

Abstract
Pyrroloquinoline quinone is a prominent redox cofactor in many prokaryotes, produced from a ribosomally synthesized and post-translationally modified peptide PqqA via a pathway comprising four conserved proteins PqqB–E. These four proteins are now fairly well-characterized and span radical SAM activity (PqqE), aided by a peptide chaperone (PqqD), a dual hydroxylase (PqqB), and an eight-electron, eight-proton oxidase (PqqC). A full description of this pathway has been hampered by a lack of information regarding a protease/peptidase required for the excision of an early, cross-linked di-amino acid precursor to pyrroloquinoline quinone. Herein, we isolated and characterized a two-component heterodimer protein from the α-proteobacterium Methylobacterium (Methylorubrum) extorquens that can rapidly catalyze cleavage of PqqA into smaller peptides. Using pulldown assays, surface plasmon resonance, and isothermal calorimetry, we demonstrated the formation of a complex PqqF/PqqG, with a KD of 300 nm. We created a molecular model of the heterodimer by comparison with the Sphingomonas sp. A1 M16B Sph2681/Sph2682 protease. Analysis of time-dependent patterns for the appearance of proteolysis products indicates high specificity of PqqF/PqqG for serine side chains. We hypothesize that PqqF/PqqG initially cleaves between the PqqE/PqqD-generated cross-linked form of PqqA, with nonspecific cellular proteases completing the release of a suitable substrate for the downstream enzyme PqqB. The finding of a protease that specifically targets serine side chains is rare, and we propose that this activity may be useful in proteomic analyses of the large family of proteins that have undergone post-translational phosphorylation at serine.
Keywords: protease, zinc, serine, enzyme catalysis, peptides, heterodimer, M16B protease, PQQ
Introduction
Pyrroloquinoline quinone (4,5-dihydro-4,5-dioxo-1H-pyrrolo-[2,3-f]quinoline-2,7,9-tricarboxylic acid (PQQ))3 is a peptide-derived redox quinocofactor, present in several bacterial dehydrogenases (1). PQQ was initially described in 1964 as the prosthetic group of a glucose dehydrogenase in Bacterium anitratum (2) and is part of a family of quinocofactors that includes trihydroxyphenylalanine quinone, tryptophan tryptophylquinone, lysyl tyrosine quinone, and cysteine tryptophylquinone. However, unlike the other members of this family, PQQ is a freely dissociable cofactor that acts as a noncovalently bound cofactor for a range of dehydrogenases (3, 4).
PQQ is mainly produced by Gram-negative bacteria, where it accumulates in the periplasmic space (5) and has been referred to as a bacterial vitamin (6). PQQ is critical for C1 metabolism in many microorganisms, whereas other species, such as Escherichia coli, that are unable to produce PQQ autonomously (7) remain able to obtain this cofactor from the medium (8). Trace amounts of PQQ have also been detected in mammalian tissues, attributed to dietary sources (9) because mammals appear incapable of its biosynthesis (10–14). Early studies showed that PQQ deficiencies in mice lead to immune system dysfunction, growth impairment, and abnormal reproductive performance (15, 16), and the health benefits of PQQ have been attributed to its antioxidant effects (17). PQQ was recently implicated as a modulator of mammalian lactate dehydrogenase in vitro (18), and Saihara et al. (19) reported evidence that PQQ stimulates mitochondrial biogenesis in mouse fibroblasts.
The dependence of numerous opportunistic pathogens on PQQ for optimal growth, together with the implied physiological and biochemical functions of PQQ (14, 17, 20), make knowledge of its biosynthetic pathway both timely and important. This pathway has been studied since 1987 (21) when the genes encoding the pathway's proteins were cloned. PQQ is derived from a ribosomally produced (PqqA) and post-translationally modified peptide (PqqA*), in a series of reactions involving several enzymes and a peptide chaperone (Fig. 1). PqqB, PqqC, PqqD, and PqqE are essential and conserved (22), whereas the protease, usually called PqqF, may or may not be present in the operon. The biosynthesis of PQQ is initiated by the formation of a C–C bond between glutamate and tyrosine residues in PqqA, with the help of the chaperone PqqD, creating a cross-linked product PqqA* (23). The steps that follow this reaction include the processing of PqqA* by a protease and the action of PqqB, which was recently found to be an iron-dependent hydroxylase that installs the quinone moiety of PQQ-forming AHQQ (24). The last reaction of the pathway is catalyzed by PqqC, the first enzyme to be characterized in this pathway (25–27). PqqC is a cofactorless oxidase that converts the isoquinoline heterocycle AHQQ into PQQ, by cyclization and overall, four stepwise two-electron, two-proton oxidative reactions.
Figure 1.

The proposed PQQ biosynthetic pathway in M. extorquens. Activity has been confirmed for PqqD/PqqE (23, 38) and PqqC (25). The reaction catalyzed by PqqB has been inferred using substrate analogs (24).
In recent years this laboratory has turned its attention to the PQQ biosynthetic pathway in Methylobacterium extorquens (Mex) AM1 (recently reclassified into a new genus, Methylorubrum (28)), because of the greater access and stability of gene products from the Mex PQQ operon. The aim of the present work has been to locate and describe (a requisite) protease for the scission of the cross-linked di-amino acid from the parent peptide (Fig. 1). The first and only biochemical characterization of a designated protease for the PQQ pathway is PqqF from the γ-proteobacterium Serratia sp. FS14. Wei et al. (29) crystallized this protein and assigned it as an M16A peptidase of the ME clan, characterized as containing the zinc-binding motif HXXEH (30). Earlier, the Lidstrom group (31) had identified two gene clusters required for the growth of Mex AM1 mutants on methanol named pqqDGCBA and pqqEF. The pqqDGCBA has been shown to correspond to the first five genes of the Klebsiella pneumoniae operon, pqqABCDE (32). Subsequent transcriptional regulation studies of growth of Mex on methanol showed induction of both pqqABC/DE and the downstream cluster (33), supporting an essential role for the latter region in PQQ production. With the publication of the Mex AM1 genome sequence in 2009 (34), the stretch of DNA corresponding to pqqEF was redefined as encoding two insulinase-like proteins that were classified as M16 proteases. In the present work we interrogate the two downstream PQQ-linked genes, showing them to encode a two-component peptidase with high activity toward PqqA, the initial peptide substrate of the PQQ pathway, from which the putative protease substrate, PqqA*, is formed by the catalytic action of PqqE with the help of PqqD. Further sequence alignment and three-dimensional threading indicate that PqqF/PqqG belong to the protease M16B family. A major unexpected result is the high specificity of cleavage by PqqF/PqqG at serine, a property that may provide a new proteomic tool for the analysis of post-translational modification of protein serine side chains in vivo.
Results
Cloning and expression of Mex pqqF and pqqG
The region of the Mex genome reported as pqqEF was originally annotated as a 709-amino acid protein (tentatively assigned to pqqE) together with a 219-amino acid protein (pqqF, sharing identity with endopeptidases) (31, 35). In this work we resequenced the part of the Mex genome where these genes are located and confirmed the presence of two ORFs encoding insulinase-like proteins, one with 460 amino acids (PqqF, accession number WP_003599161.1) and one with 427 amino acids (PqqG, accession number WP_003599163.1) (Fig. 2 and Table S1). The indicated PqqF and PqqG were cloned from Mex and expressed as His6 tag fusions in E. coli, yielding soluble protein fractions in both instances. Isolation by affinity chromatography on nickel columns led to good yields of homogeneous proteins: 25–30 mg/liter medium in the case of His6-PqqF and 11–24 mg/liter medium in the case of His6-PqqG. The His6 tags were removed by treatment with thrombin, and protein was further purified by size exclusion chromatography (in the case of PqqF) or affinity chromatography on a nickel column (in the case of PqqG). In all cases, the samples were analyzed by SDS-PAGE and by MS to confirm the protein identity and the purity (see Fig. S1 for an example of a gel of purified His-tagged and nontagged PqqF and PqqG).
Figure 2.

Arrangement of the PQQ biosynthesis protein–encoding genes in the Mex genome. The numbers indicate the basepair position in the genome. A denotes pqqA, the gene encoding the peptide precursor. pqqF and pqqG encode the proteins characterized in this paper.
The interaction of Mex PqqF and PqqG
We interrogated the possibility of complex formation between PqqF and PqqG, by analogy to the interaction of TldD with TldE described by Ghilarov et al. (36). We first used pulldown assays to investigate the protein–protein interaction (37). His6-PqqF was immobilized in a nickel affinity column and served as the “bait” protein. PqqG, the “prey” protein, was then passed through the column, and the flowthrough was examined by SDS-PAGE. Controls without bait and without prey were performed, and the aggregate results are shown in Fig. 3. Comparing lanes 7 and 8, it is clear that PqqG is immobilized in the column only when His6-PqqF is present (lane 7, no PqqG in the flowthrough). Both proteins are eluted when imidazole is added to the buffer (lane 11).
Figure 3.

Interaction between PqqF and PqqG detected by pulldown assays in an affinity nickel column (His SpinTrapTM). Lanes 1 and 9, protein ladder (10–200 kDa). Lane 2, bait protein, His6PqqF. Lanes 3 and 5, flowthrough of His6PqqF. Lane 4, flowthrough of buffer from column without bait. Lane 6, prey protein, PqqG. Lanes 7 and 10, flowthrough of PqqG solution through column with bait. Lane 8, flowthrough of PqqG solution through column without bait. Lanes 11, 12, and 13, elution with 250 mm imidazole from columns shown in lanes 7, 8, and 5, respectively. MW, molecular mass.
After observing this qualitative interaction between PqqF and PqqG, we pursued the dissociation constant (KD) for complex formation. Initially, surface plasmon resonance (SPR) was employed, because this method requires minimal protein material, and our lab has had previous success with the technique (38). However, when His6-PqqG was immobilized to a nickel–nitrilotriacetic acid gold chip, there was a concentration-dependent, nonspecific absorption of PqqF, which was apparent from the blank subtracted SPR response not returning to baseline (Fig. S2). Likewise, when His6-PqqF was immobilized to the chip, we observed nonspecific absorption of PqqG to the chip, albeit less severely.
We next turned to isothermal calorimetry (ITC), in which His6-PqqG was titrated by a concentrated stock of His6-PqqF and the heat change compared with controls where either His6-PqqF or His6-PqqG were replaced by dialysis buffer. Thermograms were obtained in triplicate, leading to a fitted KD for the PqqF/PqqG complex of 300 ± 70 nm, with an estimated molar ratio of 1:1 (Fig. 4). This submicromolar KD value suggests that the PqqF/PqqG complex will form in vivo via a strong transient (or permanent) interaction (39).
Figure 4.
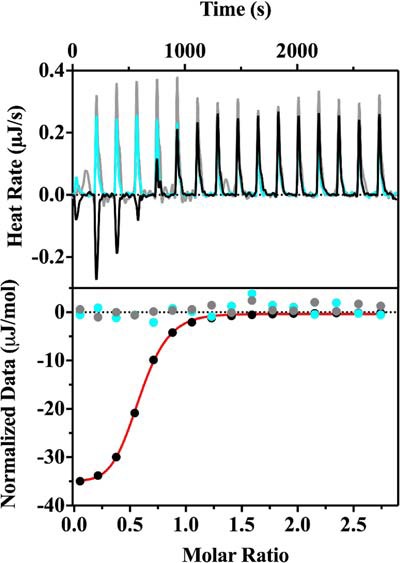
Binding of PqqF and PqqG observed by ITC. The representative raw isotherm traces (top panel) and the integral data points (bottom panel) are shown for an experiment with His6PqqF and His6PqqG (black). Controls with just His6PqqF (cyan) and just His6PqqG (gray) are also shown. As discussed under “Experimental Procedures,” both proteins were dialyzed in the same container against 50 mm Tris, pH 8, 300 mm NaCl. In a typical experiment, a sample of 20 μm His6-PqqG was titrated with a concentrated stock (200 μm) of His6-PqqF. The first injection for each experiment was omitted in the data analysis. The thermograms were analyzed by the companion software NanoAnalysis (TA Instruments), and the single-site model fitted curve generated from the experimental data is shown in red. A Kd of 300 ± 70 nm was obtained from triplicate assays.
Proteolytic activity of PqqF/PqqG toward PqqA.
Because of a limited availability of chemically synthesized Mex PqqA, our experimental strategy was to screen broadly for enzyme activity using either separate samples of PqqF or PqqG or a 1:1 complex of these two proteins. The mass spectrometry–based assays for loss of PqqA as a function of time are presented in Fig. 5A. As shown, PqqA remains undigested for up to 1 h after incubation with PqqG alone. By contrast, PqqF alone or in complex with PqqG gives rise to 50 and 90% cleavage after 10 min, respectively. From Fig. 5B, it can be seen that, at the high levels of enzyme used in the present study, an initial 2-fold increase in the PqqF/PqqG complex increases the rate of PqqA cleavage ∼2-fold and then levels off.
Figure 5.
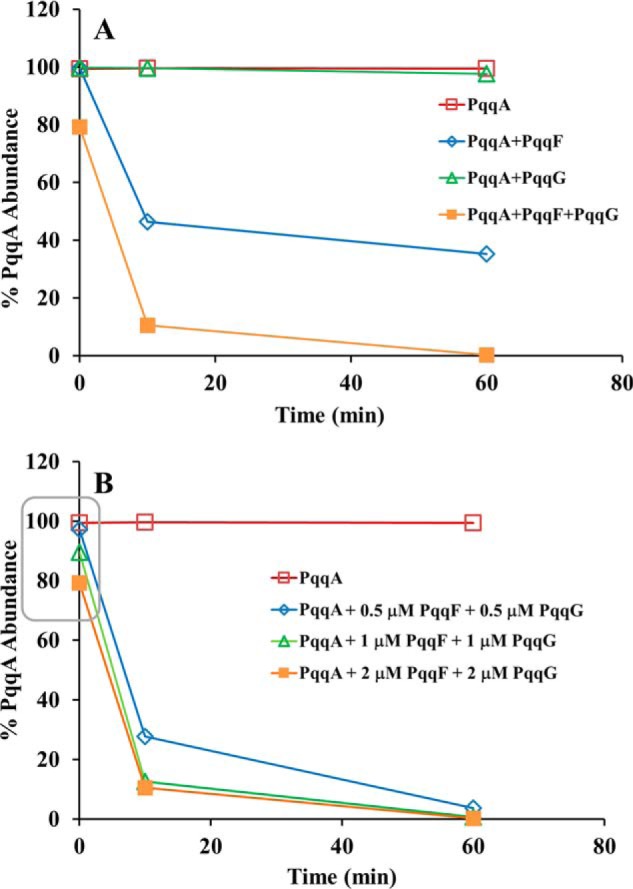
Activity of PqqF and/or PqqG with PqqA(C11S) monitored by PqqA disappearance via MS. All assays were performed in autoclaved 50 mm Tris-HCl, 300 mm NaCl, pH 8.0 buffer at room temperature. A, effect of individual enzymes and the enzyme complex. The concentration of PqqF and PqqG (1:1) was 2 μm, whereas PqqA was 20 μm. B, effect of enzyme concentration. PqqF and PqqG were used at the indicated concentrations, and PqqA was at 20 μm. The area highlighted in a rectangle clearly shows that there is an effect of the enzyme concentration on the initial reaction rate. In all experiments, a control of PqqA in buffer was performed, and the PqqA relative abundance was determined by a mass spectrometric assay, as previously described (23). All results shown are from a representative data set.
The rapid hydrolysis of PqqA in the presence of PqqF/PqqG is striking, and future structure–function analyses are being developed for assays of recombinantly produced PqqA and analogs thereof, and the cross-linked peptide PqqA* derived from the action of PqqE/PqqD. Testing an available truncated version of PqqA, EVTSY, containing the Glu and Tyr side chains that undergo cross-linking by PqqE (Fig. 1), we find a very slow and incomplete hydrolysis for up to 4 h, approximating only 1% of the initial substrate (Fig. 6).
Figure 6.
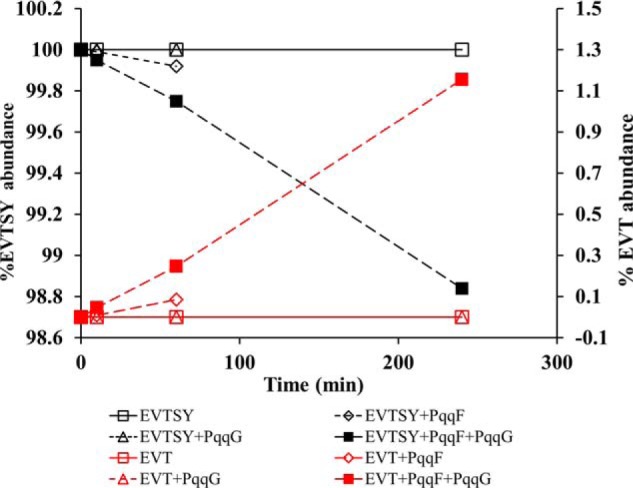
Activity of PqqF and/or PqqG with the short PqqA subpeptide EVTSY. The assays were performed in autoclaved 50 mm Tris-HCl, 300 mm NaCl, pH 8.0 buffer at room temperature. The concentration of PqqF and PqqG was 2 μm, and the concentration of EVTSY was 20 μm. Note that the y axis is greatly expanded and that the maximum amount of the product EVS is ∼1% obtained after a 250-min incubation. In all experiments, a control of EVTSY in buffer was performed, and the relative abundance of EVTSY and possible ion products was determined by the mass spectrometric assay previously described (23). The results shown are from a representative data set.
We next turned to an analysis of the time and positional dependence of products released from the full-length PqqA in the presence of PqqF/PqqG. We focused on the higher intensity peaks, corresponding to peptide products whose percentage abundance was more than 3%, and obtained the results shown in Fig. 7. Analysis of the formed peptide products indicates a preference of the PqqF/PqqG complex for serine residues, at Ser–Glu, Ser–Val, Ser–Tyr, and Glu–Ser and less efficiently at Thr-Ser. The most rapid appearance of an N-terminal peptide product KWAAPIVSEIS indicates an initial cleavage at Ser11-Val12; note that the native peptide has Cys at position 11 and is not expected to be cleaved between amino acids at positions 11 and 12. This is followed by cleavage of Ser8-Glu9 yielding KWAAPIVS, concomitant with a decrease in the abundance of KWAAPIVSEIS (Fig. 7A). After 10 min, central peptides such as VGMEVTS, VGMEVTSYE, and VGMEVT are detectable, a consequence of the cleavage at Ser11-Val12, as well as cleavage at Ser18-Tyr19, Glu20-Ser21, and Thr17-Ser18 (Fig. 7B). The C-terminal peptides YESAEIDTFN and SAEIDTFN are also detected (Fig. 7C). Cleavages at Val7-Ser8, Ile10-Ser11, and Ser21-Ala22 do not seem to happen rapidly or at all, because the corresponding peptides were not detected for up to 4 h (results not shown). Taken together, these results suggest the targeting of Mex PqqF/PqqG to both N- and C-terminal serine side chains with a likely preference for hydrogen bonding and/or negatively charged flanking side chains (Fig. 7D). The one instance in which cleavage occurred adjacent to a hydrophobic side chain was for a nonnative Ser site (position 11 in which Cys was replaced by Ser in the synthetically prepared peptide substrate).
Figure 7.

Main peptide products derived from the activity of PqqF/PqqG on PqqA. A, N terminus peptides. B, central peptides. C, C terminus peptides. D, suggested cleavage pattern by PqqF/PqqG. The results shown are from a representative data set.
Structural modeling of the PqqF/PqqG heterodimer
The M16 family of peptidases is subdivided into M16A, M16B and M16C (40), depending on the different domain organization in these peptidases (Fig. 8). A template for comparative structural modeling of the Mex PqqF/PqqG heterodimer was identified based on sequence similarity to the heterodimeric M16B protease comprised of Sph2681/Sph2682 from Sphingomonas sp. A1 (PDB code 3amj). These two monomers, displaying 41 and 32% sequence identity, respectively, with PqqF and PqqG, were the highest rated polypeptides of known structure. The best comparative model of the PqqF/PqqG heterodimer shows a high degree of structural similarity toward the template structure (root mean square deviation of 0.96 Å for the aligned carbon α atoms; Fig. 9A). In Fig. 9B, a close-up of the zinc center highlights the coordinating histidines and the catalytic R/Y pair on the B chain. The residue orientation is nearly identical in the template and target. Zinc analyses of the as-isolated proteins using ICP have shown that in the absence of any reconstitution PqqG is metal-free and that PqqF binds Zn2+ (0.2 μm).
Figure 8.
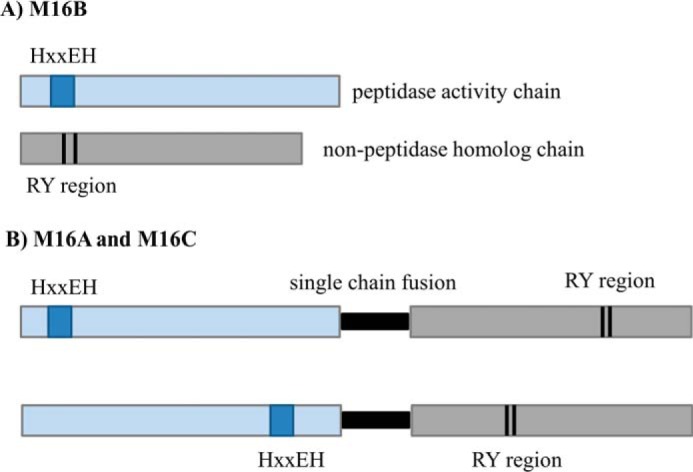
Differences between domain organization in metallopeptidases M16A, M16B, and M16C. A, the M16.019/nonpeptidase homolog heterodimer of the M16B subfamily, which is the arrangement found in PqqF/PqqG. B, single chain fusion of two tandem domains connected by a short hinge loop (in black) that characterizes the M16A and M16C subfamilies. The M16A arrangement is widely distributed among PqqF found in γ-proteobacteria.
Figure 9.

Structural model of the PqqF/PqqG heterodimer. A, alignment of the model (PqqF in gold and PqqG in blue) with the template structure (Sph2681 in green and Sph2682 in gray) with a root mean square deviation of 0.96 Å for PqqF/Sph2681 and 1.65 Å for PqqG/Sph2682. B, close-up of the metal center, showing the coordinating residues (gold and green), catalytic R/Y (blue and gray), and putative catalytic base (red and pink). The zinc ion is in teal.
The diversity of PQQ proteases in proteobacteria
In light of the present results, we proceeded to survey annotated PQQ-producing bacterial species for additional examples of the presence of PqqF and/or PqqG in pqq operons (Table S2). In the case of α-proteobacteria like Mex, we observe that genes for the two-component PqqF/PqqG system are located outside of the major portion of the pqq operon, either upstream or downstream (Fig. S3). On the other hand, γ-proteobacteria species possess an M16A-like peptidase comprised of a single-chain protein with two tandem domains of ∼500 amino acids each, and a less conserved HXXEH zinc-binding site (analogous to the PqqF found in the pqq operon of K. pneumonia (41)). This ORF can be located either within or outside the major pqq operon (Fig. S3). It is interesting to note that we did not locate any α-proteobacteria species containing pqqF within their pqq operon, even in the case of species with an M16A PqqF of similar size to the ones found in most γ-proteobacteria (e.g. Acetobacter pasteurianus, Agrobacterium radiobacter, and Gluconoacetobacter diazotrophicus; Table S2). The sequence alignments of several PqqF from α-proteobacteria (Fig. S4) and γ-proteobacteria (Fig. S5) clearly show the M16B and M16A signatures, respectively (Fig. 8). In the case of α-proteobacteria, an alignment of the PqqG sequence shows the RY region (Fig. S6) commonly found in the C terminus of the M16A PqqF from γ-proteobacteria (Fig. S5B).
Discussion
The biosynthesis of PQQ has been studied since the late 1980s (42), but a complete understanding of the details of the pathway has been elusive. The PQQ operon was identified in 1987 (21), and shortly afterward two independent groups showed that the amino acids tyrosine and glutamate contributed all of the carbon and nitrogen atoms within the final cofactor structure (43, 44). Goosen et al. (45) identified a gene encoding a 24-amino acid peptide (later called PqqA) as the precursor of PQQ biosynthesis in Acinetobacter calcoaceticus. The PqqC structure and function were solved in 2004 (27, 46), and PqqD was later shown to be a peptide chaperone interacting with PqqA and PqqE (38), a radical SAM enzyme (47). Although PqqE had been the candidate for the catalysis of the carbon-carbon bond formation between tyrosine and glutamate, evidence for this reaction did not appear until 2016, together with the elucidation of the critical role played by PqqD (23). With the recent discovery that PqqB is an iron-dependent hydroxylase (24), only the role of an essential protease remained unknown. Previous work showed that PqqF is necessary for PQQ production in Pseudomonas aeruginosa (48) and Serratia sp. FS14 (29). However, other reports indicated that PqqF did not seem to be essential for PQQ biosynthesis in select microorganisms (31). This feature, as well as the fact that PqqF is not uniformly present in the pqq operon (22, 31), has made identification of proteases targeted to PQQ biosynthesis difficult.
This study shows that in Mex AM1, the primary microorganism currently used by our group to study the PQQ biosynthetic pathway, two proteins, PqqF and PqqG, encoded by separate genes located downstream of the main pqq operon (Fig. 2), interact to form a M16B heterodimer (Figs. 3 and 4) that catalyzes the proteolysis of PqqA (Fig. 5). The pqqF and pqqG genes are different from the ones initially described by Springer et al. (31). Several prokaryotic M16B peptidases have been predicted based on sequence alignment (49), and Maruyama et al. (50) reported the first heterodimeric M16B peptidase similar to the one presented here, in Sphingomonas sp. A1. Our observation of activity for PqqF alone is in contrast with what was observed for the Sphingomonas sp. A1 Sph2681, one of the subunits of the heterodimer on which we modeled PqqF/PqqG (Fig. 9). Unlike Mex PqqF, Sph2681 with 41% sequence identity does not exhibit any peptidase activity by itself (50). The heterodimer Sph2681–Sph2682 is active toward several peptides and proteins from 5 to 51 amino acid residues, with maximum specific activity obtained for a 31-mer β-endorphin substrate. MALDI-TOF analyses of the proteolyzed products suggest a random cleavage of the peptide/protein substrates (50).
Despite the above differences in activity and specificity and an average of ≈35% sequence identity, we have constructed a comparative model of the PqqF/PqqG heterodimer based on the Sphingomonas sp. protease structure (Fig. 9). It can be seen that there is a high structural similarity between the two heterodimers, supported by good quality assessment indicators for the model and pointing to a reliable 3D prediction. Although based on the closed dimer structure present in the 3amj PDB file (chains A+B) of the Sphingomonas sp. protease, model building shows it is possible to also fit the monomers to the open conformation (3amj, chains C + D). In this conformation, a likely entry point for the cross-linked substrate PqqA* is made evident (see Fig. S7). We note that an estimate of theoretical binding energies for the formation of a closed conformation dimer of PqqF/PqqG (data not reported) are in reasonable agreement with the experimental KD for the binding of PqqF to PqqG obtained herein (Fig. 4).
From activity assays (Figs. 5 and 7), a chemically synthesized form of PqqA (identical to the native peptide with the exception of C11S) is concluded to be a robust substrate for PqqF/PqqG. Activity was followed by the decrease in PqqA abundance and the formation of peptide products, monitored by MS. Although PqqF is capable of some proteolytic activity with PqqA, the reaction catalyzed by PqqF/PqqG is much faster, with a significant decrease in the abundance of PqqA seen at t ≅ 0, when both proteins are present (Fig. 5A). Increasing the protein concentration increases the reaction rate (Fig. 5B), but initial rate studies will be needed to obtain quantitative results for initial rates versus catalyst concentration. This is expected to become possible once recombinant levels of PqqA are available and/or via modification of the assay that currently involves reaction quenching and product analysis by MS. The heterodimer did not catalyze the cleavage of the short substrate analog EVTSY (Fig. 6) to a significant extent, showing either a role for flanking amino acids for efficient hydrolysis and/or a requisite precursor relationship in cleavage.
Using the synthesized PqqA (C11S), mainly seven proteolytic products were detected by MS: the N-terminal peptides KWAAPIVSEIS and KWAAPIVS; central peptides products VGMEVTS and VGMEVTSYE and, at lower abundance, VGMEVT; and the C-terminal peptides YESAEIDTFN and SAEIDTFN. Taking these results together, a pattern of cleavage at serine residues emerged, as shown in Fig. 7D. The biologically relevant substrate of the protease is expected to be the cross-linked PqqA* product from the activity of PqqE, with the presence of a peptide chaperone (PqqD) facilitating cross-linking while possibly protecting the underivatized peptide from hydrolysis in vivo (Fig. 1). We hypothesize that one of the main products when using the cross-linked product from native Mex PqqA will be: 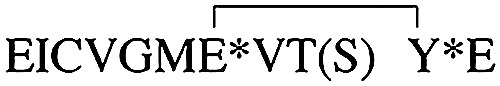 , where the asterisks denote the cross-linked residues Glu and Tyr. Subsequent trimming from the N and C termini will be required (via catalytic activity of additional protease(s)) to generate the unadorned cross-linked diamino acid as the postulated substrate of PqqB (Fig. 10). According to MEROPS, the Peptidase Database (https://www.ebi.ac.uk/merops) (69),4 171 known and putative peptidases and 36 nonpeptidase homologs have been identified in Mex AM1, and 56 of these are unassigned peptidases of unknown function. Thus, there are several potential candidates for the catalytic activity required to further process the peptide obtained from the activity of PqqF/PqqG. Interestingly, α-proteobacteria seem to be rich in prokaryotic caspase homologs, termed metacaspases (51) and, more recently, orthocaspases (52). These homologs are structurally and functionally diverse and might have roles not only in programmed cell death but also on signaling, enzyme catalysis, and protein modification (51). Caspases were initially thought to be highly specific to cleave after aspartic acid residues, but the work by Seaman et al. (53) suggested that these proteases can also cleave after glutamic acid residues (note that the putative additional cleavages of our peptide are at E residues) and, rarely, at phosphoserine residues. Using MEROPS, we identified one protease of the caspase family (clan PA, family S1) in Mex, but it has not been biochemically characterized. In summary, further work will be needed to identify the enzyme(s) responsible for the further trimming of the peptide, but several candidates are available in the Mex genome.
, where the asterisks denote the cross-linked residues Glu and Tyr. Subsequent trimming from the N and C termini will be required (via catalytic activity of additional protease(s)) to generate the unadorned cross-linked diamino acid as the postulated substrate of PqqB (Fig. 10). According to MEROPS, the Peptidase Database (https://www.ebi.ac.uk/merops) (69),4 171 known and putative peptidases and 36 nonpeptidase homologs have been identified in Mex AM1, and 56 of these are unassigned peptidases of unknown function. Thus, there are several potential candidates for the catalytic activity required to further process the peptide obtained from the activity of PqqF/PqqG. Interestingly, α-proteobacteria seem to be rich in prokaryotic caspase homologs, termed metacaspases (51) and, more recently, orthocaspases (52). These homologs are structurally and functionally diverse and might have roles not only in programmed cell death but also on signaling, enzyme catalysis, and protein modification (51). Caspases were initially thought to be highly specific to cleave after aspartic acid residues, but the work by Seaman et al. (53) suggested that these proteases can also cleave after glutamic acid residues (note that the putative additional cleavages of our peptide are at E residues) and, rarely, at phosphoserine residues. Using MEROPS, we identified one protease of the caspase family (clan PA, family S1) in Mex, but it has not been biochemically characterized. In summary, further work will be needed to identify the enzyme(s) responsible for the further trimming of the peptide, but several candidates are available in the Mex genome.
Figure 10.

Downstream region of the PQQ biosynthetic pathway showing the predicted proteolytic steps catalyzed by PqqF/PqqG (described in this work) and other possible proteases.
Currently, our group is optimizing the kinetics of formation of the cross-linked product PqqA* (catalyzed by PqqE in the presence of the chaperone PqqD), to obtain material in high yield and purity for further testing and activity comparison with PqqF/PqqG, especially concerning the reaction products. Additionally, we are developing a recombinant method to obtain a stable, nondimerizing PqqA that retains Cys at position 11. Looking to the future, the highly unexpected observation of a protease with specificity toward serine side chains at both the C and N termini of a peptide is of considerable possible interest. This type of selectivity, which is rarely observed in biology (54), may provide a novel tool for proteomic analyses of in vivo post-translational protein phosphorylation.
Experimental procedures
Materials
All restriction enzymes, the Phusion polymerase, T4 DNA ligase, and the protein ladder for SDS-PAGE were obtained from New England Biolabs (Ipswich, MA). All oligonucleotide primers were purchased from Eurofins Genomics (Louisville, KY). DNA was sequenced by the University of California Sequencing Facility (Berkeley, CA). The peptide PqqA (Uniprot number Q49148) was synthesized by CPC Scientific (Emeryville, CA) as KWAAPIVSEISVGMEVTSYESAEIDTFN, with Ser substituting for Cys11 to avoid possible dimer formation. A control subpeptide, containing the Try and Glu residues involved in PqqA cross-link (EVTSY), was synthesized by the same company.
PqqF and PqqG cloning strategies
The genome portion containing the two genes of interest, 2,400,880 to 2,403,819 bp, was amplified from Mex AM1 genomic DNA (DSMZ, Braunschweig), using different combinations of primer pairs Mex_seq_Fwd1–4 and Mex_seq_Rev1–2 (Table 1) and sequenced. After identifying the genes encoding the putative Mex proteases PqqF and PqqG, these were amplified from the same genomic DNA, using the PqqF_Clon and PqqG_Clon primer set (Table 1) with the Phusion polymerase system. The PCR products were cloned into pET28a vectors (Novagen) using the NheI and HinIII restriction sites. The plasmids were transformed into E. coli XL1 Blue competent cells (Stratagene) and selected on LB agar with 50 μg/ml kanamycin (LBKan50). The plasmids harboring the correct constructs, His6-PqqF and His6-PqqG, were cotransformed into E. coli BL21-CodonPlus(DE3)-RIPL competent cells (Agilent) and also selected on LBKan50.
Table 1.
Primers used in the amplification and sequencing procedures
NA, not applicable.
| Primer | Sequence (5′ → 3′) | Annealing temperature | Restriction sites introduced |
|---|---|---|---|
| °C | |||
| Mex_seq_Fwd1 | ATCTCGGCGGGTCGGCGAAG | 68.6 | NA |
| Mex_seq_Fwd2 | CGCGACGTGGTGCTGGAGGA | 68.6 | NA |
| Mex_seq_Fwd3 | GAGGGCGTGACCCTGGAGGCC | 72.3 | NA |
| Mex_seq_Fwd4 | CGGGCGATCTCGACTCGGAT | 66.6 | NA |
| Mex_seq_Rev1 | GCCGTGATGGAATTGGTGGAA | 62.6 | NA |
| Mex_seq_Rev2 | CACGAACAGCGAGGCGGACA | 66.6 | NA |
| PqqF_clon_Fwd | aatagctagcATGCACCTCTACCGGAAGGCC | 66.5 | NheI |
| PqqF_clon_Rev | taaaagcttATCAGGGCCGTCTCAGGCGAT | 66.5 | HindIII |
| PqqG_clon_Fwd | aatagctagcATGAATCTCGCCGAGACCGG | 64.5 | NheI |
| PqqG_clon_Rev | taaaagcttCTACAGCCCCGTGGGCCG | 69.0 | HindIII |
| PqqF_seq_Fwd | ATGCACCTCTACCGGAAGGCC | 66.5 | NA |
| PqqF_seq_Rev | ATCAGGGCCGTCTCAGGCGAT | 66.5 | NA |
| PqqG_seq_Fwd | ATGAATCTCGCCGAGACCGG | 64.5 | NA |
| PqqG_seq_Rev | taaaagcttCTACAGCCCCGTGGGCCG | 69.0 | NA |
Protein expression and purification
The His-tagged proteins were expressed in Terrific broth with 50 μg/ml kanamycin (TBKan50) supplemented with 10 μm ZnSO4. The cultures were grown at 37 °C, 220 rpm until an A600 = 0.8, and protein expression was induced with isopropyl β-d-1-thiogalactopyranoside (final concentration, 100 μm). Expression proceeded for 22 h at 17 °C, 220 rpm, and afterward the cells were harvested by centrifugation, and the pellets were frozen at −80 °C until use. Protein purification was done by affinity chromatography using His60 nickel Superflow (Clontech) columns. The cell pellets were resuspended in lysis buffer (50 mm Tris-HCl, pH 8.8, 300 mm NaCl, 10% (v/v) glycerol, 1 mm phenylmethylsulfonyl fluoride) and lysed by sonication on ice, and the cell-free extracts were obtained by centrifugation of the lysates. The extracts were loaded into the columns previously equilibrated with lysis buffer, and the columns were sequentially washed with buffer containing 5 and 20 mm imidazole, respectively, to remove nontagged proteins, and finally the tagged proteins were eluted with buffer containing 250 mm imidazole. The desired fractions were pooled and dialyzed overnight against 50 mm Tris-HCl, pH 8.8, 300 mm NaCl, 10% (v/v) glycerol at 4 °C. The dialyzed protein solutions were concentrated as needed, aliquoted, and flash frozen in liquid nitrogen. Protein concentration and purity were monitored along the process by absorbance at 280 nm, by the Bradford assay (55), and by SDS-PAGE using the method of Laemmli (56).
MS of the native and digested proteins
Small volumes (∼1.5 ml) of the purified protein solutions (predialysis) were buffer exchanged over PD-10 columns (GE Healthcare) pre-equilibrated with 50 mm Tris-HCl, pH 8.8, 300 mm NaCl, to remove the glycerol and imidazole. Aliquots, with a concentration of ≥30 μm, were used for intact protein MS. The remaining volume was concentrated by centrifugal ultrafiltration in an Amicon Ultra4 10K (Millipore). The protein was denatured by incubation with urea and DTT at 55 °C for 15 min and then digested with trypsin (in a concentration less than 50 times that of the protein being digested) in 50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 1 mm DTT, and 1 mm CaCl2, at room temperature for 4 h. Samples of intact and digested proteins were analyzed using Synapt and Orbitrap mass spectrometers, respectively, as described previously (57, 58).
Cleavage of His6 tag with thrombin
His6-tagged protein samples were defrosted on ice and incubated with biotinylated thrombin (Novagen), 2 units of thrombin/mg of tagged protein for 16 h at 14 °C in the buffer provided by the supplier. Streptavidin-agarose (Novagen) was then added to remove the thrombin, and the mixture was gently shaken for 30 min at room temperature and transferred to 0.45-μm cellulose acetate centrifugal microfilters (GVS Life Sciences) to separate the filtrate. The untagged PqqG was separated from the cleaved His tag and the uncleaved enzyme by passing the mixture through a His60 nickel Superflow (Clontech) column and collecting the flowthrough. The untagged PqqF was found to nonspecifically interact with the nickel column and had to be recovered by size-exclusion chromatography on a HiPrep 16/60 Sephacryl S-200 High resolution column (GE Healthcare) outfitted to an AKTA FPLC (GE Healthcare) with a flow rate of 0.5 ml/min at 4 °C. The mobile phase was 20 mm Tris-HCl, 0.15 m NaCl, 5% (v/v) glycerol, and the elution was monitored by absorbance at 280 nm. The fractions containing protein were collected and analyzed by SDS-PAGE, and the ones containing the untagged enzymes were pooled and concentrated by ultrafiltration (Amicon Ultra15 30 kDa). The samples were analyzed by native MS, as described previously, and the concentration was determined by absorbance at 280 nm.
ICP optical emission spectroscopy analysis
The analysis of zinc content in as purified His tag–cleaved PqqF (10 and 20 μm) was performed in at least triplicate using a PerkinElmer 5300 DV optical emission ICP instrument. Metal concentration was determined from a calibration curve for zinc, prepared from dilution of a zinc ICP standard stock solution (Sigma–Aldrich).
Protein interaction assays
Pulldown assays in nickel column
The interaction between PqqF and PqqG was studied by a pulldown assay using His SpinTrapTM columns (GE Healthcare) with a bed volume of 100 μl, using His6-PqqF (75 nmol) as the bait immobilized in the column and PqqG as the prey. The binding buffer was 20 mm Tris-HCl, pH 8.0, 0.15 m NaCl, 5% (v/v) glycerol; the wash buffer contained 10 mm imidazole; and the elution buffer had 250 mm imidazole. A nontreated column control was also run to identify and eliminate false positives caused by nonspecific binding of PqqG, as well as a control with bait (His6-PqqF) but no prey (PqqG).
SPR measurements
The interaction between PqqF and PqqG was also studied by SPR using an nitrilotriacetic acid-functionalized Dextran gold chip installed on a Biosensing Instruments BI4500 SPR instrument (Biosensing Instruments, Tempe, AZ). The temperature was set to 20 °C, and a flow rate of 60 μl/min was used for all experiments. The chip was pretreated with EDTA and a nickel solution, followed by immobilization of His6-PqqF. Following a 60-s wash step, native PqqG in concentrations from 45 nm to 3 μm was injected as the analyte. Following each binding experiment, immobilized His6-PqqF was striped with EDTA and reimmobilized prior to the next binding experiment. Experiments were run in triplicate, and the response from a reference cell without bound PqqF was subtracted. The data were analyzed using the accompanying SPR software.
Isothermal titration calorimetry
The dissociation constant between His6-PqqG and His6-PqqF was measured by ITC, in triplicate, using a NanoITC (TA Instruments). Purified His6-tagged enzymes were dialyzed in the same container against 50 mm Tris, pH 8.0, 300 mm NaCl. In a typical experiment, the sample cell was loaded with 20 μm His6-PqqG and titrated with one injection of 1 μl containing 200 μm His6-PqqF, followed by 15 more injections at 2.98 μl. The reference cell contained degassed deionized water. Control experiments were run identically except His6-PqqF or His6-PqqG was replaced by dialysis buffer. The collected thermograms were analyzed by the companion software, NanoAnalysis (TA Instruments). The data were baseline-subtracted and fitted to an independent model. All first injections were removed from data analysis.
Activity assays
The solution of chemically synthesized PqqA was freshly prepared in MilliQ water. The materials and buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl) used in these assays were autoclaved to avoid contaminating protease interference. To check for the purity and stability of PqqA, controls of PqqA by itself, in the presence of zinc and EDTA were performed. The activity was followed by determination of the percentage of abundance of PqqA as a function of time, measured by LC-MS. PqqF and/or PqqG were incubated for 10 min on ice, with or without added zinc. Unless specified otherwise the concentrations of enzymes were 2 μm, and PqqA was added to a final concentration of 20 μm. The reactions were kept at room temperature, and samples were collected at 0, 10, or 60 min and, in some cases, 240 min and were quenched with 5% (v/v) formic acid, immediately frozen in liquid nitrogen and stored at −80 °C. The activity was also tested with the small subpeptide EVTSY that contains the amino acid precursors to PQQ (Glu and Tyr).
Mass spectrometry assay
The samples quenched with formic acid were defrosted and centrifuged at 10,000 rpm for 10 min at 4 °C. The supernatants were analyzed using an Orbitrap mass spectrometer, as described previously (23).
Computational methods
Bioinformatics analysis
We checked the pqq operon in the genomes of the bacterial species reported by Shen et al. (22) to detect M16 peptidases, using the NCBI database RefSeq (59). The retrieved protein sequences were separated into M16B and M16A peptidases and were aligned using Clustal Omega, which was also used to calculated identity percentages (60). Alignment figures were obtained and annotated with Jalview (61).
Comparative modeling of the PqqF/PqqG dimer
A suitable template for the PqqF/PqqG putative heterodimer was found by searching for sequence homologs on the UNIPROT subset of protein of known structure. The highest-ranking candidate was the M16B metallopeptidase heterodimer from Sphingomonas sp. with PDB code 3amj. The two sequences in this dimer, Sph2681 and Sph2682, display, respectively, 41 and 32% identity with PqqF and PqqG; at this level of identity good comparative models can usually be built with confidence (62). Also, Sph2681 contains the conserved HXXEH zinc-binding motif, and Sph2682 contains the R/Y catalytic pair, like PqqF and PqqG (Figs. S4 and S6). Based on this template, comparative models were generated for the target using the MODELLER software (63). Run input files were prepared for MODELLER, taking into consideration the presence of the Zn2+ ion in the PqqF chain. To ensure the correct placement of the metal in the models, harmonic restraints were applied based on the coordination geometry observed in the 3amj structure. A total of 60 models were produced with very stringent optimization and refinement MODELLER parameters (maximum variation iterations 300, automatic scheduling slow, two repeat cycles, and the “very slow” refinement protocol). The model showing the best (lowest) DOPE score (64) was selected as the starting point to produce 10 models with automatic loop refinement. Best models were selected combining the normalized Z-DOPE and Swissprot QMEAN4 scores (65). These models were further analyzed with the WHATCHECK, PROSA, and MolProbity tools, revealing several steric clashes and outlier values for some bond distances and angles. To alleviate this situation, the final models were energy-minimized with the GROMACS software (66) using the Gromos54a7 forcefield (67). The minimized model showed marked improvement, producing a near-native MolProbity score and a QMEAN score of −1.6. This model was also checked with the ModFOLD6 server (68), producing an overall score 0.518 and a p value of 4.6 × 10−5 (indicative of a high-quality model).
Author contributions
A. M. M., I. B., and J. P. K. conceptualization; A. M. M., J. A. L., P. J. M., and J. P. K. formal analysis; A. M. M. and J. P. K. writing-original draft; A. M. M., J. A. L., P. J. M., I. B., A. T. I., and J. P. K. writing-review and editing; A. T. I. methodology; J. P. K. supervision.
Supplementary Material
This work was supported by National Institutes of Health Grants GM118117 (to J. P. K.) and GM124002 (to J. A. L.) and the QB3/Chemistry Mass Spectrometry Facility Grant 1S10OD020062-01. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S8.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PQQ
- pyrroloquinoline quinone (4,5-dihydro-4,5-dioxo-1H-pyrrolo[2,3-f] quinolone-2,7,9-tricarboxilic acid)
- AHQQ
- 3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid
- ICP
- inductively coupled plasma
- ITC
- isothermal titration calorimetry
- LB
- Luria–Bertani medium
- LBKan50
- Luria–Bertani medium with 50 μg/ml kanamycin
- Mex
- M. extorquens
- PqqA*
- cross-linked PqqA
- SPR
- surface plasmon resonance
- TBKan50
- Terrific broth with 50 μg/ml kanamycin
- PDB
- Protein Data Bank.
References
- 1. Klinman J. P., and Bonnot F. (2014) Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem. Rev. 114, 4343–4365 10.1021/cr400475g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hauge J. G. (1964) Glucose dehydrogenase of Bacterium anitratum: an enzyme with a novel prosthetic group. J. Biol. Chem. 239, 3630–3639 [PubMed] [Google Scholar]
- 3. Blake C. C., Ghosh M., Harlos K., Avezoux A., and Anthony C. (1994) The active site of methanol dehydrogenase contains a disulphide bridge between adjacent cysteine residues. Nat. Struct. Biol. 1, 102–105 10.1038/nsb0294-102 [DOI] [PubMed] [Google Scholar]
- 4. Oubrie A., Rozeboom H. J., Kalk K. H., Olsthoorn A. J., Duine J. A., and Dijkstra B. W. (1999) Structure and mechanism of soluble quinoprotein glucose dehydrogenase. EMBO J. 18, 5187–5194 10.1093/emboj/18.19.5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. RoseFigura J. M. (2010) Investigation of the structure and mechanism of a PQQ biosynthetic pathway component, PqqC, and a bioinformatics analysis of potential PQQ producing organisms, Ph.D. thesis, University of California, Berkeley [Google Scholar]
- 6. Sode K., Ito K., Witarto A. B., Watanabe K., Yoshida H., and Postma P. (1996) Increased production of recombinant pyrroloquinoline quinone (PQQ) glucose dehydrogenase by metabolically engineered Escherichia coli strain capable of PQQ biosynthesis. J. Biotechnol. 49, 239–243 10.1016/0168-1656(96)01540-4 [DOI] [PubMed] [Google Scholar]
- 7. Matsushita K., Arents J. C., Bader R., Yamada M., Adachi O., and Postma P. W. (1997) Escherichia coli is unable to produce pyrroloquinoline quinone (PQQ). Microbiology 143, 3149–3156 10.1099/00221287-143-10-3149 [DOI] [PubMed] [Google Scholar]
- 8. van Kleef M. A., and Duine J. A. (1989) Factors relevant in bacterial pyrroloquinoline quinone production. Appl. Environ. Microbiol. 55, 1209–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumazawa T., Seno H., Urakami T., Matsumoto T., and Suzuki O. (1992) Trace levels of pyrroloquinoline quinone in human and rat samples detected by gas chromatography/mass spectrometry. Biochim. Biophys. Acta 1156, 62–66 10.1016/0304-4165(92)90096-D [DOI] [PubMed] [Google Scholar]
- 10. Lobenstein-Verbeek C. L., Jongejan J. A., Frank J., and Duine J. A. (1984) Bovine serum amine oxidase: a mammalian enzyme having covalently bound PQQ as prosthetic group. FEBS Lett. 170, 305–309 10.1016/0014-5793(84)81333-2 [DOI] [PubMed] [Google Scholar]
- 11. Kasahara T., and Kato T. (2003) Nutritional biochemistry: a new redox-cofactor vitamin for mammals. Nature 422, 832 10.1038/422832a [DOI] [PubMed] [Google Scholar]
- 12. Harris E. D. (1992) The pyrroloquinoline quinone (PQQ) coenzymes: a case of mistaken identity. Nutr. Rev. 50, 263–267 [DOI] [PubMed] [Google Scholar]
- 13. Felton L. M., and Anthony C. (2005) Biochemistry: role of PQQ as a mammalian enzyme cofactor? Nature 433, E10–E12 10.1038/nature03322 [DOI] [PubMed] [Google Scholar]
- 14. Rucker R., Storms D., Sheets A., Tchaparian E., and Fascetti A. (2005) Biochemistry: is pyrroloquinoline quinone a vitamin? Nature 433, E10–E12 10.1038/nature03323 [DOI] [PubMed] [Google Scholar]
- 15. Steinberg F., Stites T. E., Anderson P., Storms D., Chan I., Eghbali S., and Rucker R. (2003) Pyrroloquinoline quinone improves growth and reproductive performance in mice fed chemically defined diets. Exp. Biol. Med. (Maywood) 228, 160–166 10.1177/153537020322800205 [DOI] [PubMed] [Google Scholar]
- 16. Steinberg F. M., Gershwin M. E., and Rucker R. B. (1994) Dietary pyrroloquinoline quinone: growth and immune response in BALB/c mice. J. Nutr. 124, 744–753 10.1093/jn/124.5.744 [DOI] [PubMed] [Google Scholar]
- 17. Akagawa M., Nakano M., and Ikemoto K. (2016) Recent progress in studies on the health benefits of pyrroloquinoline quinone. Biosci. Biotechnol. Biochem. 80, 13–22 10.1080/09168451.2015.1062715 [DOI] [PubMed] [Google Scholar]
- 18. Akagawa M., Minematsu K., Shibata T., Kondo T., Ishii T., and Uchida K. (2016) Identification of lactate dehydrogenase as a mammalian pyrroloquinoline quinone (PQQ)-binding protein. Sci. Rep. 6, 26723 10.1038/srep26723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saihara K., Kamikubo R., Ikemoto K., Uchida K., and Akagawa M. (2017) Pyrroloquinoline quinone, a redox-active O-quinone, stimulates mitochondrial biogenesis by activating the sirt1/pgc-1α signaling pathway. Biochemistry 56, 6615–6625 10.1021/acs.biochem.7b01185 [DOI] [PubMed] [Google Scholar]
- 20. Misra H. S., Rajpurohit Y. S., and Khairnar N. P. (2012) Pyrroloquinoline-quinone and its versatile roles in biological processes. J. Biosci. 37, 313–325 10.1007/s12038-012-9195-5 [DOI] [PubMed] [Google Scholar]
- 21. Goosen N., Vermaas D. A., and van de Putte P. (1987) Cloning of the genes involved in synthesis of coenzyme pyrrolo-quinoline-quinone from Acinetobacter calcoaceticus. J. Bacteriol. 169, 303–307 10.1128/jb.169.1.303-307.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Y. Q., Bonnot F., Imsand E. M., RoseFigura J. M., Sjölander K., and Klinman J. P. (2012) Distribution and properties of the genes encoding the biosynthesis of the bacterial cofactor, pyrroloquinoline quinone. Biochemistry 51, 2265–2275 10.1021/bi201763d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barr I., Latham J. A., Iavarone A. T., Chantarojsiri T., Hwang J. D., and Klinman J. P. (2016) Demonstration that the radical S-adenosylmethionine (SAM) enzyme PqqE catalyzes de novo carbon-carbon cross-linking within a peptide substrate PqqA in the presence of the peptide chaperone PqqD. J. Biol. Chem. 291, 8877–8884 10.1074/jbc.C115.699918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koehn E. M., Latham J. A., Armand T., Evans R. L. 3rd, Tu X., Wilmot C. M., Iavarone A. T., and Klinman J. P. (2019) Discovery of hydroxylase activity for pqqb provides a missing link in the pyrroloquinoline quinone biosynthetic pathway. J. Am. Chem. Soc. 141, 4398–4405 10.1021/jacs.8b13453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonnot F., Iavarone A. T., and Klinman J. P. (2013) Multistep, eight-electron oxidation catalyzed by the cofactorless oxidase, PqqC: identification of chemical intermediates and their dependence on molecular oxygen. Biochemistry 52, 4667–4675 10.1021/bi4003315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magnusson O. T., RoseFigura J. M., Toyama H., Schwarzenbacher R., and Klinman J. P. (2007) Pyrroloquinoline quinone biogenesis: characterization of PqqC and its H84N and H84A active site variants. Biochemistry 46, 7174–7186 10.1021/bi700162n [DOI] [PubMed] [Google Scholar]
- 27. Magnusson O. T., Toyama H., Saeki M., Rojas A., Reed J. C., Liddington R. C., Klinman J. P., and Schwarzenbacher R. (2004) Quinone biogenesis: structure and mechanism of PqqC, the final catalyst in the production of pyrroloquinoline quinone. Proc. Natl. Acad. Sci. U.S.A. 101, 7913–7918 10.1073/pnas.0402640101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Green P. N., and Ardley J. K. (2018) Review of the genus Methylobacterium and closely related organisms: a proposal that some Methylobacterium species be reclassified into a new genus, Methylorubrum gen. nov. Int. J. Syst. Evol. Microbiol. 68, 2727–2748 10.1099/ijsem.0.002856 [DOI] [PubMed] [Google Scholar]
- 29. Wei Q., Ran T., Ma C., He J., Xu D., and Wang W. (2016) Crystal structure and function of pqqf protein in the pyrroloquinoline quinone biosynthetic pathway. J. Biol. Chem. 291, 15575–15587 10.1074/jbc.M115.711226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rawlings N. D., and Barrett A. J. (2013) Metallopeptidases and their clans. In Handbook of Proteolytic Enzymes (Rawlings N. D., and Salvesen G., eds) pp. 325–370, Elsevier, Amsterdam [Google Scholar]
- 31. Springer A. L., Ramamoorthi R., and Lidstrom M. E. (1996) Characterization and nucleotide sequence of pqqE and pqqF in Methylobacterium extorquens AM1. J. Bacteriol. 178, 2154–2157 10.1128/jb.178.7.2154-2157.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morris C. J., Biville F., Turlin E., Lee E., Ellermann K., Fan W. H., Ramamoorthi R., Springer A. L., and Lidstrom M. E. (1994) Isolation, phenotypic characterization, and complementation analysis of mutants of Methylobacterium extorquens AM1 unable to synthesize pyrroloquinoline quinone and sequences of pqqD, pqqG, and pqqC. J. Bacteriol. 176, 1746–1755 10.1128/jb.176.6.1746-1755.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang M., and Lidstrom M. E. (2003) Promoters and transcripts for genes involved in methanol oxidation in Methylobacterium extorquens AM1. Microbiology 149, 1033–1040 10.1099/mic.0.26105-0 [DOI] [PubMed] [Google Scholar]
- 34. Vuilleumier S., Chistoserdova L., Lee M. C., Bringel F., Lajus A., Zhou Y., Gourion B., Barbe V., Chang J., Cruveiller S., Dossat C., Gillett W., Gruffaz C., Haugen E., Hourcade E., et al. (2009) Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS One 4, e5584 10.1371/journal.pone.0005584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goodwin P. M., and Anthony C. (1998) The biochemistry, physiology and genetics of PQQ and PQQ-containing enzymes. Adv. Microb. Physiol. 40, 1–80 10.1016/S0065-2911(08)60129-0 [DOI] [PubMed] [Google Scholar]
- 36. Ghilarov D., Serebryakova M., Stevenson C. E. M., Hearnshaw S. J., Volkov D. S., Maxwell A., Lawson D. M., and Severinov K. (2017) The origins of specificity in the microcin-processing protease TldD/E. Structure 25, 1549–1561.e5 10.1016/j.str.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Louche A., Salcedo S. P., and Bigot S. (2017) Protein–protein interactions: pull-down assays. Methods Mol. Biol. 1615, 247–255 10.1007/978-1-4939-7033-9_20 [DOI] [PubMed] [Google Scholar]
- 38. Latham J. A., Iavarone A. T., Barr I., Juthani P. V., and Klinman J. P. (2015) PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J. Biol. Chem. 290, 12908–12918 10.1074/jbc.M115.646521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Acuner Ozbabacan S. E., Engin H. B., Gursoy A., and Keskin O. (2011) Transient protein–protein interactions. Protein Eng. Des. Sel. 24, 635–648 10.1093/protein/gzr025 [DOI] [PubMed] [Google Scholar]
- 40. Liddington R. C. (2013) YMXG peptidase. In Handbook of Proteolytic Enzymes (Rawlings N. D., and Salvesen G., eds) pp. 1451–1457, Elsevier, Amsterdam [Google Scholar]
- 41. Meulenberg J. J., Sellink E., Riegman N. H., and Postma P. W. (1992) Nucleotide sequence and structure of the Klebsiella pneumoniae pqq operon. Mol. Gen. Genet. 232, 284–294 [DOI] [PubMed] [Google Scholar]
- 42. Jongejan J. A., and Duine J. A. (1989) PQQ and Quinoproteins, Kluwer Academics, Dordrecht, The Netherlands [Google Scholar]
- 43. Houck D. R., Hanners J. L., Unkefer C. J., van Kleef M. A., and Duine J. A. (1989) PQQ: biosynthetic studies in Methylobacterium AM1 and Hyphomicrobium X using specific 13C labeling and NMR. Antonie Van Leeuwenhoek 56, 93–101 10.1007/BF00822589 [DOI] [PubMed] [Google Scholar]
- 44. van Kleef M. A., and Duine J. A. (1988) l-Tyrosine is the precursor of PQQ biosynthesis in Hyphomicrobium X. FEBS Lett. 237, 91–97 10.1016/0014-5793(88)80178-9 [DOI] [PubMed] [Google Scholar]
- 45. Goosen N., Huinen R. G., and van de Putte P. (1992) A 24-amino-acid polypeptide is essential for the biosynthesis of the coenzyme pyrrolo-quinoline-quinone. J. Bacteriol. 174, 1426–1427 10.1128/jb.174.4.1426-1427.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Magnusson O. T., Toyama H., Saeki M., Schwarzenbacher R., and Klinman J. P. (2004) The structure of a biosynthetic intermediate of pyrroloquinoline quinone (PQQ) and elucidation of the final step of PQQ biosynthesis. J. Am. Chem. Soc. 126, 5342–5343 10.1021/ja0493852 [DOI] [PubMed] [Google Scholar]
- 47. Wecksler S. R., Stoll S., Tran H., Magnusson O. T., Wu S. P., King D., Britt R. D., and Klinman J. P. (2009) Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-l-methionine enzyme. Biochemistry 48, 10151–10161 10.1021/bi900918b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gliese N., Khodaverdi V., and Görisch H. (2010) The PQQ biosynthetic operons and their transcriptional regulation in Pseudomonas aeruginosa. Arch. Microbiol. 192, 1–14 10.1007/s00203-009-0523-6 [DOI] [PubMed] [Google Scholar]
- 49. Rawlings N. D., Morton F. R., Kok C. Y., Kong J., and Barrett A. J. (2008) MEROPS: the peptidase database. Nucleic Acids Res. 36, D320–D325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Maruyama Y., Chuma A., Mikami B., Hashimoto W., and Murata K. (2011) Heterosubunit composition and crystal structures of a novel bacterial M16B metallopeptidase. J. Mol. Biol. 407, 180–192 10.1016/j.jmb.2011.01.038 [DOI] [PubMed] [Google Scholar]
- 51. Asplund-Samuelsson J., Bergman B., and Larsson J. (2012) Prokaryotic caspase homologs: phylogenetic patterns and functional characteristics reveal considerable diversity. PLoS One 7, e49888 10.1371/journal.pone.0049888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klemencic M., Asplund-Samuelsson J., Dolinar M., and Funk C. (2019) Phylogenetic distribution and diversity of bacterial pseudo-orthocaspases underline their putative role in photosynthesis. Front. Plant Sci. 10, 293 10.3389/fpls.2019.00293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Seaman J. E., Julien O., Lee P. S., Rettenmaier T. J., Thomsen N. D., and Wells J. A. (2016) Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ. 23, 1717–1726 10.1038/cdd.2016.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Duncan E. A., Brown M. S., Goldstein J. L., and Sakai J. (1997) Cleavage site for sterol-regulated protease localized to a Leu-Ser bond in the lumenal loop of sterol regulatory element-binding protein-2. J. Biol. Chem. 272, 12778–12785 10.1074/jbc.272.19.12778 [DOI] [PubMed] [Google Scholar]
- 55. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 56. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 57. Lin S., Yang X., Jia S., Weeks A. M., Hornsby M., Lee P. S., Nichiporuk R. V., Iavarone A. T., Wells J. A., Toste F. D., and Chang C. J. (2017) Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 10.1126/science.aal3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Offenbacher A. R., Hu S., Poss E. M., Carr C. A. M., Scouras A. D., Prigozhin D. M., Iavarone A. T., Palla A., Alber T., Fraser J. S., and Klinman J. P. (2017) Hydrogen-deuterium exchange of lipoxygenase uncovers a relationship between distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling. ACS Cent. Sci. 3, 570–579 10.1021/acscentsci.7b00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. O'Leary N. A., Wright M. W., Brister J. R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D., Astashyn A., Badretdin A., Bao Y., Blinkova O., Brover V., et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., and Barton G. J. (2009) Jalview Version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., and Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 64. Shen M.-Y., and Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 10.1110/ps.062416606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Benkert P., Tosatto S. C., and Schomburg D. (2008) QMEAN: A comprehensive scoring function for model quality assessment. Proteins 71, 261–277 10.1002/prot.21715 [DOI] [PubMed] [Google Scholar]
- 66. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., and Lindahl E. (2015) GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 [Google Scholar]
- 67. Schmid N., Eichenberger A. P., Choutko A., Riniker S., Winger M., Mark A. E., and van Gunsteren W. F. (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 40, 843–856 10.1007/s00249-011-0700-9 [DOI] [PubMed] [Google Scholar]
- 68. Maghrabi A. H. A., and McGuffin L. J. (2017) ModFOLD6: an accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res. 45, W416–W421 10.1093/nar/gkx332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rawlings N. D., Barrett A. J., Thomas P. D., Huang X., Bateman A., and Finn R. D. (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 10.1093/nar/gkx1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.