

Abstract
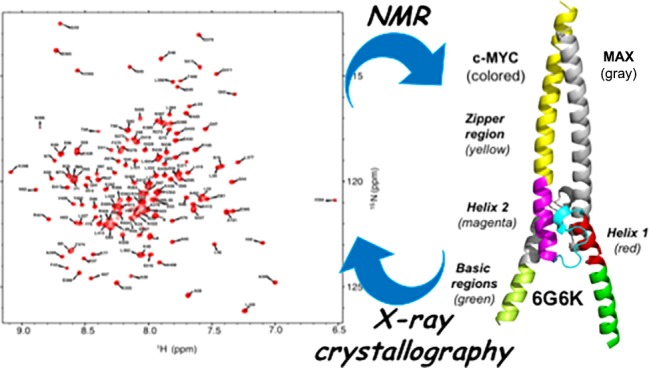
The c-MYC transcription factor is a master regulator of cell growth and proliferation and is an established target for cancer therapy. This basic helix–loop–helix Zip protein forms a heterodimer with its obligatory partner MAX, which binds to DNA via the basic region. Considerable research efforts are focused on targeting the heterodimerization interface and the interaction of the complex with DNA. The only available crystal structure is that of a c-MYC:MAX complex artificially tethered by an engineered disulfide linker and prebound to DNA. We have carried out a detailed structural analysis of the apo form of the c-MYC:MAX complex, with no artificial linker, both in solution using nuclear magnetic resonance (NMR) spectroscopy and by X-ray crystallography. We have obtained crystal structures in three different crystal forms, with resolutions between 1.35 and 2.2 Å, that show extensive helical structure in the basic region. Determination of the α-helical propensity using NMR chemical shift analysis shows that the basic region of c-MYC and, to a lesser extent, that of MAX populate helical conformations. We have also assigned the NMR spectra of the c-MYC basic helix–loop–helix Zip motif in the absence of MAX and showed that the basic region has an intrinsic helical propensity even in the absence of its dimerization partner. The presence of helical structure in the basic regions in the absence of DNA suggests that the molecular recognition occurs via a conformational selection rather than an induced fit. Our work provides both insight into the mechanism of DNA binding and structural information to aid in the development of MYC inhibitors.
The c-MYC pleiotropic transcription modulator integrates fundamental processes required for the proliferation and survival of normal cells.1−3 Acting as both a transcriptional activator and a repressor, c-MYC coordinates the expression of a large, extremely diverse set of genes in a highly context-dependent manner. These govern both intracellular functions (i.e., cell growth, cell cycle progression, biosynthetic metabolism, and apoptosis) and extracellular processes that coordinate cell proliferation with its adjacent somatic microenvironment (i.e., angiogenesis, invasion, stromal remodeling, and inflammation).4−10 c-MYC belongs to the MYC family of transcription factors that also includes N-MYC and L-MYC. In general, c-MYC is expressed in all dividing cells from embryonic and adult tissues, whereas N-MYC and L-MYC are expressed only in specific embryonic and neonatal tissues (e.g., brain, lung, liver, and kidney).11
Deregulated expression of the c-MYC protein occurs in a broad spectrum of human cancers and is particularly associated with aggressive disease and poor clinical outcome,11−14 indicating a crucial role for this oncogene in cancer progression. It has also been shown that MYC programs an immune suppressive stroma that is required for tumor progression.6 In transgenic mouse models, inactivating c-MYC halts tumor cell growth and proliferation15−17 without triggering tumor-escape pathways. These studies also have shown that somatic cells easily tolerate c-MYC inactivation, with limited side effects, which are rapidly and completely reversible. Targeting c-MYC is, therefore, regarded as a powerful approach for anticancer therapy,18−20 and it is also emerging as a promising molecular target in inflammation and heart disease.6,21,22
Although MYC physiology and pathology have been extensively studied, we still do not know how MYC works, in particular, the obligate role it appears to play in the genesis and maintenance of many, perhaps all, cancers. More practically, we need a better understanding of c-MYC structure and function to be able to target it pharmacologically.
c-MYC is an intrinsically disordered protein that belongs to the basic helix–loop–helix zipper (bHLHZip) class of transcription factors.23 It is composed of 439 amino acids (aa) and consists of an N-terminal transactivation domain (NTD), a C-terminal domain (CTD), and a central region. The N-terminal domain contains the transcription activation domain (TAD) and two highly conserved sequence elements, known as “MYC boxes” (MBI and II), which are involved in protein stability and transcription regulation. The central region also contains conserved sequences, in particular a nuclear localization signal (NLS), and MBIII and MBIV, implicated in MYC cellular transforming activity, transcription, and apoptosis. The C-terminal domain (amino acids 360–439) contains the bHLHZip motif. It plays a cardinal role in cell proliferation, transformation, and apoptosis. Upon binding to its obligatory partner MAX, also a bHLHZip protein, the C-terminal domain forms an ordered α-helical structure that extends into a left-handed coiled coil formed by the two leucine zipper motifs.24,25 This stable four-helix bundle binds to specific DNA sequences, such as CACGTG E-box motifs, in promoters and enhancers of MYC-regulated genes. The dimerization event is driven by the leucine zipper and the HLH motifs, while the basic regions interact with DNA. The helix–loop–helix region of c-MYC is the target of diverse post-translational modifications such as phosphorylation, acetylation, ubiquitination, and sumoylation.9,26−28 Furthermore, this region participates in protein–protein interactions (PPIs) that mediate and regulate c-MYC functions. To date, the only available structure of a MYC:MAX heterodimer is a c-MYC:MAX bHLHZip complex bound to DNA containing an E-box motif, tethered by an artificial disulfide bridge engineered by adding a cysteine residue at the C-terminus of the leucine zippers of both the c-MYC and MAX proteins29 [Protein Data Bank (PDB) entry 1NKP]. Nuclear magnetic resonance (NMR) has been used to study the chicken viral homologue of c-MYC (v-MYC) both free and bound to v-MAX in the absence of DNA.30 However, the sequences of the human and chicken homologues differ significantly in this region (Figure S1). In contrast to MYC proteins, MAX is expressed constitutively in the cell and can form homodimers in vitro and in vivo. MAX homodimers can also bind E-box DNA, although at physiological levels MAX homodimers do not play any role in regulating transcription.31 The crystal structure of MAX dimers bound to E-box DNA has been determined,32,33 and NMR34 has been used to determine the structure of a MAX homodimer containing mutations that increase the stability of the dimer.
Several studies have been devoted to identifying direct or indirect MYC inhibitors; however, a clinical candidate is not yet available.20,35−37 For direct targeting of MYC, inhibition of c-MYC:MAX dimerization has probably been the most “beaten path” approach. Very promising results have been obtained in vivo using a MYC-dominant-negative Omomyc protein20,38−42 (a variant bHLHZip domain with an engineered leucine zipper) that disrupts the c-MYC:MAX interaction. Recently, the crystal structures of the Omomyc homodimer in the apo form and bound to DNA have been determined.38 As in previous studies of c-MYC:MAX heterodimers, an engineered disulfide linker was used to stabilize the homodimer. The large size of the c-MYC:MAX bHLHZip interface and its lack of binding pockets make the development of c-MYC:MAX inhibitors particularly challenging.43,44 Recently, efforts have also been focused on developing molecules that bind to the c-MYC:MAX dimer to prevent it from binding to DNA.36,45
c-MYC is an intrinsically disordered protein, and the dimerization with MAX involves a coupled folding-and-binding process. The c-MYC:MAX apo complex could therefore be highly plastic and undergo significant conformational changes at the dimerization interface when bound to DNA. The structure of the basic regions in the apo form has not been established, but it is widely thought that they are unstructured and undergo an extreme example of induced fit upon binding to DNA.
c-MYC binds to diverse sites on the genome with a broad range of affinities, including high-affinity canonical (i.e., E-box) and low-affinity noncanonical DNA sequences,4,7 and it has also been proposed that a partially unfolded c-MYC:MAX heterodimer can recognize a “partial site” on the nucleosome.46
Biophysical studies of the apo form, hence, are needed to determine the conformational changes that accompany DNA binding to help to understand how these different DNA targets are recognized. The structural and biophysical information about the apo form is also relevant for the study of interactions of the c-MYC:MAX complex with cofactors that are mediated by the C-terminus, especially as some of these PPIs are mutually exclusive with DNA binding. The apo form of the c-MYC:MAX bHLHZip dimer is the target for both the dimerization inhibition and DNA binding inhibition approaches, and thus, the structure of the complex bound to DNA is limited in providing a platform for structure-based design.
To provide structural information for the design of MYC inhibitors and to gain insights into the conformational changes induced by DNA binding, we set out to study the apo form using a combination of NMR and X-ray crystallography.
Materials and Methods
Materials
Chemicals were acquired from Sigma-Aldrich or Fisher Scientific and used without further purification. Ni-NTA resin was from Qiagen. HisTrap high-performance (HP) and fast flow (FF) columns were from GE Healthcare. Amicon centrifugal units were obtained from Millipore. Polymerase chain reaction primers were obtained from IDT.
Protein Expression and Purification
[2H,13C,15N]c-MYC:MAX bHLHZip Heterodimer
The title compound (UniProt entry P01106 for c-MYC and UniProt entry P61244 for MAX) for NMR studies was produced, purified, and stored as previously described,47 and the integrity of the proteins in the complex was checked by TOF MS ES+ (Figure S2).
[13C,15N]MAX:MAX bHLHZip Homodimer for the Preparation of the Reconstituted c-MYC:MAX Complex
The sample was obtained as a byproduct of the co-expression protocol described previously.47 Although the homodimer has no His tag, due to the presence of multiple exposed histidine residues, it has an affinity for the Ni Sepharose HisTrap HP (5 mL) column and could be separated from the His-tagged c-MYC:MAX heterocomplex by careful elution using an imidazole gradient. The integrity of the protein in the complex was checked by TOF MS ES+ (Figure S2). To prepare the sample of the reconstituted c-MYC:MAX dimer, the [13C,15N]MAX:MAX complex [in phosphate-buffered saline (PBS) and 1 mM dithiothreitol (DTT)] was added to a PBS/1 mM DTT solution of unlabeled c-MYC free protein (20 μM) in a 1:1 ratio and then concentrated to 150–200 μM (as measured on a NanoDrop 2000 spectrophotometer, Thermo Fisher) for NMR studies (Figure S3).
[13C,15N]c-MYC bHLHZip Free Protein
The DNA encoding residues 352–437 of c-MYC was cloned into the BamHI and EcoRI sites of the pET24a vector to direct the expression of an N-terminally histidine-tagged protein.
Chemically competent Escherichia coli BL21 (DE3) cells were transformed with this plasmid. Cells were plated on Luria-Bertani agar supplemented with kanamycin. A single colony was used to inoculate a culture of either 2XTY broth or K-MOPS minimal medium prepared containing 15NH4Cl and [13C]glucose. c-MYC was expressed in inclusion bodies. Cells were grown at 37 °C to an OD600 of 0.8 and then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside. The cells were collected after overnight expression at 37 °C by centrifugation at 4000 rpm for 15 min and resuspended in 30 mL of ice-cold lysis buffer [20 mM Tris-HCI, 500 mM NaCl, and 1 mM DTT (pH 7.9)]. The cells were lysed via sonication, and the lysate was cleared by centrifugation at 18000 rpm for 20 min. The pellet was resuspended in a resolubilization 6 M urea binding buffer (RBB) [including 20 mM Tris-HCl, 500 mM NaCl, and 20 mM imidazole (pH 8–8.5)] and loaded onto a Ni Sepharose HisTrap FF (5 mL) affinity column, washed with a wash buffer (WB) containing 6 M urea, 20 mM Tris-HCl, 500 mM NaCl, and 50 mM imidazole (pH 8–8.5). The protein was then eluted with an elution buffer (EB) with 6 M urea containing 20 mM Tris-HCl and 500 mM NaCl (pH 8–8.5) and with a gradient from 100 to 500 mM imidazole. The eluate was collected, and a stepwise resolubilization/folding process was carried out in four steps with buffers with decreased amounts of urea from 6 M to none (i.e., 6 to 4 M, 4 to 2 M, 2 to 1 M, and 1 to 0 M urea) containing PBS (pH 7) and 1 mM DTT at 4 °C. The sample was then concentrated to 10–20 μM (measured on a NanoDrop 2000 spectrophotometer). Above this concentration, some aggregation started to appear (as seen by NMR). The integrity of the protein in the complex was checked by TOF MS ES+ (Figure S2).
c-MYC:MAX bHLHZip Homodimer for Crystallization Studies
For the crystallization studies, a c-MYC:MAX bHLHZip co-expression construct encoding the same regions of c-MYC:MAX bHLHZip (c-MYC = 352–437, MAX = 22–102) of the complex for NMR studies was used, but with a shorter c-MYC N-terminal His tag with a sequence of MHHHHHHEE. Expression and purification of the protein complex were carried as for the NMR studies that have been previously reported.47
Mass Spectrometry (MS)
Total mass analysis was performed on a Waters LCT time-of-flight mass spectrometer with electrospray ionization (Micromass) with protein solutions in PBS mixed in a 1:1 ratio with 1% formic acid in 50% MeOH. Samples were injected at a rate of 10 μL min–1, and calibration was performed in positive ion mode using horse heart myoglobin. The MS diagrams are reported in Figure S2.
NMR
The labeled c-MYC:MAX samples prepared for NMR spectroscopy experiments were typically at concentrations of 150–200 μM (measured on a NanoDrop 2000 spectrophotometer) in PBS (pH 7), 10% D2O, and 1 mM DTT. The labeled c-MYC free protein samples prepared for NMR spectroscopy experiments were typically at a concentration of 10 μM (measured on a NanoDrop 2000 spectrophotometer) in the same buffer used for the labeled c-MYC:MAX complexes [i.e., PBS (pH 7), 10% D2O, and 1 mM DTT]. NMR data acquisition was carried out at 25 °C for both MYC:MAX complex samples and c-MYC free protein samples, and in addition at 5 °C for the c-MYC samples, on either a Bruker Avance II+ 700 MHz (c-MYC free protein) or a Bruker Avance III HD 800 MHz spectrometer equipped with a cryogenic triple-resonance TCI probes. Topspin (Bruker) was used for data processing, and Sparky (SPARKY 3) for data analysis. All experiments were performed using non-uniform sampling (NUS) at a rate of 50% of complex points in the 1H, 15N, and 13C dimensions and reconstructed using compressed sensing.48 Backbone assignments were made using the following standard set of three-dimensional (3D) heteronuclear NMR experiments, i.e., HNCO, HN(CA)CO, HNCA, HNCACB, and CBCA(CO)HN, on 2H-, 13C-, and 15N-labeled samples for the c-MYC:MAX bHLHzip complex and via examination of HSQC spectra of the unlabeled c-MYC:[13C,15N]MAX bHLHZip reconstituted complex. The additional assignments for the c-MYC:MAX complex have been deposited in the Biological Magnetic Resonance Bank (see updated BMRB entry 27571). The same set of experiments was used for the 13C- and 15N-labeled samples for c-MYC free protein. The assignments for c-MYC free protein have been deposited in the Biological Magnetic Resonance Bank (BMRB entry 12033).
Crystallography
Protein Crystallization
Purified protein was buffer exchanged into 20 mM Tris and 100 mM NaCl (pH 7.0) and concentrated to 10 mg/mL, as measured on a NanoDrop 2000 spectrophotometer. The crystallization screening set of plates [22 LMB plates (LMB 01–LMB 22), 1112 conditions overall] from the UKRI Medical Research Council (MRC), Laboratory of Molecular Biology (LMB) Crystallization Facility,49 and the MORPHEUS III crystallization screen (see below for details) were applied to the c-MYC:MAX bHLHZip complex. A common approach included single drops with final volumes of 200 nL with a 1:1 ratio (100 nL of protein and 100 nL of precipitant solution). Drops were set with a nanoliter-dispensing MOSQUITO robot (TTP Labtech).
Crystals of the c-MYC:MAX bHLHZip complex were grown using the vapor diffusion method at 4 °C. Crystals were obtained under the following conditions: crystals of Collect 5/PDB entry 6G6K, 10% PEG 8000, 20% ethylene glycol, 5% EtOH, and 0.1 M MOPS/HEPES-Na (pH 7.5), with 0.075% (w/v) of each additive [0.75% menthol, 0.75% caffeic acid, 0.75% d-quinic acid, 0.75% shikimic acid, 0.75% gallic acid monohydrate, and 0.75% N-vanillylnonanamide] (plate LMB 22, an in-house test formulation of the MORPHEUS III crystallization screen); crystals of Collect 2/PDB entry 6G6J, 20% PEG 3350 and 0.2 M sodium sulfate decahydrate (pH 7) (plate LMB 05); crystals of Collect 7/PDB entry 6G6L, 15% PEG 8000 15 and 0.2 M ammonium sulfate (pH 7) (plate LMB 09).
For freezing, crystals were then immersed in the precipitant solution supplemented with 20% (v/v) glycerol for Collect 2 and 7 or with no cryoprotectant for Collect 5, prior to vitrification by direct immersion into liquid nitrogen. High-resolution data sets were collected remotely at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on beamline ID23-I for Collect 5 and at Diamond Light Source (DLS, Harwell, U.K.) on beamline I03 for Collect 2 and 7.
MORPHEUS III Crystallization Screen
The MORPHEUS III crystallization screen was formulated according to methods described elsewhere,50 with notably the ratio of volumes for the stock solutions that is fixed for each condition: 0.5 stock of cryoprotected precipitant mix, 0.1 stock of a mix of additives, 0.1 stock of the buffer system, and 0.3 water. The four cryoprotected precipitant mixes and three buffer systems were similar to the original MORPHEUS screen;51 nonetheless, alternative additive mixes were integrated (Tables S1 and S2). For this, additives were selected as PDB-derived ligands (nucleosides and cholic acid derivatives), phytochemicals (initially diluted in a 50% ethanol solution), vitamins, well-known antibiotics, and anesthetic alkaloids. To complete the formulation, dipeptides were integrated. The corresponding chemicals were ordered from Sigma-Aldrich (95–99% purity). The formulation of the resulting 96-condition screen is shown on Tables S1 and S2.
Determination of the Structure of the c-MYC:MAX bHLHZip Apo Complex
Diffraction data were indexed and integrated with XDS52 and scaled and merged with SCALA.53 Data were integrated using XDS and scaled using SCALA. The phases were determined by molecular replacement using PHASER and PDB entry 1NKB. Density modification produced experimental maps that allowed manual refinement using COOT.54 The structures were subsequently further refined using Phenix.55 The validity of all models was routinely determined using MOLPROBITY and by using the free R factor to monitor improvements during building and crystallographic refinement. Data collection and refinement statistics are listed in Table S3. Collect 2 and Collect 5 both have four molecules (two dimers) of the c-MYC:MAX complex per asymmetric unit, whereas eight molecules (four dimers) of the heterodimeric complex were found in the asymmetric unit of Collect 7. Both P1 crystals exhibited pseudo-C2 symmetry, which was subsequently resolved using space group P1.
Size Exclusion Chromatography–Multiangle Light Scattering (SEC–MALS)
Samples for SEC–MALS analysis were prepared by preincubating the c-MYC:MAX bHLHZip apo complex with the E-box DNA in a 1:1 ratio with PBS. Then 100 mL of the c-MYC:MAX bHLHZip complex bound to DNA (0.22 μm filtered with a concentration of 23 μM) was injected at a rate of 0.5 mL/min and resolved on a GE Superdex75 10/300 GL (GE Healthcare) analytical column equilibrated in PBS buffer (pH 7), which is consistent with multiangle laser light scattering using a Wyatt HELEOS-II 18-angle photometer coupled to a Wyatt Optilab rEX differential refractometer (Wyatt Technology Corp.). Molecular weight calibration was performed with bovine serum albumin (BSA), and masses were averaged in the indicated regions using a dn/dc increment of 0.1807 (as the sample is two-thirds protein and one-third DNA). Data were collected and analyzed using ASTRA software (Figure S4).
Results and Discussion
Assessment of Secondary Structure Propensities of the bHLHZip c-MYC:MAX Dimer Using NMR Chemical Shifts
Previously, we have determined assignments for the bHLHZip c-MYC:MAX dimeric complex.47 Essentially complete assignments of the HLHzip regions were obtained for both proteins, but only partial assignments of the basic region of c-MYC and no assignments for the MAX basic region were reported because of peak overlap and line broadening. In an effort to determine more assignments for these regions, NMR acquisition strategies that allow the recording of very high resolution 3D spectra were employed,48 as this is particularly useful for the analysis of highly overlapped spectra of intrinsically disordered regions and/or proteins (Figure 1). In addition, a sample was prepared in which only MAX was isotopically labeled by reconstituting the heterodimer by mixing (1:1 ratio) 15N-labeled homodimeric MAX with unlabeled c-MYC protein (Figure S3). Analysis of these spectra yielded additional assignments for both proteins (Figure 1; see updated BRMB entry 27571). Only the assignments of five residues in c-MYC (R357, T358, R367, N368, and E369) and five residues in MAX (H27, H28, R36, D37, and H38) could not be obtained due to the absence of peaks and line broadening in the 1H/15N HSQC spectra. Most of these residues are in the junction between the basic region and helix 1 in both c-MYC and MAX. Other research groups have reported that they have not been able to obtain full assignments of this region for either v-MYC-MAX30 or MAX-MAX dimers.34
Figure 1.
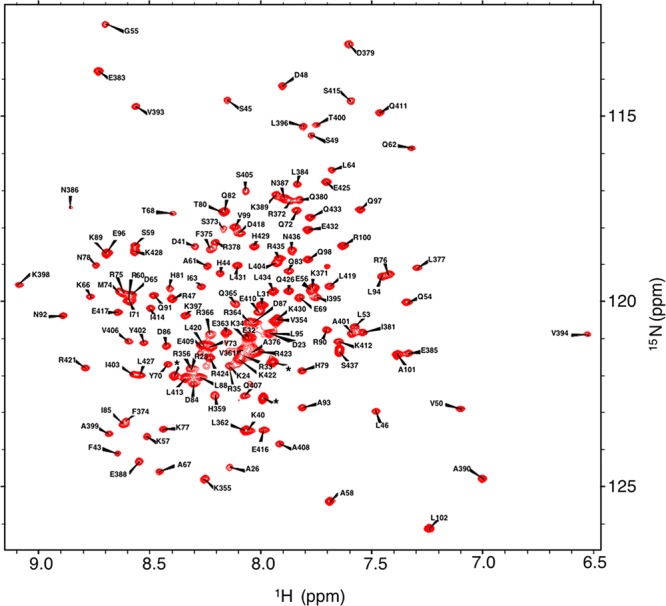
1H/15N HSQC spectra of the 2H-, 13C-, and 15N-labeled c-MYC:MAX bHLHZip apo complex. The star indicates a resonance from the histidine tag.
The availability of the additional assignments allowed us to make use of recently developed methods that use chemical shift data to assess secondary structure populations. In particular, we have employed the δ2D program developed by Vendruscolo and colleagues56 that analyzes NMR chemical shifts to provide quantitative information about the probability distributions of secondary structure elements in both folded and disordered states. As illustrated in Figures 2 and 3, the zipper region in both c-MYC and MAX is predicted to populate a nearly 100% α-helical conformation in solution.
Figure 2.

Secondary structure populations for c-MYC in the complex with MAX as determined from NMR chemical shifts using the δ2D program. The absence of a value for the α-helical secondary structure population along the Y axis indicates a lack of assignments for the residue. The program does not determine values for the first and last residues of the amino acid sequence.
Figure 3.

Secondary structure populations for MAX in the complex with c-MYC as determined from NMR chemical shifts using the δ2D program. The absence of a value for the α-helical secondary structure population along the Y axis indicates a lack of assignments for the residue. The program does not determine values for the first and last residues of the amino acid sequence.
Helix 2 in both c-MYC and MAX also populates a helical conformation at a very high percentage. In both c-MYC and MAX, a drop below 90% of helical state is predicted in the middle of the helix, at the equivalent residues K398 in c-MYC and K66 in MAX. Another significant dip is observed for residue M74 in MAX at the junction between helix 2 and the zipper region.
As expected, the loops in both proteins primarily sample a coil conformation, except for residues P382 and E383 of MYC that shows a predicted helical state of 55%.
Helix 1 shows significant differences between the two proteins: MAX has a nearly 100% populated helical conformation, while in c-MYC, a substantial drop in helical conformation can be observed. This helicity drop is centered around residue F375, a highly conserved, solvent-exposed phenylalanine, for which there is no equivalent residue in MAX. It is interesting to note that residue S373 undergoes phosphorylation that blocks dimerization.57 The fact that this region is suggested to sample a coil conformation could make this site more amenable to the post-translational modification.
The basic regions are predicted to show a varying mixture of helical and coil conformations. In c-MYC, residues in the N-terminal part of the basic region are predicted to have a small helical population while residues in the C-terminal portion of the basic region of c-MYC are predicted to have a larger helical population, reaching a maximum probability of 67% for residue R366, after which peaks could not be assigned. With regard to MAX, the equivalent residues are also predicted to adopt a significant helical conformation, but to a degree markedly lower than that in c-MYC (∼25%).
To understand whether the helical propensity of the residues in the basic region of c-MYC in the c-MYC:MAX complex is the result of the dimerization event or is the intrinsic property of the amino acid sequence, we set out to study by NMR the free form of the c-MYC bHLHZip protein.
NMR Studies of the Free Form of the c-MYC bHLHZip Protein
NMR assignments of the free form of the c-MYC bHLHZip protein were obtained using 15N- and 13C-labeled samples employing standard triple-resonance experiments. Compared to the heterodimeric complex, we observed that free c-MYC is less soluble because it is prone to aggregation at concentrations above 10–20 μM. The spectra showed a marked dependence on temperature with more peaks being visible in 15N/15N HSQC spectra recorded at 5 °C.
Complete assignments of the residues in free c-MYC, which correspond to the N-terminus of helix 2 and the loop, helix 1, and basic regions in the c-MYC:MAX complex, were obtained (Figure 4, BMRB entry 12033). The rest of helix 2 and the zipper region, which drives dimerization with MAX, could not be assigned due to the absence of peaks corresponding to these residues. Our findings have been independently confirmed by Macek et al.,58 who reported results similar to ours while this study was underway.
Figure 4.
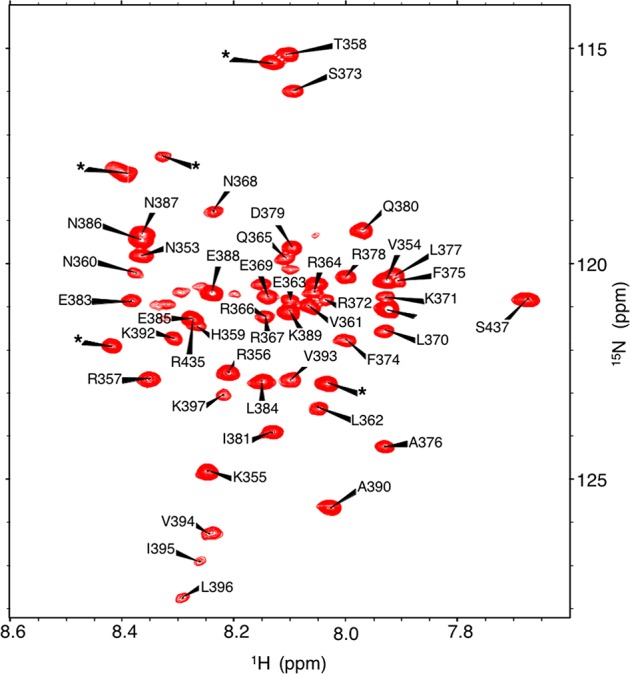
1H/15N HSQC spectra of the 13C- and 15N-labeled free c-MYC bHLHZip protein. The star indicates a resonance from the histidine tag.
The analysis of the secondary structure populations of these regions using the δ2D program predicts a level of the helical state in the region corresponding to the basic module in the c-MYC:MAX complex of ≤44% (Figure 5). The propensity to be helical is lower in the free form than in c-MYC bound to MAX, but this shows that even in the absence of dimerization, residues in the basic region populate a helical conformation.
Figure 5.

Secondary structure populations for the c-MYC free protein as determined from NMR chemical shifts using the δ2D method. The absence of a value for the α-helical secondary structure population along the Y axis indicates a lack of assignment for the corresponding residue on the X axis. The program does not determine values for the first and last residues of the amino acid sequence.
Compared to the basic region, there is then a much smaller percentage of helicity in the regions corresponding to helix 1 and even less at the N-terminus of helix 2, in contrast with the results for c-MYC when in complex with MAX.
Assignments of the NH, N, and Cα resonances of the v-MYC bHLHZip protein have been reported.59 These are insufficient to carry out secondary population analysis, but general secondary structure propensities could be inferred. Trends similar to those observed for c-MYC were found for the basic region, which has an identical amino acid sequence in v-MYC and c-MYC, and also for helix 1 and the N-terminus of helix 2, although these regions have differences in amino acid sequence. Instead, in free v-MYC the zipper region could be detected and assigned and was shown to have a significant helical propensity. One could postulate that the absence of peaks for this region in the spectra of free c-MYC is due to line broadening produced by the rate of the helix–coil transition or formation of a very low affinity, transient homodimer. The amino acid sequences of v-MYC and c-MYC differ significantly in this region (Figure S1). The differences observed between the zipper regions in c-MYC and v-MYC are likely due to differences in their amino acid sequences affecting the interconversion rate, or the ability to form homodimers. The zipper region is a target for the discovery of drugs that directly inhibit MYC. The binding of a small molecule to this region could alter the processes that affect the peak intensities in the free form, so NMR could still be used to examine interactions of molecules with this region of c-MYC. The differences observed for the zipper region, however, caution against using v-MYC as a surrogate for c-MYC for these studies.
Our NMR studies of the c-MYC:MAX complex show that even in the absence of DNA the basic region of MYC and to a lesser extent that of MAX are predicted to be able to adopt a helical conformation. We thus set out to determine if these helical conformations that are present in partial amounts can be crystallized and, if they can be, to determine their structure.
Crystal Structures of the Apo Form of the c-MYC:MAX bHLHZip Heterodimeric Complex
Due to the dynamic nature of the system and its partially disordered nature that was observed in solution by NMR, we expected that crystallization of the c-MYC:MAX bHLHZip heterodimeric complex in the absence of DNA would be challenging, so a large number of initial crystallization conditions were screened. We employed the same c-MYC:MAX bHLHZip construct used in the NMR studies (i.e., without an artificial linker) but with a shorter histidine tag. Three crystal forms were obtained at different resolutions: 2.25 Å (PDB entry 6G6J), 2.20 Å (PDB entry 6G6L), and 1.35 Å (PDF entry 6G6K) (Figure 6). Crystallization was carried out at room temperature (20 °C) and 4 °C, but crystals were obtained only at the lower temperature.
Figure 6.
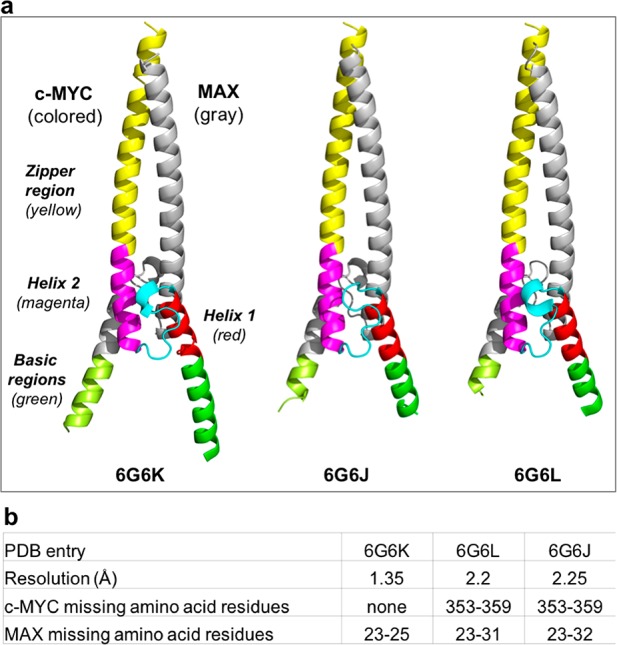
Side-by-side comparison of the cartoon representations of the three crystal structures of the c-MYC:MAX bHLHZip apo complex. (a) For each of the crystal structures, for MYC, the zipper region is colored yellow, helix 2 magenta, the loop cyan, and helix 1 red, and for MAX, the corresponding regions are all colored gray. The basic region in MYC is colored green, and that in MAX light green. (b) Table showing the missing residues in each of the free crystal forms.
Initially, we employed the screening set of LMB plates from the UKRI MRC LMB Crystallization Facility,60 which yielded the two crystal forms with lower resolution. Electron density in these forms was seen for the zipper, loop, helix 2, all helix 1 regions, and part of the basic region for both c-MYC and MAX. The residues of helix 1 and the basic regions also adopt a helical conformation even in the absence of DNA. In an attempt to improve the resolution and to see if it was possible to obtain data for the entire basic region for drug discovery purposes, we then employed advanced crystallization screening conditions that were under development at the time of our study, i.e., MORPHEUS III. This allowed us to obtain a crystal form that diffracted at 1.35 Å and to determine an apo structure of the c-MYC:MAX bHLHZip complex that contains the entire basic region of c-MYC and all but the first helical turn of MAX. For this structure, the lowest B factors are found in the helices of the HLH motif. The loops within this motif in contrast have high B factors. There is a progressive increase in B factors toward the C-terminus of the leucine zipper. Within the basic regions proceeding from the N-terminus, there is a progressive decrease in B factors, correlating with the degree to which the helical state is populated in solution from the analysis of the NMR spectroscopy data (Figure S5).
The three different crystal forms have two, two, and four c-MYC:MAX dimers within the asymmetric unit. The only significant protein–protein interaction in any of the crystal forms was between the basic regions of the two MAX proteins from each heterodimer within the asymmetric unit of the 6G6K/Collect 5 structure (1.35 Å). In the structure of the c-MYC:MAX dimer bound to DNA, packing mediated by direct protein–protein interactions was observed through the zipper regions that packed in an antiparallel fashion. This form of packing was not observed in any of the crystal forms of the apo structure. It has been suggested that the packing within the asymmetry unit of the c-MYC:MAX/DNA complex (with 1.9 Å resolution) reflects an interaction that takes place in vivo. NMR studies of our bHLHZip construct were consistent with the dimer being in a monomeric form in solution both free and bound to DNA. Furthermore, SEC–MALS analysis (Figure S4) of the DNA-bound complex of the c-MYC:MAX bHLHZip dimer used in this study shows that it is in a monomeric state.
A more detailed analysis of the three crystal structures of the apo form reveals a series of commonalities and differences between c-MYC and MAX. With regard to the zipper region, there is no significant conformational difference between the crystal forms (Figure 6). Consistent with the NMR data, the zipper region helices of both MYC and MAX extend to encompass all of the heptad repeats (even in the absence of the disulfide linker). In our construct, the residues in MYC that form the GGC linker for the disulfide bridge in the c-MYC:MAX/DNA complex are replaced with the native RNS sequence, which forms an additional helical turn. In MAX, the helix ends at R100 as in the structure with the disulfide linker (Figure 7). With regard to the zipper region, there is no significant difference among the three apo complexes or between them and the DNA-bound complex. Similarly, there are no conformational differences in helix 1 and helix 2 among the three crystal structures of the apo form and no structural differences between the apo complex and the heterodimer bound to DNA. It is important to emphasize that there is no deviation from helicity for helix 1 in MYC. This suggests that the 25% loss in the helical state observed, which is not seen for helix 1 in MAX, is likely to be due to the intrinsic instability of this region in MYC.
Figure 7.

Cartoon representations of the superposition of the crystal structures of the c-MYC:MAX complex in its apo form and bound to DNA. (a) Overall view of the superimposition of the highest-resolution structure (PDB entry 6G6K) with c-MYC colored blue and MAX colored magenta and the 1NKP structure with c-MYC colored green and MAX colored yellow. (b and c) Details of the MYC basic region with residues K355, R356, H359, E363, and R367 that make contact with DNA shown as red sticks, and the same residues are colored orange in the crystal structure of the apo form.
The loop region in c-MYC, which contains the ubiquitylation site lysine 389, adopts different conformations in the three structures of the apo form (Figure S6), all of which differ from the structure in the DNA-bound complex (Figure 7). We can observe the formation of a short 310 helix for residues P382–L384 in two of the three crystal forms (Figure 6 and Figure S6), which reflects the dynamic nature of the loop. This concurs with the NMR analysis that shows a 55% predicted population of helical conformation for residues P382 and E383. The loop in MAX, which is shorter than that in c-MYC, presents one conformation in all of the structures (Figure S6), which is identical to that seen in the structure of the DNA-bound complex (Figure 7).
The greatest differences both among the three apo structures and between them and the DNA-bound complex are seen in the basic regions. The amounts of the basic regions visible vary among the different crystal forms. Strikingly, in all of the structures, less of the basic region can be observed in MAX than in c-MYC. In the highest-resolution structure (Collect 5/6G6K), we can see the entire basic region of c-MYC. In the two other crystal forms, instead, less of the basic region of c-MYC is visible, as in both Collect 7/6G6L and Collect 2/6G6J residues N353–H359 are missing. For MAX, the full basic region is not visible in any of the crystal forms. Even in the structure with the highest resolution, residues D23, K24, and R25 are still missing (Figure 6). In the other crystal forms, even less of the basic region is visible. In Collect 7/6G6L, residues D23–L31 are missing, and in Collect 2/6G6J, the density for residues D23–E32 is also not observed.
The crystal structure of the complex bound to DNA has revealed that the E-box sequence is recognized by contacts with the DNA bases made by residues H359, E363, and R367 in MYC and by residues H28, E32, and R36 in MAX. Residues K355 and R356 in MYC and residues K24 and R25 in MAX also contact the phosphate backbone in the DNA. Compared to the structure of the complex bound to DNA (Figure 7), in the highest-resolution structure of the apo form all of the residues in MYC that make contact with the DNA bases and the phosphate backbone of the DNA are present and in a helical conformation. However, the helices deviate to enable H359 to contact the G base of the E-box motif but especially for residues R356 and K355 to contact the phosphate backbone. With regard to MAX, in this structure all of the residues contacting the DNA bases are visible and in an helical conformation, but the residues contacting the phosphate backbone are missing. Therefore, the distortion in the helical conformation is less marked.
Conclusions
The crystal structures of the c-MYC:MAX complex in its apo form in combination with NMR studies have enabled a better understanding of the conformational plasticity of this system and its relationship with DNA binding.
The basic regions have been historically assumed to be in an unstructured form prior to binding to DNA. They were thought to become helical only when bound to DNA, as part of an induced fit binding mechanism. Consequently, these regions were typically removed in other crystallization studies of apo dimeric complexes of bHLHZip proteins.38,61 Our NMR studies have shown that the apo complex is indeed a dynamic system with the basic regions adopting coil structures for a significant portion of the time, but we have also shown that these regions can also populate helical conformations. The sampling of a wide range of crystallization conditions has enabled us to capture the transiently populated more ordered states in a crystal lattice. We argue that the formation of a helix in the basic region is driven by both formation of helix 1 via dimerization and the intrinsic helical propensity of the basic region of the free c-MYC protein observed by NMR. This would result in the formation of a population of preformed helices, which include the amino acid residues contacting DNA. This implies that molecular recognition occurs via conformational selection rather than an induced fit mechanism. In fact, the only evidence of any induced fit is the small distortion observed at the beginning of the helix of the basic region of MYC that allows for optimal contacts with the DNA.
The basic regions of c-MYC and MAX behave differently in both the degree to which they populate a helical conformation, as determined by the NMR chemical shift analysis, and the crystal structures where this region in MAX is consistently less visible. This shows that in the heterodimeric complex the basic regions have distinct conformational properties that could affect the ability of the complex to recognize noncanonical DNA sequences, such as half-site recognition4 or recognition of sequences in different structural contexts.46
One feature of the spectrum of the complex is the absence of peaks at the junctions between the start of helix 1 and the end of the basic region. This is where there is a transition between highly populated and less populated helical structure, and a dynamic process associated with this transition most probably leads to line broadening to a point where the peaks are not detectable. The formation of the extended helix made by helix 1 and the basic region will be energetically unfavorable as the highly charged residues in the basic regions are brought into their proximity by the formation of helices in the heterodimeric complex. This would account for the observation that the removal of the basic regions of both c-MYC and MAX results in significant stabilization of the heterodimer.47 This destabilization may also contribute to the fraying of helix 1 where it merges with the basic region. The plastic nature of helix 1 in the apo form of c-MYC, which is primarily in a coil conformation in the free form, may be an attractive feature to exploit for targeting MYC with small molecules that trap it in a conformation that cannot bind to DNA.
In conclusion, this study shows that a combination of different structural and biophysical techniques is needed both to understand the molecular interactions and to target a complex system as c-MYC that includes both folded and disordered/partially folded regions. We now have in hand a powerful set of tools and a proper understanding of the behavior of the c-MYC protein both by itself and in complex with MAX that can underpin the development of effective chemical approaches to target MYC.
Acknowledgments
The authors thank Dr. Minmin Yu for all her help and useful discussions about X-ray crystallography and Dr. Chris Johnson for SEC–MALS analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00296.
Eight mixes of additives (44 compounds) integrated in the final formulation of MORPHEUS III (Table S1), formulation of the 96 crystallization conditions forming MORPHEUS III (Table S2), X-ray crystallography structures (Table S3), alignment of c-MYC and v-MYC (Figure S1), HSQC spectra of the reconstituted dimer with only MAX bHLHZip labeled (Figure S2), mass spectrometry diagrams (Figure S3), SEC–MALS analysis of the c-MYC:MAX complex bound to the DNA complex (Figure S4), and comparison of the loop regions in the three crystal forms of the apo complex (Figure S5) (PDF)
Accession Codes
NMR assignments of the c-MYC:MAX bHLHZip complex, BMRB entry 27571; NMR assignments of the c-MYC bHLHZip free protein, BMRB entry 12033; crystal structures of c-MYC:MAX bHLHZip complexes, PDB entries 6G6K, 6G6J, and 6G6L.
Author Contributions
S.S. and N.H. contributed equally to this work. G.Z. conceived the study. S.S., N.H., F.G., M.D.A., S.M.V.F., and G.Z. performed the experiments. M.B. and G.Z. analyzed data. F.G., M.B., and G.Z. contributed to writing of the manuscript.
This work was supported by the Karolinska Institutet (S.S., N.H., and G.Z.) and the UKRI Medical Research Council (F.G., M.D.A., S.M.V.F., and M.B.).
The authors declare the following competing financial interest(s): The authors (F.G. and G.Z.) declare the following competing financial interest: the MORPHEUS III crystallization screen is commercialized by Molecular Dimensions Ltd under an exclusive license from LifeArc (London, UK).
Supplementary Material
References
- Carroll P. A.; Freie B. W.; Mathsyaraja H.; Eisenman R. N. (2018) The MYC transcription factor network: balancing metabolism, proliferation and oncogenesis. Frontiers of medicine 12, 412–425. 10.1007/s11684-018-0650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M.; Eisenman R. N. (2008) Myc’s broad reach. Genes Dev. 22, 2755–2766. 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan G. I.; Littlewood T. D. (1993) The role of c-myc in cell growth. Curr. Opin. Genet. Dev. 3, 44–49. 10.1016/S0959-437X(05)80339-9. [DOI] [PubMed] [Google Scholar]
- Blackwell T. K.; Huang J.; Ma A.; Kretzner L.; Alt F. W.; Eisenman R. N.; Weintraub H. (1993) Binding of myc proteins to canonical and noncanonical DNA sequences. Mol. Cell. Biol. 13, 5216–5224. 10.1128/MCB.13.9.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori C.; Eisenman R. N. (1997) Myc target genes. Trends Biochem. Sci. 22, 177–181. 10.1016/S0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- Kortlever R. M.; Sodir N. M.; Wilson C. H.; Burkhart D. L.; Pellegrinet L.; Brown Swigart L.; Littlewood T. D.; Evan G. I. (2017) Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315. 10.1016/j.cell.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabò A.; Amati B. (2014) Genome Recognition by MYC. Cold Spring Harbor Perspect. Med. 4, a014191 10.1101/cshperspect.a014191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M.; McFerrin L.; Eisenman R. N. (2014) An overview of MYC and its interactome. Cold Spring Harbor Perspect. Med. 4, a014357 10.1101/cshperspect.a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W. B.; Helander S.; Pilstal R.; Hickman K. A.; Lourenco C.; Jurisica I.; Raught B.; Wallner B.; Sunnerhagen M.; Penn L. Z. (2015) Myc and its interactors take shape. Biochim. Biophys. Acta, Gene Regul. Mech. 1849, 469–483. 10.1016/j.bbagrm.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Hann S. R. (2014) MYC Cofactors: Molecular Switches Controlling Diverse Biological Outcomes. Cold Spring Harbor Perspect. Med. 4, a014399 10.1101/cshperspect.a014399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson W. C.; Weiss W. A. (2010) Myc proteins as therapeutic targets. Oncogene 29, 1249–1259. 10.1038/onc.2009.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C. V. (2012) MYC on the path to cancer. Cell 149, 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodir N. M.; Swigart L. B.; Karnezis A. N.; Hanahan D.; Evan G. I.; Soucek L. (2011) Endogenous Myc maintains the tumor microenvironment. Genes Dev. 25, 907–916. 10.1101/gad.2038411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay M.; Li Y.; Felsher D. W. (2014) MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harbor Perspect. Med. 4, a014241 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodir N. M.; Evan G. I. (2011) Finding cancer’s weakest link. Oncotarget 2, 1307–1313. 10.18632/oncotarget.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L.; Evan G. I. (2010) The ups and downs of Myc biology. Curr. Opin. Genet. Dev. 20, 91–95. 10.1016/j.gde.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L.; Whitfield J.; Martins C. P.; Finch A. J.; Murphy D. J.; Sodir N. M.; Karnezis A. N.; Swigart L. B.; Nasi S.; Evan G. I. (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castell A.; Yan Q.; Fawkner K.; Hydbring P.; Zhang F.; Verschut V.; Franco M.; Zakaria S. M.; Bazzar W.; Goodwin J.; Zinzalla G.; Larsson L.-G. (2018) A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci. Rep. 8, 10064. 10.1038/s41598-018-28107-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzalla G. (2016) Targeting MYC: is it getting any easier?. Future Med. Chem. 8, 1899–1902. 10.4155/fmc-2016-0119. [DOI] [PubMed] [Google Scholar]
- Whitfield J. R.; Beaulieu M. E.; Soucek L. (2017) Strategies to Inhibit Myc and Their Clinical Applicability. Frontiers in Cell and Developmental Biology 5, 10. 10.3389/fcell.2017.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfram J. A.; Lesnefsky E. J.; Hoit B. D.; Smith M. A.; Lee H.-g. (2011) Therapeutic Potential of c-Myc Inhibition in the Treatment of Hypertrophic Cardiomyopathy. Ther. Adv. Chronic Dis. 2, 133–144. 10.1177/2040622310393059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown M. R.; Bradner J. E. (2014) Therapeutic Strategies to Inhibit MYC. Cold Spring Harbor Perspect. Med. 4, a014266 10.1101/cshperspect.a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C. (2019) Helix-loop-helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev. 33, 6–25. 10.1101/gad.320663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B.; Littlewood T. D.; Evan G. I.; Land H. (1993) The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 12, 5083–5087. 10.1002/j.1460-2075.1993.tb06202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandori C.; Cowley S. M.; James L. P.; Eisenman R. N. (2000) The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 16, 653–699. 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- Vervoorts J.; Luscher-Firzlaff J.; Luscher B. (2006) The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 281, 34725–34729. 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- Muller J.; Eilers M. (2008) Ubiquitination of Myc: proteasomal degradation and beyond. Ernst Schering Res. Found. Workshop 1, 99–113. 10.1007/2789_2008_103. [DOI] [PubMed] [Google Scholar]
- Sun X. X.; Chen Y.; Su Y.; Wang X.; Chauhan K. M.; Liang J.; Daniel C. J.; Sears R. C.; Dai M. S. (2018) SUMO protease SENP1 de SUMOylates and stabilizes c-Myc. Proc. Natl. Acad. Sci. U. S. A. 115, 10983–10988. 10.1073/pnas.1802932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S. K.; Burley S. K. (2003) X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205. 10.1016/S0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- Baminger B.; Ludwiczek M. L.; Hoffmann B.; Kontaxis G.; Bister K.; Konrat R. (2004) Backbone assignment of the dimerization and DNA-binding domain of the oncogenic transcription factor v-Myc in complex with its authentic binding partner Max. J. Biomol. NMR 30, 361–362. 10.1007/s10858-005-1058-z. [DOI] [PubMed] [Google Scholar]
- Yin X.; Grove L.; Prochownik E. V. (1998) Lack of transcriptional repression by max homodimers. Oncogene 16, 2629–2637. 10.1038/sj.onc.1201777. [DOI] [PubMed] [Google Scholar]
- Brownlie P.; Ceska T.; Lamers M.; Romier C.; Stier G.; Teo H.; Suck D. (1997) The crystal structure of an intact human Max-DNA complex: new insights into mechanisms of transcriptional control. Structure (Oxford, U. K.) 5, 509–520. 10.1016/S0969-2126(97)00207-4. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare A. R.; Prendergast G. C.; Ziff E. B.; Burley S. K. (1993) Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363, 38–45. 10.1038/363038a0. [DOI] [PubMed] [Google Scholar]
- Sauve S.; Tremblay L.; Lavigne P. (2004) The NMR solution structure of a mutant of the Max b/HLH/LZ free of DNA: insights into the specific and reversible DNA binding mechanism of dimeric transcription factors. J. Mol. Biol. 342, 813–832. 10.1016/j.jmb.2004.07.058. [DOI] [PubMed] [Google Scholar]
- Posternak V.; Cole M. D. (2016) Strategically targeting MYC in cancer. F1000Research 5, 408–414. 10.12688/f1000research.7879.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S.; Prochownik E. V. (2015) Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta, Gene Regul. Mech. 1849, 525–543. 10.1016/j.bbagrm.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.; Sorolla A.; Cunningham P. T.; Bogdawa H. M.; Beck S.; Golden E.; Dewhurst R. E.; Florez L.; Cruickshank M. N.; Hoffmann K.; Hopkins R. M.; Kim J.; Woo A. J.; Watt P. M.; Blancafort P. (2019) Tumor penetrating peptides inhibiting MYC as a potent targeted therapeutic strategy for triple-negative breast cancers. Oncogene 38, 140–150. 10.1038/s41388-018-0421-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung L. A.; Gebhardt A.; Koelmel W.; Ade C. P.; Walz S.; Kuper J.; von Eyss B.; Letschert S.; Redel C.; d’Artista L.; Biankin A.; Zender L.; Sauer M.; Wolf E.; Evan G.; Kisker C.; Eilers M. (2017) OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 36, 1911–1924. 10.1038/onc.2016.354. [DOI] [PubMed] [Google Scholar]
- Soucek L.; Nasi S.; Evan G. I. (2004) Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 11, 1038–1045. 10.1038/sj.cdd.4401443. [DOI] [PubMed] [Google Scholar]
- Soucek L.; Jucker R.; Panacchia L.; Ricordy R.; Tato F.; Nasi S. (2002) Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 62, 3507–3510. [PubMed] [Google Scholar]
- Soucek L.; Helmer-Citterich M.; Sacco A.; Jucker R.; Cesareni G.; Nasi S. (1998) Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17, 2463–2472. 10.1038/sj.onc.1202199. [DOI] [PubMed] [Google Scholar]
- Beaulieu M.-E.; Jauset T.; Masso-Valles D.; Martinez-Martin S.; Rahl P.; Maltais L.; Zacarias-Fluck M. F.; Casacuberta-Serra S.; Serrano del Pozo E.; Fiore C.; Foradada L.; Cano V. C.; Sanchez-Hervas M.; Guenther M.; Romero Sanz E.; Oteo M.; Tremblay C.; Martin G.; Letourneau D.; Montagne M.; Morcillo Alonso M. A.; Whitfield J. R.; Lavigne P.; Soucek L. (2019) Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 11, 5012–5025. 10.1126/scitranslmed.aar5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsafou K.; Tiwari P. B.; Forman-Kay J. D.; Metallo S. J.; Toretsky J. A. (2018) Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 430, 2321–2341. 10.1016/j.jmb.2018.04.008. [DOI] [PubMed] [Google Scholar]
- Sammak S.; Zinzalla G. (2015) Targeting protein-protein interactions (PPIs) of transcription factors: Challenges of intrinsically disordered proteins (IDPs) and regions (IDRs). Prog. Biophys. Mol. Biol. 119, 41–46. 10.1016/j.pbiomolbio.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Jung K. Y.; Wang H.; Teriete P.; Yap J. L.; Chen L.; Lanning M. E.; Hu A.; Lambert L. J.; Holien T.; Sundan A.; Cosford N. D.; Prochownik E. V.; Fletcher S. (2015) Perturbation of the c-Myc-Max protein-protein interaction via synthetic alpha-helix mimetics. J. Med. Chem. 58, 3002–3024. 10.1021/jm501440q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A.; Garcia M. F.; Jaroszewicz A.; Osman N.; Pellegrini M.; Zaret K. S. (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammak S.; Allen M. D.; Hamdani N.; Bycroft M.; Zinzalla G. (2018) The structure of INI1/hSNF5 RPT1 and its interactions with the c-MYC:MAX heterodimer provide insights into the interplay between MYC and the SWI/SNF chromatin remodeling complex. FEBS J. 285, 4165–4180. 10.1111/febs.14660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazimierczuk K.; Orekhov V. Y. (2011) Accelerated NMR spectroscopy by using compressed sensing. Angew. Chem., Int. Ed. 50, 5556–5559. 10.1002/anie.201100370. [DOI] [PubMed] [Google Scholar]
- Gorrec F.; Lowe J. (2018) Automated Protocols for Macromolecular Crystallization at the MRC Laboratory of Molecular Biology. J. Visualized Exp. 131, 55790–55807. 10.3791/55790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrec F. (2015) The MORPHEUS II protein crystallization screen. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 71, 831–837. 10.1107/S2053230X1500967X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrec F. (2009) The MORPHEUS protein crystallization screen. J. Appl. Crystallogr. 42, 1035–1042. 10.1107/S0021889809042022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 58, 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- Camilloni C.; De Simone A.; Vranken W. F.; Vendruscolo M. (2012) Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry 51, 2224–2231. 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Traugh J. A.; Bishop J. M. (2004) Negative control of the Myc protein by the stress-responsive kinase Pak2. Molecular and cellular biology 24, 1582–1594. 10.1128/MCB.24.4.1582-1594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek P.; Cliff M. J.; Embrey K. J.; Holdgate G. A.; Nissink J. W. M.; Panova S.; Waltho J. P.; Davies R. A. (2018) Myc phosphorylation in its basic helix-loop-helix region destabilizes transient alpha-helical structures, disrupting Max and DNA binding. J. Biol. Chem. 293, 9301–9310. 10.1074/jbc.RA118.002709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieber W.; Schneider M. L.; Matt T.; Krautler B.; Konrat R.; Bister K. (2001) Structure, function, and dynamics of the dimerization and DNA-binding domain of oncogenic transcription factor v-Myc. J. Mol. Biol. 307, 1395–1410. 10.1006/jmbi.2001.4537. [DOI] [PubMed] [Google Scholar]
- https://www2.mrc-lmb.cam.ac.uk/groups/JYL/WWWrobots/robot-overview.html.
- Pogenberg V.; Ogmundsdottir M. H.; Bergsteinsdottir K.; Schepsky A.; Phung B.; Deineko V.; Milewski M.; Steingrimsson E.; Wilmanns M. (2012) Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 26, 2647–2658. 10.1101/gad.198192.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.