproductive events on blood counts and biomarkers of inflammation: Implications for chronic disease risk. PLoS One 2017; 12(2):e0172530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Davis GK, Newsome AD, Ojeda NB, Alexander BT. Effects of intrauterine growth restriction and female sex on future blood pressure and cardiovascular disease. Curr Hypertens Rep 2017; 19(2):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lindsay R. Hormones and bone health in postmenopausal women. ENDO 2004; 24(3):223–230. [DOI] [PubMed] [Google Scholar]
- 76. Richards JS, Liu Z, Shimada M. Chapter 22 - Ovulation. In: Knobil and Neill's Physiology of Reproduction (Fourth Edition). San Diego; Academic Press; 2015:997–1021. [Google Scholar]
- 77. Wierman ME. Sex steroid effects at target tissues: mechanisms of action. Adv Physiol Educ 2007; 31(1):26–33. [DOI] [PubMed] [Google Scholar]
- 78. Blatt J, Van Le L, Weiner T, Sailer S. Ovarian carcinoma in an adolescent with transgenerational exposure to diethylstilbestrol. J Pediatr Hematol Oncol 2003; 25(8):635–636. [DOI] [PubMed] [Google Scholar]
- 79. Titus-Ernstoff L, Troisi R, Hatch EE, Hyer M, Wise LA, Palmer JR, Kaufman R, Adam E, Noller K, Herbst AL, Strohsnitter W, Cole BF et al.. Offspring of women exposed in utero to diethylstilbestrol (DES): a preliminary report of benign and malignant pathology in the third generation. Epidemiology 2008; 19(2):251–257. [DOI] [PubMed] [Google Scholar]
- 80. Kaufman RH, Adam E. Findings in female offspring of women exposed in utero to diethylstilbestrol. Obstet Gynecol 2002; 99(2):197–200. [DOI] [PubMed] [Google Scholar]
- 81. Tournaire M, Epelboin S, Devouche E, Viot G, Le Bidois J, Cabau A, Dunbavand A, Levadou A. Adverse health effects in children of women exposed in utero to diethylstilbestrol (DES). Therapies 2016; 71(4):395–404. [DOI] [PubMed] [Google Scholar]
- 82. Manikkam M, Aque MM, Guerrero-Bosagna C, Nilsson EE, Skinner MK. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult-onset disease through the female germline. PLoS One 2014; 9(7):e102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zama AM, Uzumcu M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology 2009; 150(10):4681–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Uzumcu M, Zama AM, Oruc E. Epigenetic mechanisms in the actions of endocrine-disrupting chemicals: gonadal effects and role in female reproduction. Reprod Domest Anim 2012; 47(Suppl 4):338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gunderson EL. FDA total diet study, July 1986-April 1991, dietary intakes of pesticides, selected elements, and other chemicals. J AOAC Int 1995; 78(6):1353–1363. [PubMed] [Google Scholar]
- 86. Zhou C, Gao L, Flaws JA. Exposure to an environmentally relevant phthalate mixture causes transgenerational effects on female reproduction in mice. Endocrinology 2017; 158(6):1739–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhou C, Flaws JA. Effects of an environmentally relevant phthalate mixture on cultured mouse antral follicles. Toxicol Sci 2017; 156:217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yazdy MM, Coull BA, Gardiner JC, Aguiar A. A possible approach to improving the reproducibility of urinary concentrations of phthalate metabolites and phenols during pregnancy. J Expo Sci Environ Epidemiol 2018; 28(5):448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PLoS One 2013; 8(1):e55387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Brehm E, Rattan S, Gao L, Flaws JA. Prenatal exposure to Di(2-ethylhexyl) phthalate causes long-term transgenerational effects on female reproduction in mice. Endocrinology 2018; 159(2):795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rattan S, Brehm E, Gao L, Niermann S, Flaws JA. Prenatal exposure to di(2-ethylhexyl) phthalate disrupts ovarian function in a transgenerational manner in female mice. Biol Reprod 2018; 98(1):130–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kawano M, Qin XY, Yoshida M, Fukuda T, Nansai H, Hayashi Y, Nakajima T, Sone H. Peroxisome proliferator-activated receptor alpha mediates di-(2-ethylhexyl) phthalate transgenerational repression of ovarian Esr1 expression in female mice. Toxicol Lett 2014; 228(3):235–240. [DOI] [PubMed] [Google Scholar]
- 93. Latini G, Scoditti E, Verrotti A, De Felice C, Massaro M. Peroxisome proliferator-activated receptors as mediators of phthalate-induced effects in the male and female reproductive tract: epidemiological and experimental evidence. PPAR Res 2008; 2008:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lovekamp-Swan T, Davis BJ. Mechanisms of phthalate ester toxicity in the female reproductive system. Environ Health Perspect 2003; 111(2):139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu C, Chen JA, Qiu Z, Zhao Q, Luo J, Yang L, Zeng H, Huang Y, Zhang L, Cao J, Shu W. Ovotoxicity and PPAR-mediated aromatase downregulation in female Sprague-Dawley rats following combined oral exposure to benzo[a]pyrene and di-(2-ethylhexyl) phthalate. Toxicol Lett 2010; 199(3):323–332. [DOI] [PubMed] [Google Scholar]
- 96. Fourth Report on Human Exposure to Environmental Chemicals, Updated Tables, (January 2019) [cited 2019 April 17] 2019; Available from: https://www.cdc.gov/exposurereport/. [Google Scholar]
- 97. Mahalingam S, Ther L, Gao L, Wang W, Ziv-Gal A, Flaws JA. The effects of in utero bisphenol A exposure on ovarian follicle numbers and steroidogenesis in the F1 and F2 generations of mice. Reprod Toxicol 2017; 74:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Berger A, Ziv-Gal A, Cudiamat J, Wang W, Zhou C, Flaws JA. The effects of in utero bisphenol A exposure on the ovaries in multiple generations of mice. Reprod Toxicol 2016; 60:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Susiarjo M, Xin F, Bansal A, Stefaniak M, Li C, Simmons RA, Bartolomei MS. Bisphenol A exposure disrupts metabolic health across multiple generations in the mouse. Endocrinology 2015; 156(6):2049–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lakind JS, Naiman DQ. Bisphenol A (BPA) daily intakes in the United States: estimates from the 2003–2004 NHANES urinary BPA data. J Expo Sci Environ Epidemiol 2008; 18(6):608–615. [DOI] [PubMed] [Google Scholar]
- 101. Nilsson E, Larsen G, Manikkam M, Guerrero-Bosagna C, Savenkova MI, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PLoS One 2012; 7(5):e36129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nilsson E, Klukovich R, Sadler-Riggleman I, Beck D, Xie Y, Yan W, Skinner MK. Environmental toxicant induced epigenetic transgenerational inheritance of ovarian pathology and granulosa cell epigenome and transcriptome alterations: ancestral origins of polycystic ovarian syndrome and primary ovarian insufiency. Epigenetics 2018;13:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Newbold RR, Jefferson WN, Grissom SF, Padilla-Banks E, Snyder RJ, Lobenhofer EK. Developmental exposure to diethylstilbestrol alters uterine gene expression that may be associated with uterine neoplasia later in life. Mol Carcinog 2007; 46(9):783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med 1995; 122(10):778–788. [DOI] [PubMed] [Google Scholar]
- 105. Mascaro ML. Preconception tort liability: recognizing a strict liability cause of action for DES grandchildren. Am J Law Med 1991; 17(4):435–455. [PubMed] [Google Scholar]
- 106. Newbold RR, Hanson RB, Jefferson WN, Bullock BC, Haseman J, McLachlan JA. Increased tumors but uncompromised fertility in the female descendants of mice exposed developmentally to diethylstilbestrol. Carcinogenesis 1998; 19(9):1655–1663. [DOI] [PubMed] [Google Scholar]
- 107. Walker BE, Haven MI. Intensity of multigenerational carcinogenesis from diethylstilbestrol in mice. Carcinogenesis 1997; 18(4):791–793. [DOI] [PubMed] [Google Scholar]
- 108. Ruden DM, Xiao L, Garfinkel MD, Lu X. Hsp90 and environmental impacts on epigenetic states: a model for the trans-generational effects of diethylstibesterol on uterine development and cancer. Hum Mol Genet 2005; 14(suppl_1):R149–R155. [DOI] [PubMed] [Google Scholar]
- 109. Newbold RR, Padilla-Banks E, Jefferson WN. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology 2006; 147(6):s11–s17. [DOI] [PubMed] [Google Scholar]
- 110. Jefferson WN, Chevalier DM, Phelps JY, Cantor AM, Padilla-Banks E, Newbold RR, Archer TK, Kinyamu HK, Williams CJ. Persistently altered epigenetic marks in the mouse uterus after neonatal estrogen exposure. Mol Endocrinol 2013; 27(10):1666–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sato K, Fukata H, Kogo Y, Ohgane J, Shiota K, Mori C. Neonatal exposure to diethylstilbestrol alters expression of DNA methyltransferases and methylation of genomic DNA in the mouse uterus. Endocr J 2009; 56(1):131–139. [DOI] [PubMed] [Google Scholar]
- 112. Bruner-Tran KL, Gnecco J, Ding T, Glore DR, Pensabene V, Osteen KG. Exposure to the environmental endocrine disruptor TCDD and human reproductive dysfunction: translating lessons from murine models. Reprod Toxicol 2017; 68:59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bruner-Tran KL, Osteen KG. Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reprod Toxicol 2011; 31(3):344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ding T, McConaha M, Boyd KL, Osteen KG, Bruner-Tran KL. Developmental dioxin exposure of either parent is associated with an increased risk of preterm birth in adult mice. Reprod Toxicol 2011; 31(3):351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bruner-Tran KL, Duleba AJ, Taylor HS, Osteen KG. Developmental toxicant exposure is associated with transgenerational adenomyosis in a murine model. Biol Reprod 2016; 95(4):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Bruner-Tran KL, Resuehr D, Ding T, Lucas JA, Osteen KG. The role of endocrine disruptors in the epigenetics of reproductive disease and dysfunction: potential relevance to humans. Curr Obstet Gynecol Rep 2012; 1(3):116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vogel CF, Matsumura F. Interaction of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) with induced adipocyte differentiation in mouse embryonic fibroblasts (MEFs) involves tyrosine kinase c-Src. Biochem Pharmacol 2003; 66(7):1231–1244. [DOI] [PubMed] [Google Scholar]
- 118. Ziv-Gal A, Wang W, Zhou C, Flaws JA. The effects of in utero bisphenol A exposure on reproductive capacity in several generations of mice. Toxicol Appl Pharmacol 2015; 284(3):354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Vasiliu O, Muttineni J, Karmaus W. In utero exposure to organochlorines and age at menarche. Hum Reprod 2004; 19(7):1506–1512. [DOI] [PubMed] [Google Scholar]
- 120. Rattan S, Brehm E, Gao L, Flaws JA. Di(2-ethylhexyl) phthalate exposure during prenatal development causes adverse transgenerational effects on female fertility in mice. Toxicol Sci 2018; 163(2):420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhou C, Gao L, Flaws JA. Prenatal exposure to an environmentally relevant phthalate mixture disrupts reproduction in F1 female mice. Toxicol Appl Pharmacol 2017; 318:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Titus L, Hatch EE, Drake KM, Parker SE, Hyer M, Palmer JR, Strohsnitter WC, Adam E, Herbst AL, Huo D, Hoover RN, Troisi R. Reproductive and hormone-related outcomes in women whose mothers were exposed in utero to diethylstilbestrol (DES): a report from the US National Cancer Institute DES third generation study. Reprod Toxicol 2019; 84:32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhou C, Gao L, Flaws JA. Prenatal exposure to an environmentally relevant phthalate mixture disrupts reproduction in F1 female mice. Toxicol Appl Pharmacol 2017; 318:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]