

Abstract
Oxytetracycline is an antibiotic for the treatment of the infections caused by Gram-positive and Gram-negative microorganisms. Among novel formulations applied for damaged skin, hydrogels have shown to be superior as they can provide a moist environment for the wound. The purpose of this study was to prepare and evaluate the hydrogels of oxytetracycline consisted of polyvinyl alcohol (PVA) and chitosan polymers. A study design based on 4 factors and 3 levels was used for the preparation and evaluation of hydrogels formed by freeze-thaw (F-T) cycle using PVA and chitosan as a matrix-based wound dressing system. Furthermore, an experimental design was employed in order to study the effect of independent variables, namely drug amount (X1, 500-1000 mg), the amount of PVA (X2, 3.33-7.5%), the amount of chitosan (X3, 0.5-1%), and F-T cycle (X4, 3-7 cycles) on the dependent variables, including encapsulation efficiency, swelling index, adsorption of protein onto hydrogel surface, and skin permeation. The interaction of formulation variables had a significant effect on both physicochemical properties and permeation. Hydrogel microbial tests with sequential dilution method in Muller-Hinton broth medium were also carried out. The selected hydrogel (F6) containing 5% PVA, 0.75% chitosan, 1000 mg drug, and 3 F-T cycles was found to have increased encapsulation efficiency, gel strength, and higher skin permeation suitable for faster healing of wounds. Results showed the biological stability of oxytetracycline HCl in the hydrogel formulation with a lower dilution of the pure drug. Thus, oxytetracycline-loaded hydrogel could be a potential candidate to be used as a wound dressing system.
Keywords: Chitosan, Hydrogel, Oxytetracycline, Polyvinyl alcohol, Skin
INTRODUCTION
Hydrogels are three dimensional, hydrophilic, and polymeric lattice which are appropriate for absorbing large amounts of water or biological liquids (1). Due to their high water content, porosity, and soft consistency, hydrogels closely simulate natural living tissue more than any other type of synthetic biomaterials (2). Hydrogels are also partly deformable and may adapt to the shape of the surface to which they are applied. Hydrogels can be chemically stable or they can degrade and finally disintegrate and dissolve (3). Their highly spongy structure may simply be tuned by regulating the density of cross-links in the gel matrix and the tendency of the hydrogels for the humid environment in which they are swollen (4). Their porosity also allows entrapment of drugs within the gel matrix and further drug release at a rate associated with the diffusion coefficient of a small molecule or a macromolecule by the gel network (5). Hydrogels are also usually highly biocompatible, which can be related to the high amounts of water in patches.
Biodegradability or dissolution in case of patches may be obtained by enzymatic, hydrolytic, or environmental (e.g. pH, temperature, or electric field) pathways. Depending on the state of hydration of the tissue, hydrogels can absorb water from or donate it to the wound environment (6). Hydrogels leave no residue, are malleable, and improve re-epithelialization of wounds (7). Moist wound bed is widely accepted as an ideal environment for effective wound healing (8). Many clinical studies have testified the facilities of moist wound healing and shown that hydrogel dressings lead to faster healing, less pain, and cheaper cost when compared to saline dressings (9). Many hydrogels are prepared by physical methods (e.g. repeated freezing and thawing), chemical methods using a covalent cross-linking agent, or radiation methods (10). In applying these freeze-thawed gels for many pharmaceutical and medical purposes, it is important to characterize the stability of the system over long-time periods. Many of the difficulties related to polyvinyl alcohol (PVA) gels, manufactured by freeze/thaw (F-T) techniques, consist of the dissolution of PVA chains, melting out of crystallites, and additional crystallization over-long time periods, or secondary crystallization. These problems can considerably change the behavior of the gels over time and therefore they require to be taken into account for any long-term utilization. Peppas et al. (10) reported the crystalline nature of freeze/thawed gels during swelling. A primary reduction in the degree of crystallinity was considered as smaller crystalline regions melted out. This step was pursued by an increase in the crystallinity over longer time periods. This increase was related to additional crystallite production, due to aging. Wan et al. (11) tested alteration in the crystalline structure of freeze/thawed PVA gels over time. They considered the syneresis or solvent extraction due to an increase in the crystallinity levels. Freeze/thawing is one of the physical hydrogel production methods in which repeated freezing and thawing cycles result in higher degrees of crystallization of PVA chains and production of a highly elastic hydrogel. As the number of cycles increases, the percentage of crystalline regions within the hydrogel also increases, resulting in a stiffer structure (12). Hydrogels produced by this method have been shown to have mechanical properties similar to those of the aortic root. The hydrogels produced by the F-T method were similar to human tissue that had anti-inflammatory effects (13). In this method, the chemical substance was not used as the cross-linking agent. The polymers in hydrogel made ice crystals by freezing. Ice crystals became coarser with the frequency of freezing times and caused the formation of the gel structure. At each thawing (melting of ice), the cavities inside the crystal structure were empty and filled with polymers (14).
Chitosan and PVA create a matrix-like nanocomposite that fills inside the crystals. This matrix lattice is formed on a large and uniform surface. The sequential freezing-thawing of PVA, in addition to the formation of the gel state, creates a good tensile strong effect on the dosage form, which can be due to the distribution of electric charge proportional to the crystal level.
Oxytetracycline is a product of the metabolism of Streptomyces rimosus and is one of the categories of tetracycline antibiotics. Oxytetracycline is firstly bacteriostatic and is explained to exert its antimicrobial effect by the prevention of the protein synthesis (15).
PVA is a semi-crystalline copolymer of vinyl acetate and vinyl alcohol that has been widely utilized in the chemical and medical industries because of its biocompatibility, non-toxicity, hydrophilicity, fiber/patch-forming ability, chemical resistance, and protein adsorption.
Chitosan has fungicide activity and is shown to elevate the function of leukocytes, macrophages, and fibroblasts for increasing granulation and rebuilding tissue. It is also biocompatible and nontoxic. Generally, it provides antifungal activity, enhances normal tissue rebirth, and regains the original tensile strength of the wound by expediting the fibroblast synthesis of collagen.
Response surface methodology (RSM) is a group of statistical methods for examining the effects of different independent variables (16).
The Box-Behnken designs include lesser design points and are cheaper compared to central composite designs with the similar number of factors. In the present study, we aimed to statistically study the expansion of an effectual oxytetracycline-loaded wound dressing with an increased healing effect. The cross-linked hydrogel was prepared with PVA and chitosan using the F-T method. Their gel properties such as gel fraction, swelling, water vapor transmission test, morphology, tensile strength, and thermal property were studied. In vitro protein adsorption test, in vivo wound healing test, and histological examination in rat was also performed. To this end, a 4-factor, 3-level Box-Behnken design was used to derive a polynomial equation and construct contour plots to predict responses.
MATERIALS AND METHODS
Materials
PVA (MW = 72000; 98-99% hydrolyzed), propylene glycol, calcium chloride anhydrous, glacial acetic acid, Mueller-Hinton agar, and Mueller-Hinton broth were purchased from Merck company (Merck Schuchardt OHG, Germany). Chitosan (medium grade) was purchased from Sigma-Aldrich Company (St. Louis, MO, USA). Oxytetracycline was kindly supplied from Pfizer Corporation (Brussels, Belgium). Total protein kit was purchased from Pars Azmoon (Pars Azmoon Inc., Tehran, I.R. Iran).
Preparation of hydrogel
PVA/chitosan hydrogels were prepared by the F-T cycle (17). The Box-Behnken design was applied for the preparation of oxytetracycline hydrogels. Four variables were investigated at three levels with three repeats at the central points. In brief, the solutions containing PVA (3.33, 5 and 7.5% w/v), chitosan (0.5, 0.75 and 1% w/v), and oxytetracycline (500, 750, and 1000 mg) were prepared in distilled water (Table 1). The aqueous solution of PVA and acidic solution of chitosan are mixed together and then the oxytetracycline powder was wetted into propylene glycol (5% w/w) and added to the aqueous mixture.
Table 1.
Compositions of polyvinyl alcohol and chitosan hydrogels with oxytetracycline hydrochloride.
| Formulations | Independent variables levels | |||
|---|---|---|---|---|
| X1 (mg) | X2 (% w/v) | X3 (% w/v) | X4 (n) | |
| F1 | 750 | 3.33 | 0.75 | 3 |
| F2 | 750 | 5 | 1 | 3 |
| F3 | 750 | 7.5 | 0.75 | 3 |
| F4 | 750 | 5 | 0.5 | 3 |
| F5 | 500 | 5 | 0.75 | 3 |
| F6 | 1000 | 5 | 0.75 | 3 |
| F7 | 750 | 7.5 | 0.5 | 5 |
| F8 | 750 | 3.33 | 0.5 | 5 |
| F9 | 750 | 7.5 | 1 | 5 |
| F10 | 500 | 3.33 | 0.75 | 5 |
| F11 | 1000 | 7.5 | 0.75 | 5 |
| F12 | 500 | 5 | 1 | 5 |
| F13 | 500 | 7.5 | 0.75 | 5 |
| F14 | 750 | 3.33 | 1 | 5 |
| F15 | 500 | 5 | 0.5 | 5 |
| F16 | 1000 | 3.33 | 0.75 | 5 |
| F17 | 1000 | 5 | 0.75 | 5 |
| F18 | 750 | 5 | 0.5 | 5 |
| F19 | 1000 | 3.33 | 0.75 | 7 |
| F20 | 750 | 5 | 0.75 | 5 |
| F21 | 750 | 5 | 1 | 7 |
| F22 | 500 | 5 | 0.75 | 7 |
| F23 | 750 | 5 | 0.5 | 7 |
| F24 | 1000 | 5 | 0.75 | 7 |
| F25 | 750 | 7.5 | 0.75 | 7 |
X1, drug amount; X2, amount of polyvinyl alcohol; X3, amount of chitosan; X4, freeze-thaw cycle.
The solutions with different proportions of PVA and chitosan were mixed by vortexing for 1 h and then poured into a glass plate. Afterward, they were frozen at -20 °C for 18 h and then thawed at room temperature for 6 h for 3 consecutive cycles.
Blank hydrogels were formulated according to the same procedure without oxytetracycline.
Hydrogel characterization
Morphological analysis
The morphology of surface and internal structure of oxytetracycline hydrogels were examined via a high-resolution scanning electron microscopy (SEM, MIRA3 TESCAN, Czech Republic). The specimens were removed and mounted on a metal stub using a double-sided carbon adhesive tape and coated with platinum/palladium alloy under vacuum.
Differential scanning calorimetry
The physical state of the drug in the patch was analyzed by a differential scanning calorimeter (Shimadzu, Japan). The thermograms were obtained at a scanning rate of 10 °C/min conducted within the temperature range of 25-300 °C and air atmosphere.
Determination of minimum inhibitory concentration and minimum bactericidal concentration
To determine the minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) of oxytetracycline, a suspension of Staphylococcus aureus with the turbidity equivalent to 0.5 McFarland standard was prepared and then diluted 1:100 with Mueller-Hinton broth medium (18).
Using the serial dilution method, 12 test tubes containing different drug concentrations were prepared, in such a way that the concentration in each tube was half of the concentration in the previous tube. Then, 1 mL of Staphylococcus aureus suspension was added to each of these tubes. The tubes were incubated at 37 °C for 24 h, following which their turbidity was examined against light according to the Clinical and Laboratory Standard Institute guidelines (19).
The lowest concentration at which the tube did not show turbidity was considered as MIC. To determine MBC, 20 μL of medium was taken from the tube considered the MIC and the next tubes and inoculated on Mueller-Hinton agar plates. The lowest concentration at which no colony growth was observed on the plate was considered as MBC.
Comparison of the antibacterial effects of different patches
According to the MBC, 5 μL of the microbial suspension (1:500 dilution of the 1 McFarland standard), inoculated on 1 × 1 cm2 cuts of the hydrogel patch was poured into the test tube (20). Then, 500 μL trypticase soy broth (TSB) was added to the patches. Immediately after adding TSB (0 times) and after 4 and 8 h, 50 μL of the medium was taken and inoculated into 20 mL Mueller- Hinton melt agar (Sigma; USA) medium using the pour plate technique. After 18-24 h of incubation at 37 °C, the colonies were counted.
Determination of gel fraction
After 3 F-T cycles, the obtained oxytetracycline hydrogel patches (2 ×2 cm2) were dried in a vacuum oven at 50 °C for 24 h and weighed (W0), then they were soaked in distilled water for 24 h up to an equilibrium swelling weight (Ws) for removing the leachable or soluble oxytetracycline parts from the patches. The hydrogel patches were then dried at 50 °C in a vacuum oven and weighed again (We). The gel fraction (GF%) was calculated by equation 1:
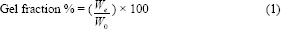
Determination of swelling behavior
In order to measure the swelling degree of oxytetracycline hydrogel, samples were cut into 2 × 2 cm2 pieces and dried at 50 °C in a vacuum oven for 6 h up to an equilibrium swelling weight, and the weight of the dried samples was determined (Wa). The dried samples were soaked in phosphate buffer saline (PBS, pH = 7.4), maintained and incubated at 37 °C, then weighted (Ws) at specific interval times. The water uptake of patches or swelling ratio was estimated by equation 2 (21):
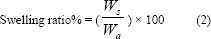
Study of protein adsorption onto hydrogel surface
Five pieces of each sample of hydrogel patches cut into 1 × 1 cm2 were immersed in 10 mL PBS (pH 7.4) and incubated at 37 °C for 24 h until they reached equilibrium swelling weight. Then, 100 μL protein (30 mg/mL solution) was poured into swollen hydrogel tubes and shaken for 4 h at 37 °C. After protein adsorption, the hydrogel pieces were gently removed. The protein adsorption of each sample was calculated by the difference between protein concentrations before and after immersing the hydrogel pieces in protein/PBS solution using total protein kit (Pars Azmoon, I.R. Iran) at 630 nm using a UV spectrophotometer (UV-Visible 160, Shimadzu, Japan).
Mechanical property measurements
After 3 F-T cycles, the maximum tensile strength and the elongation degree to break oxytetracycline hydrogel membranes was conducted using a tensile test machine SANTAM STM (BANG SHIN Company, DBBP-20 model, Korea). Hydrogel patches were cut into a specific dog-bone shape (6 cm long, 2 cm wide at the ends, and 1 cm at the middle). The thickness of membrane samples was measured with a digital vernier caliper (Mitutoyo, Japan) before the examination (22). The analysis was performed at stretching rate of 20 mm/min with pre-load of 1.3 ± 0.25 N to determine load for each sample (23). Ultimate tensile strength (UTS) and elongation at break (EB%) were estimated by equations 3 and 4:
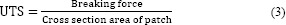

Water vapor transmission test
The water vapor transmission tests were performed using calcium chloride anhydrous desiccators. Five pieces of the hydrogel (2 × 2 cm2) were transferred on desiccators and placed in an incubator of 90% relative humidity at 40 °C (24). The water vapor transmission rate (WVTR) was determined by equation 5:
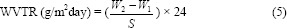
In this equation, W1 and W2 are the weights of the whole cup at the first and second hours, respectively, and S is the surface area of the water vapor transmission from the calcium chloride sample.
Permeation studies
The ex-vivo permeation study of the oxytetracycline hydrogel through rat abdominal skin was performed using Franz diffusion cell at 37 ± 0.2 °C. The freshly obtained skin was localized between the donor and receptor compartments in such a way that the skin faced the donor compartment. The patches were positioned on the skin and the compartments were clenched together. The donor compartment was filled with 0.5 mL PBS, pH 5.5. The receptor compartment was filled with 22-25 mL PBS, pH 5.5, and stirred with a magnetic bead at 700 rpm (25). Three mm of the sample were withdrawn at predetermined time intervals and analyzed for the drug at 275.6 nm.
In vivo wound healing test
Male rats weighing approximately 250-280 g were used to evaluate the in vivo wound healing ability of hydrogels. The present study was carried out according to the guidelines for the Care and Use of Laboratory Animals of Tabriz University of Medical Sciences, Tabriz, I.R. Iran (National Institutes of Health Publication No. 85-23, revised 1985).
The dorsal hair of each animal was shaved with an electric razor. After creating one wound area (2 × 2 cm2) by excising the dorsum, 70% ethanol was used for sterilization. Each wound was covered with sterile gauze (control), the hydrogel without the drug, and the hydrogel with the drug, respectively. All materials were fixed with an elastic adhesive bandage. All rats were separately kept in individual cages. At the 1st, 3rd, 6th, 9th, 12th, and 15th days after the operation, each wound size was measured using a digital camera.
Histological process
The wounded area of skin containing dermis and hypodermis was sampled and crossly trimmed. The tissue was fixed with 10% formalin, routinely processed, and embedded in paraffin. On glass slides, the sections were cut and stained with hematoxylin and eosin (26). A pathologist, blinded to the study, detected any damage to the tissue and examined the sections on the light microscope.
The experimental procedure of the hydrogels
RSM is known as a very useful statistical technique for the optimization of different pharmaceutical formulations. In this study, 4 factors were evaluated, each at 3 levels and experimental attempts were carried out at all 24 possible formulations. The amounts of drug (X1), the volume of PVA (X2), the amount of chitosan (X3), and times of F-T cycle were considered as independent variables. The weight (W), thickness (T), GF, swelling index (SI), folding times (F), absorbance protein (Pr), WVTR, production yield (PY), drug content (DC), and flux were attended as dependent variables (Table 2).
Table 2.
Effect of different levels of independent variables on the weight, thickness, gel fraction, swelling ratio, folding, absorbance protein, water vapor transmission rate, production, drug content and flux.
| F | Variable levels | W (g) | T (mm) | GF (%) | SI (%) | Pr (mg/mL) | F (n) | WVTR (%) | PY (%) | DC (%) | Flux (mg/cm2/min) * 10-5 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X1 | X2 | X3 | X4 | |||||||||||
| F6 | 1000 | 5 | 0.75 | 3 | 0.027 | 0.207 | 64 | 61. | 269.6 | > 300 | 0 | 103 | 20.52 | 2 |
| F7 | 750 | 7.5 | 0.5 | 5 | 0.018 | 0.195 | 57.9 | 61.2 | 439.1 | > 300 | 0 | 97 | 20.50 | 2 |
| F17 | 1000 | 5 | 1 | 5 | 0.036 | 0.257 | 72 | 58.7 | 173.9 | > 300 | 0 | 92.62 | 26 | 3 |
| F21 | 750 | 5 | 1 | 7 | 0.026 | 0.166 | 37.8 | 65.6 | 126.1 | > 300 | 0 | 81.60 | 17.04 | 2 |
X1, drug amount; X2, amount of polyvinyl alcohol; X3, amount of chitosan; X4, freeze-thaw cycle; W, Weight; T, thickness; GF, gel fraction; SI, swelling index; F, folding; Pr, absorbance of protein onto hydrogel surface; WVTR, water vapor transmission rate; PY, production; DC, drug content.
Regression analysis
RSM is applied as a suitable statistical technique helpful for improving processes. The widest applications of RSM are in conditions where various input variables potentially affect some performance measure or quality properties of the formulation or process. Box-Behnken designs are one category of the experimental designs for RSM. The number of experiments (N) needed for the improvement of Box-Behnken designs is explained as N = 2k (k-1) + C0, (where, k is the number of factors and C0 is the number of central points).
The Box-Behnken design was particularly chosen, because it needs fewer runs than a central composites design, in cases of four variables. A design matrix that consists of 25 experimental runs was created, for which the nonlinear computer-generated quadratic model is supported as:
Y = b0 + b1X1+ b2 X2 + b3 X3 + b11 X12 + b22 X22 + b33 X32 + b12 X1X2 + b13 X1X3 + b23 X2X3……………………(6)
In this equation, Y is the predicted response; b0 is the intercept; b1-b3 are linear effects; b12, b13, and b23 are interaction terms; X1, X2, and X3 are independent variables. The major effects (X1-X3) show the average result of altering one factor at a time from its low to high value. The interaction terms (X1X2 and X1X3) display how the response changes when three factors are altered together. The polynomial terms (X1X1, X2X2, and X3X3) are included to report nonlinearity. Three-dimensional surface (3D) plots were attracted to exhibit the major and interactive effects of the independent variables on W, T, GF, SI, folding times (F), absorbance protein (Pr), WVTR, PY, DC and flux. The optimum values of the chosen variables were provided from the software and also from the response surface plots.
Polynomial equations provided from the factorial design are applied to deduce that the coefficients of the independent variables and the mathematical (positive and negative) symbols are utilized. High correlation coefficients for dependent variables against independent variables showed that these two groups of dependent and independent variables are well-adjusted or not. Moreover, the equations are applied to approximate the variance of the errors caused by the repeat of the trials. The coefficients for independent variables (X1 to X3) are utilized to predict dependent variables. The correlation coefficients inferred from multiple linear regression analysis (such as response surface regression) exhibit the degree of arithmetic and dependence between two dependent and independent variables.
Statistical analysis
Where appropriate, results were evaluated using one-way ANOVA at the 0.05 level of significance. P < 0.05 and P > 0.05 were considered statistically significant and insignificant, respectively.
RESULTS
Hydrogels characteristics
According to the optical and SEM studies of the patches, it was observed that they appeared integrated, without any crack and fracture, without any symptom of drug migration on the surface of the patch, smooth, non-sticky, and yellow (Figs. 1-3).
Fig. 1.

Optical microscopic photograph of oxytetracycline hydrogel.
Fig. 3.

Scanning electron microscopy of F6 hydrogel with (A-D) different magnitudes; 2, 5, 20, and 50 μm, respectively.
The aluminum foil was applied for the patches prepared at all freezing and thawing temperatures to prevent the hydrolysis of the drug. No bubble was observed on the hydrogel surfaces, due to continuous pouring of hydrogel solution by syringes on a glass plate. Based on the results, the weight of hydrogels increased with the increase of the concentration of drug (X1, 750-1000 mg), PVA (X2, 5-7.5% w/w), chitosan (X3, 0.5-1% w/w), and F-T cycles (X4, 3-7). Besides, weight varied in the range of 0.018-0.036 g. The changes in the thickness of the patches (1 × 1 cm2) were in the range of 0.018-0.36 g and 0.66-1.26 mm, respectively. The folding values of formulations were higher than 300 times, which had the highest flexibility and tolerance to the breakage and rupture of the patches during contact with the hand (Table 2). The moisture absorption results showed that none of the hydrogels absorbed moisture. Furthermore, the WVTR indicated that the sufficient level of moisture was provided to prevent excessive dehydration of the hydrogel patches, leading to secretion on the wound area (Table 2). The SI of patches varied depending on the composition of patches. There was no significant difference in the hydrogels (Table 2). The actual drug loading in the hydrogels (F6, F7, F17, and F21) was in the range of 17.04-20%, indicating a uniform loading with a high degree of repeatability of the preparation of hydrogel patches. The drug loading efficiency was observed within the range of 83.83-104%, indicating a high uniformity of oxytetracycline distribution in the hydrogel patch form. The production of the formulations varied within the range of 81.60-103%, which indicated less wasting of the patches during the preparation of hydrogels (Table 2).
The effect of chitosan and drug is shown on the gel fraction in Table 2. In accordance with the regression analysis of the experimental data, the relationship of the independent variables on dependent variables (Y1-Y8) was shown by equations and summary of results of regression analysis for responses, respectively in (Tables 3, 4 and Fig. 4).
Table 3.
Equations response surface regression.
| The equations of regression analysis | |
|---|---|
| W = 0.073 - 0.093X1 - 0.076X2 - 0.0001X3 -0.063X4 + 0.079X1X1 + 0.00059 X2X2 - 0.03 X3X3 - 0.008X4X4 + 0.14X1X2 + 0.006X1X3 + 0.093X1X4 - 0.0002X2X3 + 0.004X2X4 + 0.003X3X4 | W vs X1-X4 |
| T = 0.19 + 0.025X1 + 0.007X2 - 0.01 X3 - 0.02X4 - 0.007X1X1 - 0.007X2X2 + 0.002X3X3 - 0.005 X4X4 - 0.009X1X2 - 0.023X1X3 + 0.02X1X4 - 0.009X2X3 - 0.002X2X4 - 0.001X3X4 | T vs X1-X4 |
| GF = 40.23 + 0.63X1 -2.40X2 -7.66X3 - 0.85X4 + 12.50X1X1 + 1.099X2X2 + 1.80X3X3 -0.81X4X4 + 9.75 X1X2 + 15.48X1X3 -3.50X1X4 - 4.38X2X3 + 1.32X2X4 - 1.55X3X4 | GF vs X1-X4 |
| SI = 39.63-3.27X1 - 2.24X2 - 2.42X3 -1.61X4 + 10.24X1X1 + 3.25X2X2 + 5.97X3X3 + 5.93X4X4 + 10.38X1X2 + 7.32X1X3-5.27X1X4 - 1.39X2X3 - 0.65X2X4 + 7.16X3X4 | SI vs X1-X4 |
| Pr = 141.48 - 15.24X1 + 0.66X2 - 40.31X3 - 31.06X4 + 41.51X1X1 + 45.64X2X2 + 30.57X3X3 - 10.38X4X4 + 50.28X1X2 + 29.43X1X3 - 39.75X1X4 - 71.08X2X3 - 0.92X2X4 + 5.27X3X4 | Pr vs X1-X4 |
| PY = 62.79 - 9.75X1 - 7.84X2 - 4.56X3 - 1.23X4 + 26.85X1X1 + 3.22X2X2 + 3.66X3X3 + 5.58X4X4 + 29.68X1X2 + 13.30X1X3 - 13.49X1X4 -7.71X2X3 - 0.11X2X4 + 4.15X3X4 | PY vs X1-X4 |
| DC = 10.85 + 1.008X1 + 0.21X2 - 1.84X3 + 0.26X4 + 7.87X1X1 + 0.88X2X2 + 1.95X3X3 + 0.46X4X4 + 1.26X1X2 + 4.00X1X3 - 0.76X1X4 - 1.68X2X3 + 0.024X2X4 + 0.51X3X4 | DC vs X1-X4 |
| Flux = 0.94-0.004X1 - 0.088X2 - 0.38X3 + 0.25X4 + 0.76X1X1 - 0.075X2X2 + 0.21X3X3 - 0.04X4X4 + 0.51X1X2 + 01.00X1X3 - 0.50X1X4 - 0.32X2X3 + 0.25X3X4 | Flux vs X1-X4 |
X1, drug amount; X2, amount of polyvinyl alcohol; X3, amount of chitosan; X4, freeze-thaw cycle.
Table 4.
Summary of results of regression analysis for responses Y1-Y10.
| Response/Coefficients | DF | SS | MS | F(cal) | R2 | P value | MPE (%) |
|---|---|---|---|---|---|---|---|
| Weight | 14 | 0.039 | 0.003 | 1.56 | 68.57 | 0.243 | 0.042 |
| Thickness | 14 | 0.006 | 0.0004 | 0.74 | 50.98 | 0.703 | 0.024 |
| Gel fraction | 14 | 862.93 | 61.64 | 1.56 | 68.65 | 0.241 | 6.28 |
| Swelling index | 14 | 918.20 | 65.59 | 3.22 | 63.51 | 0.371 | 7.26 |
| Absorbance of protein onto hydrogel surface | 14 | 79265 | 5661.78 | 1.57 | 68.77 | 0.238 | 60.00 |
| Production | 14 | 2690.82 | 192.202 | 1.92 | 72.89 | 0.151 | 10.00 |
| Drug content | 14 | 251.47 | 17.96 | 4.94 | 87.37 | 0.008 | 1.91 |
| Flux | 14 | 4.13 | 0.30 | 1.58 | 68.84 | 0.24 | 0.43 |
DF, Degrees of freedom; SS, sum of squares; MS, mean of squares; F(cal), degrees of freedom calculated; R2, coefficient of determination; MPE, mean percent regression.
Fig. 4.

Response contour plot showing the effect of (A and B) formulation variables (X2, polyvinyl alcohol; X3, chitosan; and X4, F-T cycles) on drug content and response surface plot showing the effect of (C and D) formulation variables (X1, drug; X3, chitosan; and X4, F-T cycles) on drug content. PVA, polyvinyl alcohol; F-T, freeze-thaw; DC, drug content.
Differential scanning calorimetry analysis
The oxytetracycline hydrochloride differential scanning calorimetric (DSC) thermograms had no sharp melting endotherms (Fig. 5). Thermogram of oxytetracycline hydrochloride exhibited one endothermic peak at 60 °C and an endothermic event at 202.25 °C and exothermic event at 217.60 °C, attributed to a fusion followed by thermal decomposition and oxidation of the evolved products. PVA thermogram exhibited relatively a large and sharp melting endothermic peak at 220 °C and a broad peak at 62.27 °C corresponds to melting temperature and glass transition temperature, respectively. Chitosan thermogram demonstrated a wide peak within the range of 75-95 °C and the peak of destruction at 209.32 °C. In all of the hydrogels (F6, F7, F17, and F21), a broad peak was observed at 69.90 °C, 77.25 °C, 75.03 °C, and 87.10 °C, respectively.
Fig. 5.
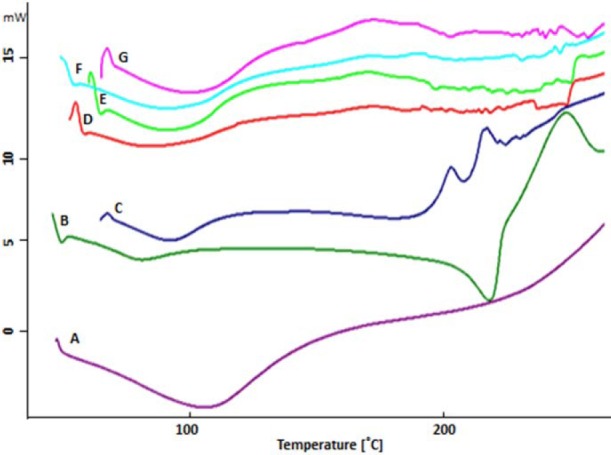
Differential scanning calorimeteric thermogram of (A) oxytetracycline, (B) polyvinyl alcohol, (C) chitosan, (D) hydrogels of F6, (E) F7, (F) F17, and (G) F21.
Mechanical characteristics
For a selected formulation such as F6 (containing PVA and chitosan, 5 and 0.75% w/v, respectively), the value of Young's modulus was at peak, 26.80 and break, 26.47, with a low value indicating the smooth of the surface of the hydrogel film (Table 5).
Table 5.
Results for the amount of force entered, tensile strength, elongation, elongation after break and modulus of elasticity of F6 hydrogel patch.
| Results | Force (N ± SD) | Extension (mm ± SD) | Stress (MPa ± SD) | Elongation (% ± SD) | Elongation after break | Module (MPa ± SD) |
|---|---|---|---|---|---|---|
| Peak | 1.30 ± 0.25 | 64.31 ± 16.08 | 1.30 ± 0.25 | 160.86 ± 40.25 | 34.97 ± 37.39 | 26.80 ± 44.97 |
| Break | 0.86 ± 0.58 | 67.39 ± 0.59 | 0.86 ± 0.58 | 129.81 ± 6.81 | 81.20 ± 73.23 | 26.47 ± 10.61 |
Microbial studies
The in vitro results of microbial culture and MIC for a hydrogel patch containing no drug against Staphylococcus aureus showed that the antimicrobial activity of oxytetracycline hydrochloride in the formed patch was reduced in comparison to the pure drug. MIC results showed that the pure drug and hydrogel were 64 and 128 μg/mL, respectively (Fig. 6) (P < 0.05).
Fig. 6.
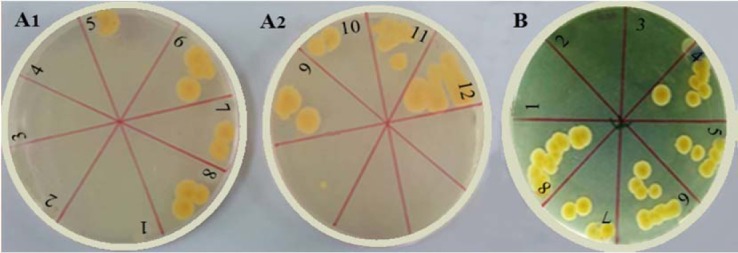
Linear culture of serial dilution tubes of (A1 and A2) oxytetracycline powder and (B) F6 hydrogel in minimum inhibitory concentration test. Serial dilutions: 1, 1 μM; 2, 2 μM; 3, 4 μM; 4, 8 μM; 5, 16 μM; 6, 32 μM; 7, 64 μM; 8, 128 μM, 9, 256 μM; 10, 512 μM; 11, 1024 μM; and 12, 2048 μM.
Permeation investigation
The highest amount of flux (the amount of drug permeated through the skin area per minute) in F6 and F7 formulations were shown to be exactly 2 × 10-5 mg /cm2/min (Table 2). In the graphs, the time of the latency was one h and after 240 min, they approached a linear state that indicated a state of equilibrium.
In vivo wound healing study
As shown in Fig. 7, it was clearly illustrated that after being treated with drug hydrogel, the wound healing rate was higher in the F6 hydrogel. Furthermore, wound healing was faster than treatment with blank hydrogel and without treatment.
Fig. 7.
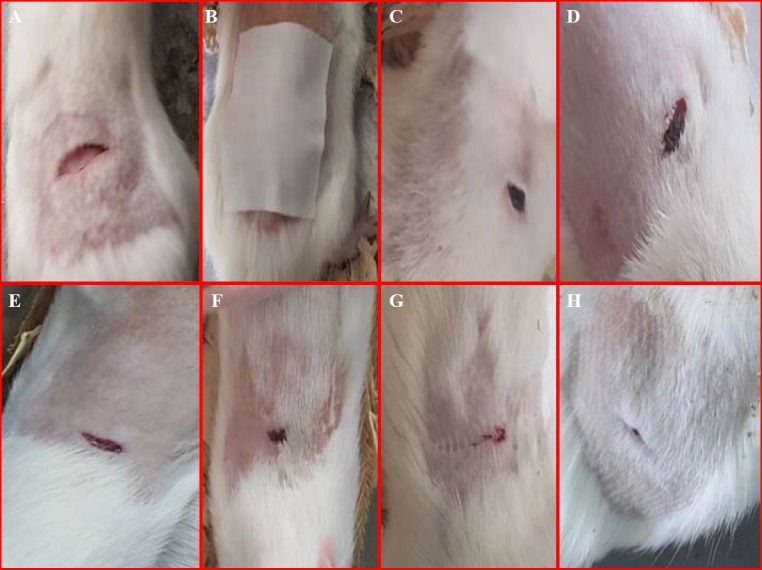
In vivo study of (A) wound appearance, (B) wound dressing, (C and D) control group (treated without drug), (E and F) blank group (treated with blank hydrogel), and (G and H) drug of group (treated with F6 hydrogel) in rats.
Histological study
Histological observations of the skin of the dorsal region of the rats showed that after the application of oxytetracycline hydrogel (for 1 week), the skin of the dorsal region of the rat was healed (Fig. 8).
Fig. 8.
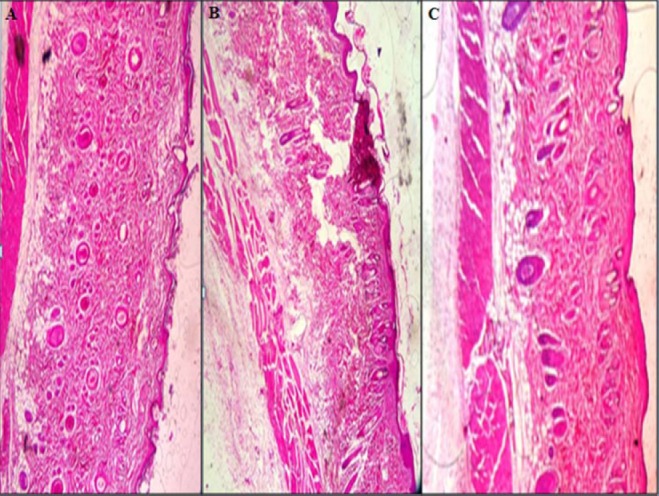
Histopathological evaluation of the abdominal skin of rat, (A) untreated, (B) treated with blank hydrogel, (C) and treated with oxytetracycline hydrogel (magnification × 10 ).
DISCUSSION
A transdermal patch or skin patch is a medicated adhesive patch that is placed on the skin to deliver a specific dose of medication through the skin, directly into the bloodstream. Skin patch uses a special membrane to control the rate at which the liquid drug encompassed in the reservoir and the patch can pass through the skin, into the bloodstream.
Transdermal formulation maintains drug concentration within the therapeutic window for a prolonged period of time and controls blood levels for a longer period of time and in predetermined manner in order to increase the therapeutic efficacy of the drug and reduce side effects of the drug. Delivery of transdermal patches increases the patient compliance and reduces the side effects and inter- and intra-patient variability. The pores of the surface of the hydrogel are made with solvent diffusion from the surface of the patch. It was observed that, despite the use of different amounts of PVA polymers, chitosan, and alternating F-T cycles, the size and depth of the pores on the surface of the hydrogels differed at nanometric dimensions. At higher concentrations of chitosan, the size of the hydrogel pore is increased. The results suggest that the bonding between PVA and chitosan is reduced (6). Chitosan can reduce network densities because it affects the relative porosity and creates a stable 3- dimensional polymer network. The results of weight and thickness showed a significant difference between the four formulations (F6, F7, F17, and F21), which was related to the difference in various polymer ratios, which had an effect on the weight of the patches (27). The average weight in the hydrogels has a negative relationship with the amount of drug, PVA and chitosan polymers, and F-T cycles (the sign of the coefficients of the independent variables is negative). Besides, the weight of the resulting hydrogels is dependent on independent variables with a regression coefficient of 68.57 and has an inverse relationship with each other (Table 4). In addition, the results of folding values showed that the hydrogel patches were not broken and their shape and integrity were not affected by normal folding of the skin when used. Data from the content studies of the chosen hydrogels are shown in Table 3 and Fig. 2. The freezing-thawing method is regarded the best and the preferred method for obtaining physically crosslinked PVA hydrogel without using any traditional toxic chemical crosslinking agent while, the obtained mechanical properties of physically cross-linked PVA hydrogel are a tunable structure and can be adjusted by the concentration of PVA or the cycle numbers of the freeze-thawing times. Increased freezing-thawing cycles lead to further crystal formation and therefore increased physical cross-linking. The ensuing physical network seems to be relatively stable. Increase of drug content in PVA hydrogel may reduce the cross-linking reaction and consequently the gelation process is clearly reduced. Initially, the degree of crystallinity increased with an increase in PVA concentration and three cycles of freezing and thawing. In the region of interest, the formation of stable crystalline regions became more probable with increased concentration due to an increase in the overlap of PVA chains as well the promotion of polymer chain folding. Higher chitosan concentrations correspond an increment in the swelling degree, enhanced porosity with a higher permeability.
Fig. 2.

Optical microscopic photograph of blank hydrogels.
The average drug content in the formulation of hydrogels is negatively correlated to the amount of chitosan (the sign of the coefficients of the independent variables is negative). Drug and PVA concentrations and F-T cycles were the most effective factors on the drug content (R2 = 87.84, Table 4). At WVTR, hydrogel rapidly dries the wounds and causes scars. In addition, with a lower WVTR, secretions are collected, which may slow down the healing process and increase the risk of bacterial growth. An increase in the amount of chitosan was associated with a lower reduction in the gel fraction (28). The gel fraction was relatively high in low chitosan proportions, suggesting that it would be approximately thoroughly cross-linked with PVA. In hydrogels F17 and F21, high levels of chitosan (1%) reduced the hydrogel gel fraction to 72% and 37.8%, respectively (Table 2). Moreover, in F21 during high F-T cycles (7 times), the cross-link strength of chitosan was weaker than that of PVA, even though chitosan had a weak cross-link with PVA in the gel, and the strength and elasticity of hydrogel became weak and usually caused the reduction of the gel fraction (29). While the F17 hydrogel, with 1% chitosan and 5% PVA, was similar to F21, with a lower frequency of F-T (5 times), the highest gel fraction was 72%. In the preparation of hydrogels, chitosan can be used by controlling the gel fraction because it creates a cross-linking reaction between the polymers in the gel. R Square given in Table 4 means that 68.65% of the total variation in the GF from hydrogels may be ascribed to the investigated experimental factors. This value is lower than expected where an increase of X1 (drug) increased the GF, but the increase of X2 (PVA), X3 (chitosan), and X4 (F-T cycles) decreased the GF, and the interaction of X1X2, X1X3, and X2X4 led to the increase of GF. Results of the swelling index, in the hydrogels of F17 and F21 with 1% chitosan and 5% PVA did not show much change compared to the lower concentrations of chitosan 0.75% and 0.5% (as F6 and F7 hydrogels with 61.56% and 61.17%, respectively). The swelling index of gels decreased with increasing the drug, polymers concentration and F-T cycles. All of the independent variables were the most effective factors on the swelling index (R2 = 63.51) (Table 4). As a result, chitosan in hydrogel caused cross-linking to be lesser than that caused by PVA. Hydrogels having fewer cross-links led to more water uptake because gel structure with lower cross-links could not uptake or absorb more water into the sustained release gel structure (30).
Blood compatibility with the hydrogel was evaluated with the amount of protein adsorbed on the surface of the hydrogel (Table 2). When the external substance was placed in contact with blood, the adsorption of the protein occurred on the surface, leading to the adhesion and activation of the platelets (31), because protein uptake on synthetic surfaces can limit platelet activation and increase clot formation. Generally, when the amount of protein uptake increases, the number of absorbed platelets decreases (439.33 mg/mL of F7 hydrogel). An increase in the amount of PVA increased the amount of protein adsorbed, whereas an increase in the concentrations of drug, chitosan and times of F-T cycles caused a decrease in the amount of protein adsorbed, since the coefficient b2 bears a negative sign (Tables 3 and 4). PVA and chitosan had high water-solubility that created cross-link reactions as a dressing system on the wounds. The broad peaks of PVA and chitosan represented the dehydration process, leading to water evaporation, which was related to hydrophilic groups in polymers and water-polymer interference. The presence of dehydration process suggested non-exit of some of the binding water from the hydrogel patch after drying.
In the observed hydrogels, the width and intensity of the drug and polymers peak were reduced. This was caused by gradual dissolution of the crystalline drug in the molten polymers and probably by conversion to the amorphous state during the DSC heating process. Moreover, it could be related to a change in the water binding capacity of the polymers (PVA and chitosan). Water binding capacity of chitosan polymer was higher than that of PVA. In the hydrogels, endothermic melting transition and degradation peaks of PVA were not observed due to a molecular change that might be related to the interference of the molecular chains between chitosan and PVA (32).
Elongation break or stretching length led to rupture and cracking at the surface of the hydrogel patch (EB%), an essential mechanical parameter that was measured to describe the elasticity of polymer films. Hydrogels with high EB% exhibited resistance and tolerance against the force and compression that occurred when moving and displacement in the skin. Tensile strength is an indicator of the resistance against the abrasion of hydrogels (8).
The in vitro antimicrobial activity of antibiotics has always been a conflict in sustained release forms. The results of the sustained release hydrogels showed an increase in MIC dilution that could be related to a negative charge of drug and positive charge of chitosan interactions in the formulation (33).
The ex-vivo permeability of the oxytetracycline was less than drug release. This reduction was due to the diffusion of the drug from the skin barrier in permeability studies. This barrier reduced the rate of water uptake by hydrogel and reduced the disruption of the hydrogel matrix in the release time (34).
An increase in the amount of drug, PVA, and chitosan decreased the flux, whereas an increase of F-T cycles led to the increase in flux since the coefficient b2 bears a negative sign. F17 hydrogel showed high permeation than other formulations. Drug, PVA, and chitosan concentrations affect the flux of hydrogels (R2 = 68.84, Table 4).
In wounds, oxytetracycline antibiotic prevents from secondary infections (burst release and sustained release). Also, chitosan contained in the patch has an anti-microbial effect and help to anti-microbial efficacy of oxytetracycline. After use of drug formulation, wound healing was perfect therefore the possibility drug released slowly and exposure on the surface of wound for long-term. Also, in comparison with blank hydrogel and pure drug reduced use times, plasma flocculation of drug, and improvement of therapeutic effects. Wound healing with oxytetracycline hydrogel was better compared to the treatment with blank hydrogel and without treatment. Contact and placement of hydrogel had a significant effect on the dorsal skin of rat compared with non-treated (control) skin and treatment with blank hydrogel (without medication containing two PVA and chitosan polymers). Oxytetracycline hydrogel appeared to be appropriate for administration in a transdermal manner. PVA and chitosan were rapidly hydrated in contact with water and the gelatin layer was formed around the hydrogel film. In addition, the polymer content was considered to be different in each selected hydrogel. The rate of penetration of the drug from the hydrogel depended on various factors such as polymers, drug, number of F-T cycles, weight, thickness, gel fraction, swelling, protein adsorption on the patch surface, and production efficiency (35).
CONCLUSION
Oxytetracycline hydrogels were prepared by F-T cycles, using the polymers chitosan, PVA, and propylene glycol plasticizer. Various ratios of chitosan polymer, PVA, oxytetracycline, under various F-T cycles were prepared. The results of physicochemical studies of hydrogel patches (weight, thickness, loading rate, production efficiency, gel fraction, and protein adsorption on the surface) showed that the best formulation was related to F6 which contained 1000 mg oxytetracycline, 5% w/v PVA, and 0.75% w/v chitosan, which was obtained by 3 F-T cycles.
The permeability of hydrogel was lower, which might be due to the presence of a dermal barrier. This barrier reduced the speed of water uptake by hydrogel film and reduced the swelling of hydrogel matrix site at release time. The degree of penetration and release of the drug from the transdermal patch was controlled by the chemical properties of the drug and the physiological and physicochemical properties of the biological membrane. In general, it can be concluded that F-T technique is a suitable method for the preparation of appropriate hydrogel patches which are effective in treating and healing skin ulcers.
ACKNOWLEDGMENTS
This work was financially supported by the Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, I.R. Iran.
REFERENCES
- 1.Park H, Park K, Shalaby WSW. CRC Press; 1993. Biodegradable Hydrogels for Drug Delivery; pp. 59–84. [Google Scholar]
- 2.Fernandez JG, Ingber DE. Bioinspired chitinous material solutions for environmental sustainability and medicine. Adv Func Mat. 2013;23(36):4454–4466. [Google Scholar]
- 3.Tokarev I, Minko S. Stimuli Jorgensen JH, Turnidge JD. Susceptibility test methods: dilution and disk diffusion methods. Manual Clin Microbiol, Eleventh Edition: American Society of Microbiology; 2015. p. 1253-73. responsive porous hydrogels at interfaces for molecular filtration, separation, controlled release, and gating in capsules and membranes. Adv Mater. 2010;22(31):3446–3462. doi: 10.1002/adma.201000165. [DOI] [PubMed] [Google Scholar]
- 4.Welzel PB, Grimmer M, Renneberg C, Naujox L, Zschoche S, Freudenberg U, et al. Macroporous starPEG-heparin cryogels. Biomacromolecules. 2012;13(8):2349–2358. doi: 10.1021/bm300605s. [DOI] [PubMed] [Google Scholar]
- 5.Hoare TR, Kohane DS. Hydrogels in drug delivery: progress and challenges. Polymer. 2008;49(8):1993–2007. [Google Scholar]
- 6.Bhattarai N, Gunn J, Zhang M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev. 2010;62(1):83–99. doi: 10.1016/j.addr.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 7.Anumolu SS, Menjoge AR, Deshmukh M, Gerecke D, Stein S, Laskin J, et al. Doxycycline hydrogels with reversible disulfide crosslinks for dermal wound healing of mustard injuries. Biomaterials. 2011;32(4):1204–1217. doi: 10.1016/j.biomaterials.2010.08.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boateng JS, Matthews KH, Stevens HN, Eccleston GM. Wound healing dressings and drug delivery systems: a review. J Pharm Sci. 2008;97(8):2892–2923. doi: 10.1002/jps.21210. [DOI] [PubMed] [Google Scholar]
- 9.Leaper DJ. Silver dressings: their role in wound management. Int Wound J. 2006;3(4):282–294. doi: 10.1111/j.1742-481X.2006.00265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hassan CM, Peppas NA. Structure, and morphology of freeze/thawed PVA hydrogels. Macromolecules. 2000;33(7):2472–2479. [Google Scholar]
- 11.Wan W, Bannerman AD, Yang L, Mak H. Poly (Vinyl Alcohol) Cryogels for Biomedical Applications. Springer. 2014:283–321. [Google Scholar]
- 12.Ricciardi R, D’Errico G, Auriemma F, Ducouret G, Tedeschi AM, De Rosa C, et al. Short-time dynamics of solvent molecules and supramolecular organization of poly (vinyl alcohol) hydrogels obtained by freeze/thaw techniques. Macromolecules. 2005;38(15):6629–6639. [Google Scholar]
- 13.Amin MA, Abdel-Raheem IT. Accelerated wound healing and anti-inflammatory effects of physically cross-linked polyvinyl alcohol-chitosan hydrogel containing honey bee venom in diabetic rats. Arch Pharm Res. 2014;37(8):1016–1031. doi: 10.1007/s12272-013-0308-y. [DOI] [PubMed] [Google Scholar]
- 14.Kathuria N, Tripathi A, Kar KK, Kumar A. Synthesis and characterization of elastic and macroporous chitosan–gelatin cryogels for tissue engineering. Acta Biomater. 2009;5(1):406–418. doi: 10.1016/j.actbio.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Petković H, Cullum J, Hranueli D, Hunter IS, Perić- Concha N, Pigac J, et al. Genetics of Streptomyces rimosus, the oxytetracycline producer. Microbiol Mol Biol Rev. 2006;70(3):704–728. doi: 10.1128/MMBR.00004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Delf Loveymi B, Jelvehgari M, Zakeri-Milani P, Valizadeh H. Statistical optimization of oral vancomycin-eudragit rs nanoparticles using response surface methodology. Iran J Pharm Res. 2012;11(4):1001–1012. [PMC free article] [PubMed] [Google Scholar]
- 17.Părpăriţă E, Cheaburu CN, Paţachia SF, Vasile C. Polyvinyl alcohol/chitosan/montmorillonite nanocomposites preparation by freeze/thaw cycles and characterization. Acta Chemica Iasi. 2014;22(2):75–96. [Google Scholar]
- 18.Tabatabaei Yazdi F, Alizadeh Behbahani B, Mortazavi A. Investigating the minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) of the Lavandula stoechas L. and Rosmarinus officinalis L. extracts on pathogen bacterias “in vitro”. J Paramed Sci. 2014;5(2):91–101. [Google Scholar]
- 19.Jorgensen JH, Turnidge JD. Susceptibility test methods: dilution and disk diffusion methods. Manual Clin Microbiol, Eleventh Edition: American Society of Microbiology. 2015:1253–73. [Google Scholar]
- 20.Hemmila MR, BAKER JJ, Dolgachev VA, Ciotti SM, Wang S. Topical nanoemulsion therapy for wounds. Google Patents. 2015 [Google Scholar]
- 21.Pendekal MS, Tegginamat PK. Formulation and evaluation of a bioadhesive patch for buccal delivery of tizanidine. Acta Pharm Sin B. 2012;2(3):318–324. [Google Scholar]
- 22.Doulabi AH, Mirzadeh H, Imani M, Samadi N. Chitosan/polyethylene glycol fumarate blend film: physical and antibacterial properties. Carbohydr Polym. 2013;92(1):48–56. doi: 10.1016/j.carbpol.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Mehdinavaz Aghdam R, Shakhesi S, Najarian S, Malek Mohammadi M, Ahmadi Tafti SH, Mirzadeh H. Fabrication of a nanofibrous scaffold for the in vitro culture of cardiac progenitor cells for myocardial regeneration. Int J Polym Mater Polym Biomaterials. 2014;63(5):229–239. [Google Scholar]
- 24.Sharma N, Jain S, Sardana S. Muccoadhesive drug delivery system: A Review. J Adv Pharm Edu Res. 2013;3(1):9–12. [Google Scholar]
- 25.Sintov AC, Botner S. Transdermal drug delivery using microemulsion and aqueous systems: influence of skin storage conditions on the in vitro permeability of diclofenac from aqueous vehicle systems. Int J Pharm. 2006;311(1-2):55–62. doi: 10.1016/j.ijpharm.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 26.Sung JH, Hwang MR, Kim JO, Lee JH, Kim YI, Kim JH, et al. Gel characterization and in vivo evaluation of minocycline-loaded wound dressing with enhanced wound healing using polyvinyl alcohol and chitosan. Int J Pharm. 2010;392(1- 2):232–240. doi: 10.1016/j.ijpharm.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 27.Chen MC, Ling MH, Lai KY, Pramudityo E. Chitosan microneedle patches for sustained transdermal delivery of macromolecules. Biomacromolecules. 2012;13(12):4022–4031. doi: 10.1021/bm301293d. [DOI] [PubMed] [Google Scholar]
- 28.Sakai T, Matsunaga T, Yamamoto Y, Ito C, Yoshida R, Suzuki S, et al. Design and fabrication of a high-strength hydrogel with ideally homogeneous network structure from tetrahedron-like macromonomers. Macromolecules. 2008;41(14):5379–5384. [Google Scholar]
- 29.Van Kampen E. USA: University of Kansas; 2016. Controlling Protein Permeability in Hydrogels for Drug Delivery Applications. A dissertation. [Google Scholar]
- 30.Chapman RG, Ostuni E, Liang MN, Meluleni G, Kim E, Yan L, et al. Polymeric thin films that resist the adsorption of proteins and the adhesion of bacteria. Langmuir. 2001;17(4):1225–1233. [Google Scholar]
- 31.Tran NQ, Joung YK, Lih E, Park KD. In situ forming and rutin-releasing chitosan hydrogels as injectable dressings for dermal wound healing. Biomacromolecules. 2011;12(8):2872–2880. doi: 10.1021/bm200326g. [DOI] [PubMed] [Google Scholar]
- 32.Yaa A. Development of a chitosan-based glucose responsive nanoparticulate insulin delivery system: University of Nottingham. 2014 [Google Scholar]
- 33.Kong M, Chen XG, Xing K, Park HJ. Antimicrobial properties of chitosan and mode of action: a state of the art review. Int J Food Microbiol. 2010;144(1):51–63. doi: 10.1016/j.ijfoodmicro.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Salatin S, Barar J, Barzegar-Jalali M, Adibkia Kh, Kiafar F, Jelvehgari M. Development of a nanoprecipitation method for the entrapment of a very water soluble drug into Eudragit RL nanoparticles. Res Pharm Sci. 2017;12(1):1–14. doi: 10.4103/1735-5362.199041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasanpouri A, Lotfipour F, Ghanbarzadeh S, Hamishehkar H. Improvement of dermal delivery of tetracycline using vesicular Nanostructures. Res Pharm Sci. 2018;13(5):385–393. doi: 10.4103/1735-5362.236831. [DOI] [PMC free article] [PubMed] [Google Scholar]