

Abstract
F-box protein 31 (FBXO31) is a reported putative tumor suppressor, and its inactivation due to loss of heterozygosity is associated with cancers of different origins. An emerging body of literature has documented FBXO31's role in preserving genome integrity following DNA damage and in the cell cycle. However, knowledge regarding the role of FBXO31 during normal cell-cycle progression is restricted to its functions during the G2/M phase. Interestingly, FBXO31 levels remain high even during the early G1 phase, a crucial stage for preparing the cells for DNA replication. Therefore, we sought to investigate the functions of FBXO31 during the G1 phase of the cell cycle. Here, using flow cytometric, biochemical, and immunofluorescence techniques, we show that FBXO31 is essential for maintaining optimum expression of the cell-cycle protein cyclin A for efficient cell-cycle progression. Stable FBXO31 knockdown led to atypical accumulation of cyclin A during the G1 phase, driving premature DNA replication and compromised loading of the minichromosome maintenance complex, resulting in replication from fewer origins and DNA double-strand breaks. Because of these inherent defects in replication, FBXO31-knockdown cells were hypersensitive to replication stress-inducing agents and displayed pronounced genomic instability. Upon entering mitosis, the cells defective in DNA replication exhibited a delay in the prometaphase–to–metaphase transition and anaphase defects such as lagging and bridging chromosomes. In conclusion, our findings establish that FBXO31 plays a pivotal role in preserving genomic integrity by maintaining low cyclin A levels during the G1 phase for faithful genome duplication and segregation.
Keywords: protein degradation, cell cycle, DNA replication, genomic instability, tumor suppressor gene, anaphase-promoting complex, F-box protein 31 (FBXO31), LOH, MCM complex, SCF complex
Introduction
FBXO31 is a putative tumor suppressor, and its inactivation due to loss of heterozygosity is reported in many cancers, including gastric, hepatocellular, melanoma, and breast carcinoma. FBXO31 acts as substrate-recognizing subunit for SCF E3 ubiquitin ligase complex (1). Recent reports have elucidated the involvement of FBXO31 at several steps in maintaining genomic integrity (2). Upon genotoxic stress induction, it degrades cyclin D1 to arrest cells at the G1 phase (3) and also degrades MDM2 for a robust stabilization of p53 to initiate appropriate DNA damage–response pathway (4). FBXO31 oscillates throughout the cell cycle with its maximum levels from G2/M to early G1 phases, and its optimum level is maintained by APC/C or SCFFBXO46 (1, 5, 6). In the G2 phase, FBXO31 targets the DNA replication licensing protein Cdt1 for its proteasomal degradation to prevent re-replication (7). Also, FBOX31 degrades a master transcription factor FOXM1 specifically at the G2/M boundary, failure of which results in mitotic aberrations such as lagging and bridging chromosomes (8). Previous reports suggest that the chromosome missegregation can arise during two crucial events (9): first, during mitosis, while segregating sister chromatids when they are still bound to each other through cohesin or while segregating abruptly condensed chromosomes (9); and second, a profound source of chromosome missegregation originates from defects during DNA replication. Structures such as unresolved replication intermediates, incompletely replicated loci, or intertwined sister chromatids that arise during replication and if persisting until mitosis may interfere with chromosome condensation and subsequently its segregation leading to genomic instability (9, 10). Interestingly, the preparations for DNA replication begin as soon as the cells exit mitosis and continue until late G1 phase (11). One of the important events for DNA replication is licensing of origins (11). It involves loading the MCM3 complex onto chromatin with the help of ORC, Cdc6/18, and Cdt1, which is required for an efficient DNA replication (12). It is a highly orchestrated process, and any defect can generate breeding grounds for genomic instability.
Most of the existing studies have explored the important roles of FBXO31 during the G2/M phase. However, as the level of FBXO31 is also high during the early G1 phase of the cell cycle, we hypothesized that FBXO31 might also be having some crucial role during the G1 phase. In this study, we used stable knockdown cells of FBXO31 (FBXO31KD) to investigate its impact on cell-cycle progression. We found that FBXO31 facilitates proteasomal degradation of cyclin A. Absence of FBXO31 resulted in the unscheduled activity of cyclin A during the G1 phase leading to compromised chromatin loading of the MCM complex. Additionally, an elevated level of cyclin A stimulated FBXO31KD cells to enter the S phase prematurely with a reduced number of licensed origins leading to replication stress resulting in DNA damage during replication. Because of the prevalence of endogenous replication stress, FBXO31KD cells are hypersensitive to replication stress inducers. Upon exposure to replicative stress inducers, the FBXO31KD cells exhibited delayed S to G2/M progression and also delayed prometaphase to metaphase transitions. Additionally, an increased population of FBXO31KD cells showed aberrant anaphase with lagging and bridging chromosomes. Interestingly, all the phenotypic defects were rescued in FBXO31 and cyclin A co-depleted cells. These observations suggest that FBXO31 is essential to maintain optimum expression of cyclin A to allow coordinated cell-cycle progression and to preserve genomic stability.
Results
FBXO31KD results in early S-phase entry of cells
We have investigated the role of FBXO31 in cell-cycle regulation following an effective and stable depletion of FBXO31 in MCF7 cell lines using two independent shRNAs against FBXO31. Scrambled shRNA was used as a nonspecific (NS) control to rule out the off-target effects. Both the shRNAs against FBXO31 showed a significant down-regulation of FBXO31 (Fig. S1A). To investigate the contribution of FBXO31 in cell-cycle regulation, we monitored the cell-cycle profile of these cells by synchronizing them at prometaphase using nocodazole for 16 h and then releasing them to progress into different phases of the cell cycle. It was observed that FBXO31KD cells showed an early progression into S phase (Fig. 1A and Fig. S1B). In particular, at 12 h post-release from nocodazole when only 25% of NS cells could mark their entry into S phase, around 35% of FBXO31KD cells have already entered S phase (Fig. 1A). Hence, compared with NS cells, around 40% more of FBXO31KD cells progressed to the S phase (calculations were done considering 25% of NS cells as 100% and compared with that the percentage of FBXO31KD cells in S phase was determined and then the difference in percentage was calculated). To further validate the early commitment of FBXO31KD cells to DNA replication, we performed the BrdU incorporation assay. Interestingly, we found a noticeable increase in BrdU incorporation in FBXO31KD cells when compared with the NS cells, clearly suggesting an early S-phase entry of FBXO31KD cells (Fig. 1B). These observations suggest that FBXO31 may play a crucial role in regulating G1 to S-phase transition during cell-cycle progression.
Figure 1.

FBXO31 is essential for an error-free replication process and mitosis. A, graphical representation of percentage of S phase in MCF7 cells stably expressing NS and shFBXO31-1 and shFBXO31-2 at different time points following nocodazole release. Cells were grown in nocodazole-containing medium for 16 h; following that, the cells were allowed to re-enter the cell cycle in fresh medium and were collected at the indicated time points for flow cytometric analysis. Data points represent the mean of two independent experiments. B, cell-cycle profile of MCF7 cells stably expressing NS and shFBXO31-1 and shFBXO31-2 at different time points following nocodazole release. Cells were grown in nocodazole-containing medium for 16 h; following that, the cells were allowed to re-enter the cell cycle in fresh medium and were collected at the indicated time points for flow cytometric analysis. Cells were chased for BrdU incorporation for 2 h before harvesting, and then the cells were stained and analyzed for BrdU incorporation using flow cytometry. C, graphical representation of percentage of NS and shFBXO31 cells in different cell cycle phases at the indicated time points. Cells were grown in the presence of 0.25 mm HU for 22 h, then released by washing twice with media followed by culturing in fresh media, and then were collected at the indicated time points. Population of cells at different cell cycle phases was assessed using flow cytometry. Error bars represent S.E. from three independent experiments. D, graphical representation of percentage of NS and shFBXO31 cells in the G2 and different mitotic phases after 12 h of hydroxyurea release. Cells were grown and collected as described in C and then stained with pH 3 Ser-10 antibody to score the number of cells at the G2 and different phases of mitosis. Error bars represent S.E. from three independent experiments. E, graphical representation of percentage of NS and shFBXO31 cells having lagging and bridging chromosomes after 12 h of hydroxyurea release. Cells were grown and collected as described in C and were stained with pH 3 Ser-10 antibody to score the number of anaphase cells, and the percentage of cells was calculated with bridging and lagging chromosomes. Error bars represent S.E. from three independent experiments. F, graphical representation of percentage of sub-G1 population of NS and shFBXO31 cells determined by flow cytometry following three times of repeated HU exposure. Cells expressing either NS or FBXO31 shRNA were exposed to replication stress using HU for 24 h, and then cells were released for 24 h; this cycle was repeated for three consecutive times. Error bars represent S.E. from three independent experiments. G, cells were grown as described in F and then the released cells were allowed to grow in fresh medium for the next 10 days. Colonies were then stained with crystal violet dye. Data are representative images of three independent experiments. H, graphical representation of percentage of γH2AX-positive NS and shFBXO31 cells as in G. n.s. represents nonsignificant; *, p ≤ 0.05; **, p < 0.01; ***, p < 0.001. Error bars represent S.E. from three independent experiments.
FBXO31 is essential for an error-free cell-cycle progression following release from replicative stress-induced cell synchronization
Early entry into S phase may lead to defects during DNA replication due to initiation of DNA replication with insufficient preparation (13). Such cells having inherent defects arising during replication are sensitive toward replication stress inducers (14). So, we postulated that FBXO31KD cells might have increased propensity toward inherent replicative stress, which makes them sensitive toward replication stress inducers. To assess this possibility, we synchronized the NS and FBXO31KD cells with hydroxyurea (HU) for 22 h. HU synchronizes the cells at the entrance to S phase by inhibiting DNA replication and thereby induces replication stress (15). Subsequent to HU synchronization, cells were washed repeatedly to allow cell-cycle progression, and then cells were collected at the indicated time points (Fig. 1C and Fig. S1C). We observed that compared with NS cells, around 30% of FBXO31KD cells were slower in the transition from S to G2/M phase (Fig. 1C and Fig. S1C). For instance, only 40% of FBXO31KD cells could reach from S to G2/M phase, and at the same time around 55% of NS cells progressed to the G2/M phase (calculations were done considering 40% of NS as 100% and compared with that the percentage of FBXO31KD cells in S phase was determined and then the difference in percentage was calculated). Additionally, FBXO31KD cells also showed delay in progression from G2/M to G1 phase as compared with NS cells (Fig. 1C and Fig. S1C). For instance, around 50% of NS cells progressed from G2/M to G1 phase, and at the same time only 30% of FBXO31KD cells could progress to G1 phase (Fig. 1C and Fig. S1C). To further assess this delay in transition from S to G2/M phase, we proceeded for BrdU chase experiment (Fig. S1D). Data from BrdU incorporation studies clearly showed that a significant population of FBXO31KD cells was halted in S phase and showed delay in transition from S to G2/M phase. Importantly, the delay persisted in FBXO31KD cells even up to 12 h post-HU release. These observations clearly indicate that FBXO31KD cells are sensitive toward replication stress inducers and might accumulate defects as cells advance through the cell cycle leading to a delay in cell-cycle phase transitions.
Challenges encountered during DNA replication may result in defects such as unresolved DNA intermediates or intertwined sister chromatids that can interfere with chromosome condensation and segregation (9). As FBXO31KD cells encounter increased replication stress, we speculated that these defects might lead to mitotic aberrations. To investigate that, both NS and FBXO31KD cells were stained for pH 3 Ser-10 at 12 h post-release from HU synchronization. Immunofluorescence analysis revealed ∼1.5-fold increase in the population of prometaphase cells upon FBXO31 depletion as compared with NS cells (Fig. 1D and Fig. S1E). Additionally, FBXO31KD cells have around 2-fold increased levels of lagging and bridging chromosomes (Fig. 1E and Fig. S1E). These observations suggest that absence of FBXO31 results in defects during DNA replication and segregation resulting in increased genomic instability.
FBXO31-depleted cells are sensitive toward replication stress inducers
The above-described results suggested that FBXO31 might be having a crucial role in replicative stress response. To evaluate the importance of FBXO31 in replication stress management, NS and FBXO31KD cells were exposed to HU for 24 h followed by a release for 24 h. This cycle was repeated three times to induce sufficient replicative stress in the cells, and a series of experiments was performed with these cells. Flow cytometric analysis revealed a 10% increase in the sub-G1 population upon FBXO31 depletion indicating that FBXO31KD cells are sensitive to repeated replicative stress and undergo death (Fig. 1F). This suggests that FBXO31 has an important role in relieving replicative stress arising during DNA replication.
Next, we were interested to evaluate the role of FBXO31 on the long-term survival of cells exposed to repeated replicative stress. To understand that, we allowed NS and FBXO31KD cells to re-enter the cell cycle after withdrawing replicative stress-inducing agent and followed them for 10 more days. Interestingly, most of the FBXO31KD cells died resulting in a significantly reduced number of colonies as compared with NS cells (Fig. 1G). Additionally, when these colonies were stained for γ-H2AX to check the extent of DNA damage (16), we observed that FBXO31KD cells showed about a 2-fold increase in the number of cells positive for γ-H2AX foci (Fig. 1H and Fig. S1F). These results taken together suggest that FBXO31 is essential for maintaining precise genome replication and replication stress management.
FBXO31 alters the turnover kinetics of cyclin A
The phenotypic defect of hastened S-phase entry and defective mitosis that we observed upon FBXO31 depletion have been attributed mainly to the altered levels of cyclin A. Importantly, cyclin A–CDK complex is the rate-limiting factor for G1 to S transition and is required until prometaphase (17–20). Therefore, any error in cyclin A regulation may create havoc in the cell cycle and compromised genomic integrity. We anticipated that depletion of FBXO31 may alter cyclin A stability resulting in genomic instability. Interestingly, knockdown of FBXO31 using two independent shRNAs showed stabilization of cyclin A at the protein level (Fig. 2A). As the major activities pertaining to cyclin A have been reported in the nucleus (21), we were interested to examine which pool of cyclin A was being stabilized upon FBXO31 depletion. Nuclear and cytoplasmic fractionation assays revealed an increased level of cyclin A in both the fractions of FBXO31KD cells. However, a prominent accumulation of cyclin A was observed in the nuclear fraction, indicative of high cyclin A–CDK activity (Fig. 2B). However, FBXO31 depletion does not alter the mRNA level of cyclin A (Fig. 2C).
Figure 2.

FBXO31 regulates cyclin A expression at the proteasomal level. A, immunoblot monitoring the level of cyclin A and FBXO31 in MCF7 cells expressing either NS or FBXO31 shRNAs. Whole-cell protein extracts were immunoblotted to probe for the indicated proteins. B, cytoplasmic and nuclear fractions of cells expressing either NS or FBXO31 shRNA were immunoblotted with the indicated antibodies. Tubulin and Lamin B1 were used as cytoplasmic and nuclear loading controls, respectively. C, real time RT-PCR was performed to monitor cyclin A mRNA expression in NS and shFBXO31 cells. Cyclin A mRNA level was normalized to the GAPDH mRNA level. Error bars represent S.E. from three independent experiments. D, immunoblots monitoring the turnover profile of cyclin A in NS and shFBXO31 cells following cycloheximide (CHX) (40 μg/ml) chase for the indicated time periods. E, quantification of levels of cyclin A in D. Expression of cyclin A was normalized with tubulin, and then cyclin A expression at 0 h was considered as 100% relative to the values of other time points that were plotted. F, immunoblot monitoring the turnover of cyclin A in cells expressing either empty vector or myc-FBXO31 following cycloheximide (40 μg/ml) chase for the indicated periods. Cells were transfected with the indicated constructs for 36 h and following that whole-cell protein extracts were immunoblotted to probe for the indicated proteins. G, quantitation of half-life of cyclin A from F. Expression of cyclin A was normalized with tubulin, and the degradation profile was plotted considering the expression of cyclin A at 0 h as 100%. H, cells were transfected either with empty vector or increasing concentrations of myc-FBXO31 for 48 h. Cells were then harvested, and whole-cell protein lysates were immunoblotted to probe for indicated proteins. I, cells were transfected with either empty vector or myc-FBXO31 for 36 h and were grown in the presence or absence of 10 μm proteasome inhibitor MG132 for 6 h and were immunoblotted and probed with the indicated antibodies. Error bars where shown represent S.E. from three independent experiments, and n.s. represents nonsignificant; *, p ≤ 0.05; **, p < 0.01; ***, p < 0.001.
Cyclins are short-lived proteins that can undergo proteasomal degradation once their function is over (22). So, we hypothesized that FBXO31 being an E3 ubiquitin ligase might be regulating cyclin A at the post-translational level via the proteasomal pathway. To test this, we incubated NS and FBXO31KD cells with the protein synthesis inhibitor cycloheximide for the indicated time points and then collected the cells to analyze the half-life of cyclin A. It was observed that depletion of FBXO31 led to a significant elevation in cyclin A half-life (from t½ = 0.9 h to t½ = 1.6 h) (Fig. 2, D and E). Furthermore, ectopic expression of FBXO31 significantly decreased the half-life of cyclin A (from t½ = 0.75 h to t½ = 0.25 h) (Fig. 2, F and G). Additionally, ectopic expression of increasing concentrations of FBXO31 resulted in a concomitant reduction in the levels of cyclin A in a dose-dependent manner (Fig. 2H). To confirm FBXO31-mediated proteasomal degradation of cyclin A, we monitored the expression of cyclin A upon overexpression of FBXO31 in the presence and absence of the proteasome inhibitor MG132. It was observed that FBXO31 could reduce the cyclin A level when expressed alone; however, it failed to reduce the level of cyclin A in the presence of proteasome inhibitor MG132 (Fig. 2I). These results taken together suggest that FBXO31 regulates the stability of cyclin A in a proteasome-dependent manner.
FBXO31 interacts with cyclin A to promote its Lys-48–linked polyubiquitination through SCF complex
In general, F-box proteins form a functional SCF complex through the interaction of their F-box motif with the SKP1 subunit of the core SCF complex to promote the ubiquitylation of its targets (23, 24). Therefore, we were interested to know whether FBXO31 regulates protein levels of cyclin A through the SCF complex. Immunoblotting data showed that the WT FBXO31 (myc-FBXO31) significantly reduced cyclin A expression, whereas the F-box motif-deleted FBXO31 mutant (myc-ΔF-FBXO31) failed to do so, suggesting the involvement of an intact SCF complex in FBXO31-mediated degradation of cyclin A (Fig. 3A). Furthermore, we were interested to investigate whether the failure of ΔF-FBXO31 to degrade cyclin A was because of perturbed interaction between the ΔF-FBXO31 and SCF core complex. We therefore performed co-immunoprecipitation experiment following overexpression of either empty vector or myc-FBXO31 or myc-ΔF-FBXO31 along with cyclin A (Fig. 3B). Immunoblot analysis revealed that both myc-FBXO31 and myc-ΔF-FBXO31 were able to interact with cyclin A. Interestingly, FBXO31 but not ΔF-FBXO31 was able to interact with the SKP1 and Cullin1, the core components of the SCF complex, demonstrating that FBXO31 facilitates proteasomal degradation of cyclin A through the SCF complex (Fig. 3B). Next, we checked the interaction of FBXO31 with cyclin A at the endogenous level through co-immunoprecipitation assay. Results demonstrated that FBXO31 interacts with cyclin A even at the normal physiological level (Fig. 3C).
Figure 3.
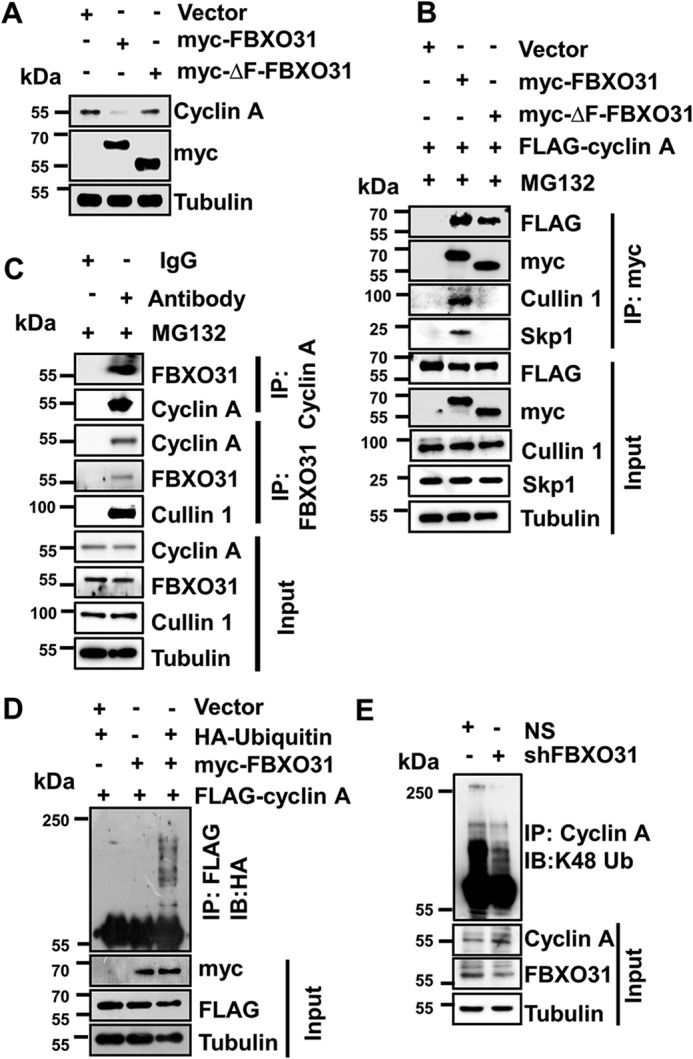
FBXO31 interacts with cyclin A to promote its polyubiquitylation through SCF complex. A, immunoblots monitoring the expression of cyclin A in MCF7 cells expressing either vector or myc-FBXO31 or F-box–deleted mutant of FBXO31 (myc-ΔF-FBXO31). Cells were transfected with the indicated plasmids for 48 h, and then whole-cell protein extracts were analyzed by immunoblotting. B, MCF7 cells were co-transfected with the indicated plasmids for 36 h, and transfected cells were then grown in the presence of 10 μm MG132 for 6 h. Whole-cell protein extracts were co-immunoprecipitated using anti-myc antibody. Immunoprecipitates and input protein extracts were immunoblotted for the indicated proteins. C, immunoblot monitoring the interaction between FBXO31 and cyclin A at the endogenous level in MCF7 cells treated with MG132 (10 μm for 6 h) in a co-immunoprecipitation assay. Whole-cell protein extracts were immunoprecipitated with the indicated antibodies as described under “Materials and methods.” Immunoprecipitates and input protein extracts were immunoblotted for the indicated proteins. D, MCF7 cells were transfected with FLAG–cyclin A in combination with either HA–ubiquitin or myc-FBXO31, or both, for 36 h. Transfected cells were then incubated with 10 μm MG132 for 6 h and harvested, and the whole-cell lysates were immunoprecipitated with anti-FLAG antibody. Immunoprecipitates and input protein extracts were immunoblotted for the indicated proteins. E, immunoblots monitoring the lysine 48 (Lys-48) (IB) linkage-specific ubiquitylation of cyclin A in MCF7 cells expressing NS or FBXO31 shRNA. Whole-protein extracts were immunoprecipitated using the anti-cyclin A antibody. Immunoprecipitates and input protein extracts were immunoblotted to probe for the indicated proteins.
F-box proteins promote polyubiquitylation of condemned proteins to facilitate their degradation via 26S proteasome (25). Therefore, we asked whether FBXO31 can polyubiquitinate cyclin A. To verify this, FLAG–cyclin A was co-transfected along with either HA–ubiquitin or myc-FBXO31 or in a combination of both and performed immunoprecipitation using anti-FLAG antibody. Immunoblot analysis revealed the appearance of high mass ladder of cyclin A when co-expressed along with FBXO31 and ubiquitin, suggesting that FBXO31 promotes polyubiquitination of cyclin A (Fig. 3D). Lysine 48-linkage–specific polyubiquitylation is an important event for dictating the proteasomal degradation of many proteins. In this regard, we observed that depletion of FBXO31 resulted in a dramatic reduction in Lys-48–linked polyubiquitination of cyclin A (Fig. 3E). To further confirm that the stabilization of cyclin A in FBXO31KD cells is mainly due to the reduction in polyubiquitination-mediated degradation of cyclin A, we ectopically expressed myc-FBXO31 and myc-ΔF-FBXO31 in FBXO31KD cells and examined for cyclin A polyubiquitination and degradation (Fig. S2, A and B). Of note, FBXO31 but not ΔF-FBXO31 was able to promote cyclin A polyubiquitination-mediated degradation when expressed in FBXO31KD cells, clearly attributing the role of FBXO31 as an important regulator of cyclin A stability (Fig. S2, A and B). These findings together suggest that FBXO31 interacts with cyclin A and promotes its Lys-48–linked polyubiquitination through SCF complex to direct its proteasomal degradation.
FBXO31 knockdown results in accumulation of cyclin A in the G1 phase leading to compromised chromatin loading of MCM4
It has been reported previously that overexpression of cyclin A can drive cells into S phase (26–28). Our results so far have shown that FBXO31 is essential in maintaining an optimum level of cyclin A, and its altered expression interferes with the turnover kinetics of cyclin A. So, we were interested in examining the effect of FBXO31 depletion on the physiological level of cyclin A during the cell-cycle progression. To address this, we probed for cyclin A in whole-cell extracts of NS and FBXO31KD cells released from nocodazole block at the indicated time points (Fig. 4, A and B, and Fig. S3A). Interestingly, we observed an increased accumulation of cyclin A in both mitosis and G1 phase of the cell cycle (Fig. 4, A and B). We then examined the interaction of FBXO31 and cyclin A at the G1, S, and mitosis phase (Fig. S3B). Immunoblotting results revealed that FBXO31 was able to interact with cyclin A at all the phases of the cell cycle in the absence and presence of MG132 (Fig. 4C and Fig. S3C). Previously, CDH1 has been reported to promote proteasomal degradation of cyclin A during G1 phase through anaphase-promoting complex (APC/C) (13). Interestingly, CDH1 could interact with cyclin A only at the G1 phase of the cell cycle, indicating that FBXO31 might regulate cyclin A in a CDH1-independent manner (Fig. 4C). To confirm whether cyclin A degradation by FBXO31 is independent of CDH1, we examined the expression levels of cyclin A in APC2, a core component of the APC/C complex, knockdown cells following overexpression of FBXO31. Results showed that ectopically expressed FBXO31 efficiently reduced the levels of cyclin A in APC2 knockdown cells, suggesting that FBXO31 could regulate cyclin A independent of CDH1 (Fig. S3D).
Figure 4.
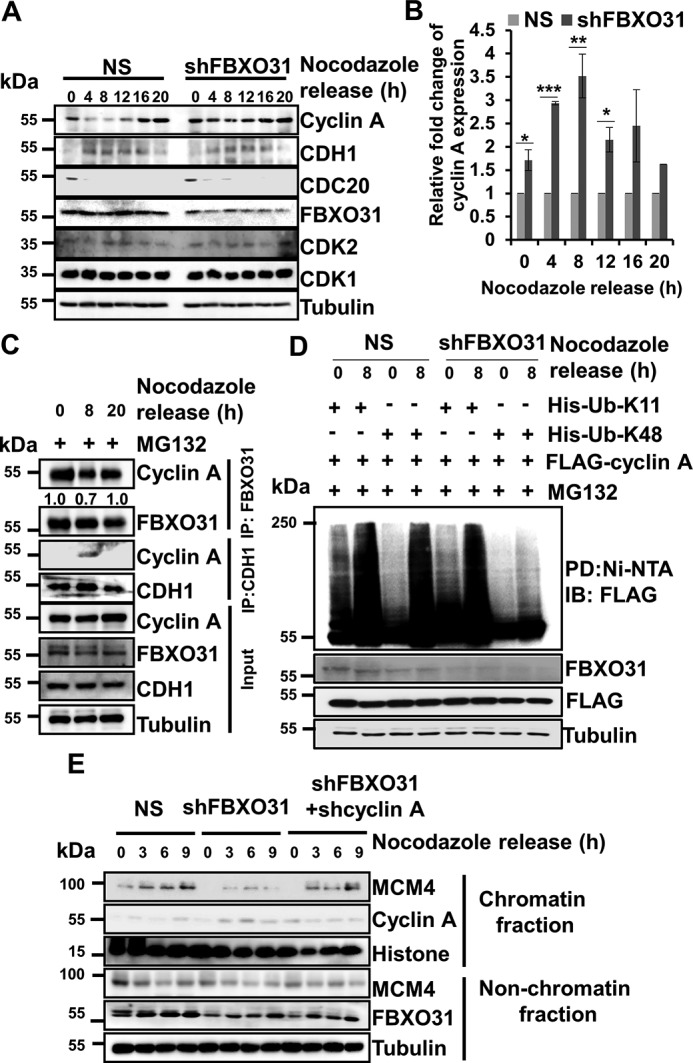
Accumulation of cyclin A during G1 phase leads to compromised chromatin loading of MCM4 in FBXO31-depleted cells. A, NS or shFBXO31-expressing MCF7 cells were synchronized using nocodazole for 16 h. Synchronized cells were then released and collected at the indicated time points. Whole-cell protein extracts were immunoblotted to probe for the indicated proteins. B, quantitation of cyclin A expression from A. Cyclin A expression was normalized with respective loading control tubulin at the corresponding time points and then normalized to 100% at each time point for NS cells. Then, cyclin A levels in shFBXO31-expressing cells at the indicated time points were quantified with respect to corresponding NS cells. Error bars represent S.E. from three independent experiments. C, immunoblot monitoring the interaction between FBXO31–cyclin A and CDH1–cyclin A at the endogenous levels at different phases of the cell cycle. MCF7 cells were synchronized using nocodazole for 16 h. Synchronized cells were then released and allowed to grow in fresh medium until harvested. MG132 (10 μm) was added to the medium for 2 h before harvesting the cells at the indicated time points. Whole-cell protein extracts were immunoprecipitated with the indicated antibodies. The immunoprecipitates and input protein extracts were immunoblotted and probed for the indicated proteins. D, MCF7 cells expressing either NS or FBXO31 shRNA were transfected with FLAG–cyclin A in combination with either His–Lys-11–only ubiquitin or His–Lys-48–only ubiquitin for 24 h, and transfected cells were then incubated with 10 μm MG132 for 2 h. Cells were then harvested, and whole-cell lysates were incubated with Ni-NTA beads. The pulled down fractions and input protein lysates were immunoblotted for the indicated proteins. E, immunoblot (IB) analysis monitoring MCM4 level in chromatin and nonchromatin fractions during mitosis and G1 phase of NS, FBXO31KD, and FBXO31–cyclin A knockdown cells. Error bars where shown represent S.E. from three independent experiments, and n.s. represents nonsignificant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Previous reports suggest that APC/C promotes Lys-11–linked polyubiquitination of its substrates (29, 30). In this study, we found that FBXO31 promotes Lys-48–linked polyubiquitination of cyclin A (Fig. 3E). So, we were interested to evaluate the nature of polyubiquitination that gets hampered in the absence of FBXO31 during mitosis and G1 phase. To investigate that, we co-expressed FLAG–cyclin A with either His–Lys-11-only ubiquitin or His–Lys-48-only ubiquitin in NS and FBXO31 KD cells. Transfected cells were then synchronized using nocodazole and were collected at 0 and 8 h post-release. The whole-cell lysates were pulled down using Ni-NTA beads and probed for cyclin A polyubiquitination. Immunoblot analysis of immunoprecipitates demonstrated increased Lys-11- and Lys-48–linked polyubiquitination of cyclin A at 0 and 8 h in NS cells (Fig. 4D). However, a comparable decrease in Lys-48- but not Lys-11–linked polyubiquitination of cyclin A was observed in FBXO31KD cells (Fig. 4D). These observations indicate that FBXO31 regulates cyclin A by promoting its Lys-48–linked polyubiquitination. Collectively, our results suggest that FBXO31 facilitates cyclin A degradation independent of the APC/C complex.
Cyclin A–CDK activity during G1 phase interferes with chromatin loading of MCM protein complex (31). Chromatin loading of MCM complex is very important for origin licensing which begins during telophase and continues until late G1 phase and is essential for an efficient DNA replication (11–13, 32). As the knockdown of FBXO31 increases cyclin A expression during G1 phase, we were interested to investigate whether the defects in DNA replication in FBXO31KD cells arise from reduced chromatin loading of MCM complex. In this regard, we probed for the chromatin-bound fraction of MCM4 (one of the crucial members of the core MCM complex). As expected, we found a drastic decrease in the chromatin loading of MCM4 during the G1 phase in FBXO31KD cells (Fig. 4E and Fig. S3E). Interestingly, chromatin loading of MCM4 was restored similar to normal upon co-depletion of cyclin A along with FBXO31 (Fig. 4E and Fig. S3, E and F). This observation suggests that the absence of FBXO31 results in the unscheduled activity of cyclin A in G1 cells that interfere with the loading of MCM complex onto the chromatin resulting in defective DNA replication.
FBXO31 knockdown leads to premature S-phase entry and reduced DNA replication accompanied by increased DNA damage
Compromised origin licensing reduces the efficiency of DNA replication by interfering with replication fork progression resulting in DNA breaks (13, 33). As FBXO31KD cells enter S-phase early with a reduced number of licensed origins, we were interested to investigate whether FBXO31KD cells initiate DNA replication prematurely with fewer origins leading to DNA damage during replication. To visualize the defects in replication, we utilized the PCNA immunostaining method. PCNA is DNA polymerase processivity factor in eukaryotes, and during S phase it actively localizes to the sites of DNA replication (34, 35). The distribution pattern of PCNA in cells varies in different stages of the cell cycle and hence is used to determine different stages of the cell cycle and more precisely the various substages of S phase (36). During G1 and G2/M phases, PCNA is located mainly in the nucleus and to a lesser extent in the cytoplasm without forming a distinct punctate structure. In the early-S phase, PCNA is distributed homogeneously in the form of puncta within the nucleus. During mid-S phase, distinct PCNA puncta can be observed at the rim of the nucleus. During the late-S phase, there is a decrease in PCNA accumulation at the rim of the nucleus; however, patches of PCNA puncta are observed within the nucleus (36). To understand whether FBXO31KD cells initiate DNA replication prematurely, NS and FBXO31KD cells were stained for PCNA after 12 h of release from nocodazole block. It was observed that 34% of NS cells entered into S phase while 45% of FBXO31KD cells entered S phase demonstrating that extra 30% of FBXO31KD cells entering early into S phase (Fig. 5, A–C). We compared PCNA staining intensity on a per cell basis among the S-phase cells, and a significant decrease in mean PCNA intensity was observed in FBXO31KD cells with respect to NS cells, suggesting DNA replication from fewer origins upon FBXO31 depletion (Fig. 5, D and E). Interestingly, co-staining of NS and FBXO31KD cells for PCNA and γ-H2AX showed an increase in the number of γ-H2AX foci in replicating FBXO31KD cells, indicative of endogenous DNA damage arising from error-prone replication process (Fig. 5, D and F).
Figure 5.
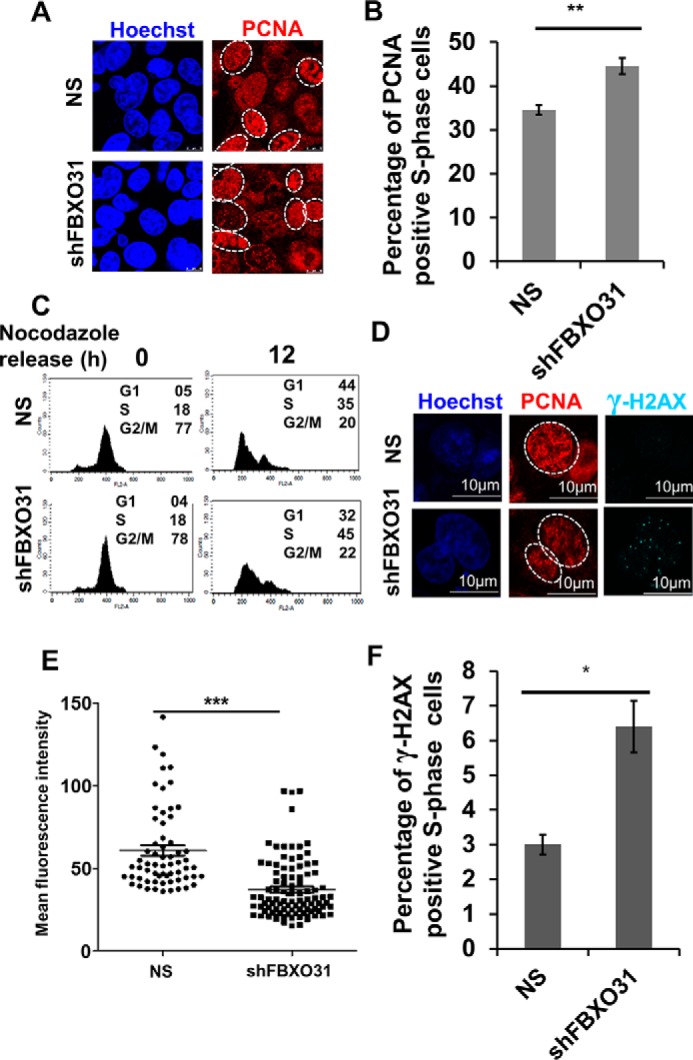
Depletion of FBXO31 leads to premature S-phase entry and reduced DNA replication accompanied by increased DNA damage. A, immunofluorescence analysis of number of cells showing S-phase–specific pattern of PCNA in NS and shFBXO31 cells. Both NS and shFBXO31-expressing cells were synchronized using nocodazole, and then cells were allowed to grow in the absence of nocodazole for 12 h. Cells were fixed and stained for PCNA (red) and Hoechst (blue). The experiment was repeated three times, and 10 random fields were observed in each time. Image represents one of the fields. B, percentage of S-phase cells was quantified from A. C, flow cytometric representation of cell-cycle profile of NS and shFBXO31-expressing cells at 12 h post-nocodazole release. D, immunofluorescence analysis of DNA damage foci represented by γ-H2AX (cyan) in S-phase cells. Both NS and shFBXO31-expressing cells were synchronized by nocodazole, and then cells were allowed to grow in the absence of nocodazole for 12 h. Cells were fixed and stained for γ-H2AX (cyan), PCNA (red), and Hoechst (blue). E, graphical representation of mean fluorescence intensity of PCNA of S phase in NS and shFBXO31-expressing cells. Region of interest was drawn at the periphery of nucleus displaying S-phase–specific PCNA distribution. Experiment was repeated three times, and five random fields were observed each time. F, quantitation of NS and shFBXO31 cells having γ-H2AX foci in S-phase cells (n = 100). Error bars represent S.E. from three independent experiments, and n.s. represents nonsignificant; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Elevated levels of cyclin A in FBXO31KD cells result in premature S-phase entry, mitotic delay, and defects
In this study, we have witnessed the stabilization of cyclin A during G1 and mitosis in FBXO31KD cells; therefore, we were interested to understand whether restoring cyclin A levels similar to that observed in NS cells could prevent premature S-phase entry. To examine this possibility, we depleted cyclin A in FBXO31KD cells (Fig. S3F). FACS analysis revealed that the observed premature S-phase entry in FBXO31KD cells was blocked upon co-depletion of cyclin A in FBXO31KD cells (Fig. 6A). Similarly, we found that reduced chromatin loading of MCM4 in FBXO31KD cells was recovered close to normal following knockdown of cyclin A in FBXO31KD cells (Fig. 4E and Fig. S3E).
Figure 6.

Elevated level of cyclin A in FBXO31-depleted cells is responsible for premature S-phase entry, replication stress, and mitotic delay and defects. A, percentage of S-phase cells in NS, shFBXO31, and co-expressing shFBXO31 and shcyclin A cells at 8 h post-nocodazole release. B, percentage of NS, shFBXO31, and shFBXO31 and shcyclin A co-depleted cells in different phases of the cell cycle at 0, 9, and 12 h post-HU release. C, graphical representation of cells in G2 phase and different mitotic phases following 12 h of hydroxyurea release. Cells were fixed and stained with Hoechst and pH 3 Ser-10 antibody to score G2 and different phases of mitotic cells. D, percentage of NS, shFBXO31, and co-expressing shFBXO31 and shcyclin A cells, harboring anaphase bridges following 12 h of HU release. Cells were stained as described in Fig. 1D, and anaphase cells were scored to calculate the percentage of cells with bridging chromosomes. E, percentage of NS, shFBXO31, and co-expressing shFBXO31 and shcyclin A cells harboring lagging chromosomes following 12 h of HU release. Cells were stained as in Fig. 1D, and anaphase cells were scored to calculate the percentage of cells with lagging chromosomes. Error bars represents S.E. from three independent experiments, and n.s. represents nonsignificant; *, p ≤ 0.05; **, p < 0.01; ***, p < 0.001.
Next, we were interested to investigate whether the sensitivity toward the replication stress inducer that we observed upon FBXO31 depletion can be reconciled upon co-depleting cyclin A in FBXO31KD cells. Flow cytometric analysis revealed a relief from HU-mediated stress-induced cell cycle delay of FBXO31KD cells following the depletion of cyclin A in FBXO31KD cells (Fig. 6B). This suggests that elevated cyclin A levels in FBXO31KD cells predispose the cells toward replicative stress, making them sensitive toward replication stress inducers (Fig. 6B). Interestingly, HU treatment-mediated prometaphase to metaphase delay as well as defects such as anaphase bridges and lagging chromosomes in FBXO31KD cells were also rescued in FBXO31 and cyclin A co-depleted cells (Fig. 6, C–E). These observations clearly suggest that the mitotic defects observed in FBXO31KD cells are exacerbated by defects in DNA replication due to unscheduled cyclin A accumulation.
DDL motif of FBXO31 at residues 297, 298, and 299 is essential for its interaction with cyclin A
Having confirmed that the elevated levels of untimely accumulated cyclin A as the major source of defects in cell-cycle progression and genomic instability in FBXO31KD cells, we wanted to identify the important residues within FBXO31 that are responsible for promoting polyubiquitination-mediated proteasomal degradation of cyclin A. To understand that, we performed molecular docking between cyclin A and FBXO31 followed by molecular dynamics (MD) simulation. The N-terminal region of cyclin A is necessary for the presentation and selective recognition of the destruction box by specific class of cellular proteins, which pave the way for their ubiquitination and proteasomal degradation (37). Unfortunately, the crystal structure of cyclin A in the PDB library encompasses only 171–432 amino acids. Therefore, we developed the full-length homology models of cyclin A in i-TASSER to delineate the importance of the N terminus in the ubiquitin-degradation program. The key advantage to the model development is that it can retrieve the template proteins (Tables S1 and S2) from the PDB library with highest sequence homology and enable ab initio modeling of the N-terminal regions by replica exchange Monte-Carlo simulations, for which no appropriate template is found (Figs. S4 and S5). FBXO31 acts as substrate-recognition adapter of the SCF (SKP1/Cullin/F-box protein) class of E3 ubiquitin ligases and actively participates in the covalent transfer of ubiquitin onto the substrates (1, 3). Earlier studies have shown that FBXO31 interacts with the core SCF complex through its F-box motif at its N-terminal region. FBXO31 interacts with its substrates such as cyclin D1 through its C-terminal substrate–binding domain to promote their ubiquitin-mediated degradation (38). Based on the evidence, we applied repulsive biasing constraints (masking) over the N-terminal F-box motif in FBXO31, which is involved in specific interactions with the SKP1 protein before carrying out the docking experiments with cyclin A. Thus, we narrowed the search space for more accurate docking poses reducing the number of false positives. We established the docking benchmarks by using two docking algorithms PIPER and Z-DOCK and clustered the similar binding poses reproduced independently by the different docking modules. Furthermore, to evaluate the quality of the docking poses, we simulated the complex for 40 ns in explicit solvent, which gives a more realistic picture of the physical contacts between FBXO31 and cyclin A retaining specific solvent interactions like that of the cellular systems. To monitor the structural changes during MD simulation, RMSD for the backbone atoms in the binding pockets of FBXO31-bound cyclin A was calculated with respect to the unbound states (Fig. S6). We observed that the complex attained a stable state over the 40-ns simulation run (Fig. S6B), and the structure converged to steady RMSD values at a range of 1.5–2.5 Å (Fig. S6C). We found that the physical interactions between FBXO31 and cyclin A mostly involve the amino acid residues proximal to the destruction box and are stabilized by an extended network of electrostatic interactions and weak hydrogen bonds. The carboxylic group of Asp-297 in FBXO31 makes a stable hydrogen bond interaction with the –NH2 group of the carboxamide side chain in Asn-93 of cyclin A. In addition, Asp-298 and Leu-299 residues of FBXO31 are tethered to Lys-91 of cyclin A by hydrogen bonding, which might serve as the ubiquitin-docking site and enable the formation of cyclin A-ubiquitin conjugates in the ubiquitin-dependent proteasomal degradation (Fig. S6A).
Taking these parameters into consideration, we generated a site-directed mutant of FBXO31 where we substituted all three residues, i.e. Asp-297, Asp-298, Leu-299 to alanine (D297A/D298A/L299A); henceforth, in this work this triple mutant of FBXO31 will be referred as Mut-FBXO31. To test the capacity of this triple mutant of FBXO31 in promoting degradation of its known substrates, we ectopically expressed MCF7 cells with either empty vector or myc-FBXO31 or myc-Mut-FBXO31 (Fig. S7A). It was observed that FBXO31, as expected, could efficiently promote the degradation of its known substrates, but the myc-Mut-FBXO31 could not promote cyclin A degradation (Fig. S7A). Interestingly, the Mut-FBXO31 could efficiently promote the degradation of other known substrates of FBXO31. Furthermore, Mut-FBXO31 failed to interact with cyclin A and also could not facilitate its Lys-48–linked polyubiquitination (Fig. S7, B and C).
To confirm our observations of enhanced cyclin A expression in FBXO31KD cells as one of the major contributors of the observed cell cycle delay and defects, we extended our studies toward ectopic expression of cyclin A. Ectopically expressed cyclin A induces accelerated S-phase entry (Fig. 7A), replicative stress (Fig. 7B), and defects in MCM loading (Fig. 7C). Interestingly, these defects were resolved to normal following co-expression of WT FBXO31 but not with Mut-FBXO31 (Fig. 7, A–C). Interestingly, mitotic progression defects in FBXO31KD cells were resolved following expression of WT FBXO31, but the cyclin A-binding defective FBXO31 mutant failed to do so (Fig. 7, D–F). Similarly, the F-box motif-deleted FBXO31 mutant, incompetent to promote degradation of cyclin A, could also not resolve the phenotypic defects observed in FBXO31KD cells (Fig. 7, D–F). Collectively, these observations clearly confirm that FBXO31 is an important E3 ubiquitin ligase that governs cyclin A expression during cell-cycle progression to facilitate coordination in cell cycle phase transitions and maintains genomic stability.
Figure 7.

FBXO31 controls the progression of mitosis and G1 phase through maintaining the optimum levels of cyclin A. A, image representing flow cytometric analysis of BrdU-pulsed MCF7 cells ectopically expressing cyclin A alone or along with either WT FBXO31 or mutant FBXO31, released from nocodazole block at the indicated time periods. Cells were pulsed with BrdU for 2 h before harvesting and following that the cells were collected by trypsinization, stained, and analyzed by flow cytometry. B, image representing flow cytometric analysis of BrdU-pulsed MCF7 cells, ectopically expressing cyclin A alone or along with either WT FBXO31 or mutant FBXO31. Transfected cells were released from HU block. Released cells were pulsed with BrdU for 2 h before harvesting. Pulsed cells were collected by trypsinization for FACS analysis. C, immunoblot analysis monitoring MCM4 level in chromatin and nonchromatin fractions during mitosis and G1 phase of MCF7 cells expressing either vector or cyclin A, or co-expressing cyclin A and WT FBXO31, or co-expressing cyclin A and mutant FBXO31. Transfected cells were synchronized with nocodazole and then released for the indicated time periods. D, graphical representation of percentage of G2 and different mitotic phases of NS and FBXO31KD cells expressing either vector or WT FBXO31 or cyclin A-binding defective FBXO31 mutant or F-box motif-deleted FBXO31 mutant following 12 h of hydroxyurea release. Cells were grown and collected as described in Fig. 1C. Cells were then stained with pH 3 Ser-10 antibody to score the number of cells at the G2 and different phases of mitosis. Error bars represent S.E. from three independent experiments. E, representative image of anaphase defects as observed in NS and FBXO31KD cells expressing either vector or WT FBXO31 or cyclin A-binding defective FBXO31 mutant or F-box motif–deleted FBXO31 mutant following 12 h of hydroxyurea release. Arrow (white) indicates cells are either depleted for FBXO31 or expressing cyclin A degradation-defective FBXO31 mutant or F-box motif–deleted FBXO31 mutant, and arrow (yellow) indicates cells are having WT FBXO31 expression. F, graphical representation of percentage of lagging and bridging chromosomes in NS and FBXO31KD cells expressing either vector or WT FBXO31 or cyclin A-binding defective FBXO31 mutant or F-box motif–deleted FBXO31 mutant following 12 h of hydroxyurea release. Cells were grown and collected as described in Fig. 1C and were stained with pH 3 Ser-10 antibody to score the number of anaphase cells and calculated the percentage of cells with bridging and lagging chromosomes. Error bars represent S.E. from three independent experiments. Error bars represents S.E. from three independent experiments, and n.s. represents nonsignificant; *, p ≤ 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Tumor suppressor FBXO31 acts as a crucial cell cycle checkpoint protein upon induction of genotoxic stresses (2–4). Recent reports have depicted the importance of FBXO31 in regulating the G2/M phase of the cell cycle to prevent genomic instability (7, 8). In addition, it has been shown that its level oscillates during the cell cycle with its high expression from late-G2 to the early-G1 phase of the cell cycle (1). However, why its expression level oscillates and what are the roles it plays during the cell-cycle progression of unstressed cells remain elusive. In this study, we have elucidated that FBXO31 assists the proteasomal degradation of cyclin A during mitosis and G1 phase to allow appropriate MCM4 loading on chromatin for an efficient licensing of origins, and thereby it regulates fidelity of DNA replication and segregation. Depletion of FBXO31 interferes with timing and efficiency of DNA replication consequently leading to endogenous replicative stress and DNA damage. These cells are sensitive toward replicative stress inducers and encounter difficulties during DNA segregation resulting in lagging chromosomes and anaphase bridges.
Cyclin A is eminent among other cyclin members as it can regulate two crucial events of the cell cycle, i.e. DNA replication and mitosis (17). Both these events are highly orchestrated, and any error may increase the susceptibility of the cells toward genomic instability (9). So, a tight regulation of cyclin A activity is paramount for maintaining the genomic integrity. Its expression is maintained at a low level from prometaphase until late G1 phase by APC/C-mediated proteasomal degradation (13). In this work, we have shown that SCFFBXO31 regulates cyclin A expression during G1, S, and mitosis (Fig. 4C). However, an unscheduled accumulation of cyclin A is observed in early and mid-G1 phases upon FBXO31 depletion suggesting an increased propensity of FBXO31 to degrade cyclin A during these phases. This is very important in order to maintain an environment of low cyclin A–CDK activity during the G1 phase to facilitate MCM loading on chromatin. Increased propensity of FBXO31 to facilitate cyclin A degradation during the G1 phase might be guided by certain specific post-translational modifications either on cyclin A or on FBXO31 during the G1 phase to enhance the polyubiquitination-mediated proteasomal degradation of cyclin A. There are ample of reports that have shown stage-specific changes in post-translation modifications such as phosphorylation, acetylation, etc., guiding the fate of multiple proteasome targets. Alterations in these post-translational modifications might not disturb the interaction between the substrates and the F-box proteins; however, it can significantly alter the polyubiquitination status of substrates (6). Therefore, future studies are needed to identify the cellular signaling mechanisms regulating the proteasomal degradation of cyclin A by FBXO31 at the G1 phase of the cell cycle.
We observed that cyclin A regulation by FBXO31 is independent of APC/CCDH1 as the levels of CDH1 in FBXO31 knockdown (KD) cells remain similar to that of the control cells; however, increased accumulation of cyclin A in FBXO31KD cells is detected during G1 phase suggesting that CDH1 alone is not sufficient to maintain the low levels of cyclin A during G1 phase. Additionally, ectopic expression of FBXO31 can promote proteasomal degradation of cyclin A in APC2KD cells indicating that FBXO31-mediated regulation of cyclin A is independent of APC/C complex (Fig. S3D). Moreover, FBXO31 promotes Lys-48–linked polyubiquitination, whereas APC/C promotes Lys-11–linked polyubiquitination of cyclin A. Based on these observations, we anticipate the existence of parallel pathways regulating the abundance of cyclin A during the G1 phase of the cell cycle to maintain low cyclin A–CDK activity. Neither CDH1 nor FBXO31 can compensate for each other's loss suggesting that both CDH1 and FBXO31 are essential for maintaining an optimum level of cyclin A for an unperturbed DNA replication. Because of the existence of two parallel regulators of cyclin A during G1 progression, depletion of any resulted in cell cycle delay and defects; however, it did not block cell-cycle progression. Interestingly, cells co-depleted of both CDH1 and FBXO31 showed retarded proliferation and were difficult to propagate in cultures (data not shown). In fact, here we are reporting, for the first time, the existence of an SCFFBXO31 E3 ubiquitin ligase that can promote the degradation of cyclin A independent of APC/C.
Enhanced level cyclin A during G1 phase interferes with the loading of MCM proteins to chromatin (31). In this study, we have observed that FBXO31 depletion prevented chromatin loading of MCM complex during the G1 phase, as represented by hampered loading of MCM4, which is an important component of the MCM complex (Fig. 4E). Interestingly, restoring cyclin A expression to normal physiological levels either by co-depletion of cyclin A in FBXO31KD cells (Fig. 4E and Fig. S3E) or by ectopic expression of functional FBXO31 in cyclin A–overexpressing cells (Fig. 7C) allowed the normal loading of MCM4 on chromatin demonstrating the role of FBXO31 in maintaining low expression levels of cyclin A during the G1 phase for efficient MCM4 loading on chromatin.
Previous reports have shown that the cyclin A–CDK2 complex controls the progression of S phase (19). Additionally, ectopic expression of cyclin A is reported to promote S-phase entry (26–28). In this study, we observed that a significant percentage of FBXO31KD cells enter S phase prematurely when the NS cells were still at G1 phase (Fig. 1B and Fig. S1B). These FBXO31KD cells showed an increased number of cells positive for S-phase–specific pattern of PCNA distribution when compared with NS cells. However, the mean PCNA intensity per cell was quite low in replicating FBXO31KD cells (Fig. 5, D and E) suggesting that FBXO31KD cells enter S phase prematurely with a reduced number of PCNA foci per cell, indicative of DNA replication utilizing a reduced number of origins. These observations are in agreement with a previous report explaining that entry into S phase with insufficient licensed origins leads to firing from fewer origins and thus reducing the efficiency and fidelity of DNA replication (13). We also observed increased γ-H2AX foci in cells harboring S-phase–specific PCNA foci indicative of DNA damage arising during DNA replication (Fig. 5, D and F). Increased DNA damage following premature DNA replication may arise due to inefficient DNA replication resulting in an increased unresolved replication intermediate. Such type of replication intermediates hampers fork progression and results in DNA double-strand breaks that are significantly higher in the S-phase population of FBXO31KD cells, evident in the form of γ-H2AX foci (Fig. 5, D and F). Interestingly, co-depletion of cyclin A in FBXO31KD cells prevented FBXO31KD cells from entering S phase prematurely (Fig. 6A). Additionally, ectopic expression of functional FBXO31 in cells overexpressing cyclin A could significantly prevent premature DNA replication (Fig. 7A). Thus, our study demonstrates that FBXO31 is an important regulator of unperturbed DNA replication.
Although the depletion of FBXO31 resulted in a reduced level of MCM loading and affected the timing and efficiency of DNA replication, FBXO31KD cells proliferated at the same rate as that of NS cells (data not shown). This might be due to the utilization of dormant origins by the FBXO31KD cells. However, despite the use of dormant origins, cells defective in MCM loading accumulate genomic instability and more profoundly when exposed to replicative stress (14). Similarly, when FBXO31KD cells were exposed to hydroxyurea-mediated replication stress, we found them to be sensitive toward the replication stress inducer. In response to exposure to hydroxyurea, FBXO31KD cells showed a significant delay in the progression from S to G2/M phase and subsequently in G2/M to G1 phase. Interestingly, we also observed that upon exposure to repeated replicative stress, FBXO31KD cells underwent massive cell death. This is particularly important because it indicates that FBXO31KD cells are sensitive to replicative stress inducers, which can be harnessed for designing therapeutic strategies against tumors lacking FBXO31. Interestingly, this anomaly of FBXO31KD cells toward replication stress was eliminated following co-depletion of cyclin A in FBXO31KD cells indicating that enhanced cyclin A levels in FBXO31KD cells predisposes FBXO31KD cells toward replication stress.
It has been well-documented that inefficient DNA replication results in persistence of complicated replication intermediates until mitosis, generation of intertwined sister chromatids, and incompletely replicated loci (9). Such defects interfere with DNA segregation during mitosis leading to lagging and bridging chromosomes (9). In the same line, we observed a significant increase in the percentage of lagging and bridging chromosomes in FBXO31KD cells released from HU treatment, which was resolved in FBXO31 and cyclin A co-depleted cells (Fig. 6, D and E). Additionally, ectopic expression of WT FBXO31 in FBXO31KD cells efficiently prevented the occurrence of defects during mitosis (Fig. 7, D–F). During prometaphase, cyclin A is reported to prevent stable interactions between kinetochore and microtubule that act as a surveillance mechanism for preventing improper kinetochore and microtubule attachment (39). However, failure to degrade cyclin A at the end of prometaphase delays prometaphase to metaphase transition (18). Similarly, we observed a delay in prometaphase to metaphase transition in FBXO31KD cells, which was resolved upon co-depleting cyclin A in FBXO31KD cells (Fig. 6C). Similarly, ectopic expression of WT FBXO31 in FBXO31KD cells prevented mitotic delay (Fig. 7D). These observations showed that limiting the scheduled degradation of cyclin A during prometaphase progression in FBXO31KD cells prevents prometaphase to metaphase transition suggesting the requirement of FBXO31 to prevent occurrence of mitotic progression defects. Thus, an imbalance in cyclin A levels allows creeping of error both during DNA replication and mitosis. Hence, FBXO31 by regulating cyclin A stability monitors the accuracy of cell-cycle progression both during DNA replication and its segregation.
Previous reports have shown that FBXO31 maintains genomic integrity by regulating the post-translational level of Cdt1 and FOXM1 (7, 8). Transient depletion of FBXO31 resulted in the enhanced transcriptional activity of FOXM1 during G2/M progression leading to mitotic delay and defects. Interestingly, the defects in mitosis were rescued upon co-depletion of FBXO31 along with FOXM1 but not the delay in mitosis, indicating the involvement of additional important substrate(s) of FBXO31. Additionally, previous reports even show that cyclin A–CDK complex phosphorylates and monitors the activity of FOXM1, and high cyclin A–CDK activity can concomitantly result in high FOXM1 activity (40). This indicates that FBXO31 might be capping FOXM1 activity by dual mechanisms: directly by directing its polyubiquitination and indirectly via cyclin A. Interestingly, co-depletion of cyclin A in FBXO31KD cells eliminated the cell-cycle delay as well as mitotic defects that were observed upon FBXO31 depletion and deciphering why co-depletion of FOXM1 in the FBXO31KD cells could not resolve the mitotic delay. Thus, this study reports the underlying cause of delay observed upon FBXO31 depletion is due to an elevated cyclin A level. Importantly, we are the first to report that the defects observed upon FBXO31 depletion arise not only during mitosis but begin accumulating during initiation of the cell cycle right from the G1 phase and also during the S phase, the consequence of which can be visualized in the form of mitotic defects. These recent studies and our present study emphasize that FBXO31 plays major roles during the progression of different phases of cell cycle.
In a nutshell, our observations have demonstrated that FBXO31 facilitates scheduled degradation of cyclin A to maintain genomic stability by safeguarding the fidelity of DNA replication and mitosis (Fig. 8). Our study has focused on one aspect of FBXO31 in regulating faithful DNA replication; however, detailed studies focusing on the role of FBXO31 during S phase, replicative stress, and repair will be of immense importance to pursue. Importantly, FBXO31KD cells are sensitive toward replication stress and fail to tolerate repeated replicative stress and eventually undergo death. Therefore, therapeutic regimes involving replication stress inducers either alone or in combination with other genotoxic agents might prove helpful toward the management of tumors with diminished FBXO31 expression.
Figure 8.

Proposed model depicts the role of FBXO31 in coordinated cell-cycle progression. Polyubiquitination of cyclin A by FBXO31 facilitates its degradation during mitosis and G1 phase thus maintaining low cyclin A–CDK activity during G1. This environment is favorable for MCM complex loading and licensing of origins and efficient DNA replication. Cells lacking FBXO31 accumulate cyclin A during mitosis and throughout G1 phase resulting in high cyclin A–CDK activity, which promotes dissociation of MCM complex from origins and hence hampers origin licensing. High level of cyclin A being a rate-limiting factor for S-phase progression stimulates S-phase progression with reduced number of licensed origins resulting in inefficient DNA replication and DNA damage. Segregation of damaged DNA during mitosis results in genomic instability in the form of lagging and bridging chromosomes.
Materials and methods
Cell culture and generation of stable knockdown cells
HEK-293T cells and MCF7 cells (a kind gift from Prof. Michael R. Green, University of Massachusetts Medical School) were cultured in DMEM (Gibco, 12800-017) containing 10% fetal bovine serum (Gibco 16000044) and 0.1% penicillin and streptomycin solution (Life Technologies, Inc., 15140-122s) at 37 °C with 5% CO2 under humid conditions. Cells were periodically checked for mycoplasma.
Knockdown of FBXO31 (FBXO31KD) and APC2 (APC2KD) were generated in MCF7 cells using lentiviral transduction as described previously (3). Briefly, lentivirus particles were generated in HEK-293T cells upon transfection of shRNA (shRNA-FBXO31-1 clone ID V2LHS_157523, shRNA-FBXO31-2 clone ID TRCN0000141663, shAPC2 clone ID V2LHS_64696, shCyclin A-1 clone ID V2LHS_47803, and shCyclin A-2 clone ID V3LHS_364609) and lentivirus packaging plasmids (pPAX2 and pMD2.G). After 48 h of transfection, media containing virus particles were collected and filtered through 0.45-μm syringe filter, and then host cells were transduced with virus particles in the presence of 8 μg/ml Polybrene (Sigma, TR-1003). Virus-infected MCF7 cells were selected by growing them in the presence of puromycin (1 μg/ml, Sigma, P8833) to obtain stable clones. Knockdown efficiency was tested by RT-PCR as well as Western blotting. Cells infected with virus containing scramble shRNAs were taken as WT control cells (NS). Cells for immunofluorescence analysis were seeded on poly-l-lysine (Sigma, P4707)-coated coverslips.
Cell synchronization
For HU (Sigma, H8627)-mediated synchronization, MCF7 cells were grown in the presence of 0.25 mm HU for 22 h. For nocodazole (Sigma, M1404)-mediated prometaphase block, MCF7 cells were treated with 125 nm nocodazole for 16 h. Subsequently, cells were released by washing three times with 0.5% fetal bovine serum containing DMEM and then were allowed to re-enter the cell cycle by growing in complete media without synchronizing agents. Synchronized cells were then collected at the indicated time points in two parts: one part by flushing out in PBS for Western blot analysis, and the other part was harvested by trypsinization followed by ethanol fixation for flow cytometric analysis.
Transfections for overexpression
Cells were grown in complete medium for 24 h before transfection at 60–70% confluency. The next day, cells were transfected using Lipofectamine 2000 reagent (Invitrogen, 15338100) according to the manufacturer's protocol. After 12 h of transfection, media were replaced with complete DMEM, and then cells were harvested at specified time points (12–48 h) for further studies. Cells were treated with proteasome inhibitor MG132 (10 μm, Calbiochem, 474790) for 6 h wherever required. For cell cycle-related immunoprecipitation (IP) experiments, cells were treated with MG132 for 2 h before harvesting the cells.
Flow cytometry–based cell cycle analysis
Cells were collected by trypsinization and were fixed in 95% ethanol for at least 24 h. On the day of acquisition, cells were washed twice with PBS and incubated with 1 ml of staining solution (50 μg/ml propidium iodide (Sigma, P4170), 50 μg/ml RNase A (Invitrogen, 12091-039), and 2 mm MgCl2 in PBS) in the dark at 37 °C for 30 min. Cells were then passed through a cell strainer (BD Biosciences) and acquired using FACSCalibur flow cytometry (BD Biosciences). Data were analyzed using BD CellQuest Pro software (BD Biosciences).
BrdU chase assay to monitor cell proliferation
BrdU pulse-chase assay was carried according to the manufacturer's protocol for BrdU staining kit (BD Pharmingen, 552598). Briefly, 106 cells were seeded and pulsed with 1 mm BrdU solution for 2 h. The cells were then collected by trypsinization and fixed in BD Cytofix/Cytoperm buffer for 30 min in dark at 25 °C. Then the cells were washed with 1 ml of 1× BD Perm/Wash buffer by centrifugation at 300 × g. Cell pellet was then resuspended in BD Cytoperm Permeabilization Buffer Plus for 10 min on ice. Permeabilized cells were then washed in BD Perm/Wash buffer as mentioned previously. The cells were again re-fixed in BD Cytofix/Cytoperm buffer for 5 min at 25 °C. Fixed cells were again washed and incubated with 300 μg/ml of DNase solution for 1 h at 37 °C. Next, the cells were incubated with APC-conjugated anti-BrdU antibody and then washed in BD Perm/Wash buffer. The cells were then stained with propidium iodide to probe for total DNA levels and acquired in a BD Canto II machine. The data were analyzed using FlowJo software.
Western blotting
Protein extracts were prepared as described previously (3). Briefly, cells were harvested, washed twice in PBS, and lysed for 20 min on ice in cell lysis buffer (50 mm Tris, pH 7.4, 250 mm NaCl, 50 mm NaF, 0.5 mm Na3VO4, 0.5% Triton X-100, 5 mm EDTA, and freshly added protease inhibitor mixture). Then, the lysates were centrifuged at 21,000 × g for 20 min and the supernatants were collected. Protein concentration was measured by the Bradford method using BSA as a standard (41). Proteins (24 μg/lane) were separated on SDS-PAGE, transferred onto the polyvinylidene difluoride membrane followed by 1 h of blocking in 3% skimmed milk, and then incubated with primary antibody overnight at 4 °C. Next day, membranes were incubated with the respective horseradish peroxidase–conjugated secondary antibody for 1 h at 25 °C. Membranes were washed three times in TBS with 0.1% Tween 20, and protein expression was detected using either X-ray film or LAS Image Quant 4000 (GE Healthcare) using an enhanced chemiluminescent kit from Pierce (West Pico-34578 and West Femto-34096).
IP
Whole-cell lysates were prepared as described above. IP was set in 600 μl of IP lysis buffer (50 mm Tris, pH 7.4, 250 mm NaCl, 50 mm NaF, 0.5 mm Na3VO4, 0.1% Triton X-100, 5 mm EDTA, and freshly added protease inhibitor mixture) containing 800 μg of total protein and 2–3 μg of primary antibody. Lysate/antibody mixtures were incubated overnight in IP rotor with gentle rotation at 4 °C. The next day, washed protein G–agarose beads (Thermo Fisher Scientific 22852) were added to the mixture and incubated at 4 °C for an additional 1.5 h on an IP rotor. Then, beads were spun down at 250 × g for 1 min, and the supernatant was discarded. Again, the beads were washed three times in IP lysis buffer. Immunoprecipitated proteins were eluted in Laemmli buffer (1×) by boiling the beads for 5 min and then spun down at 2700 × g for 2 min, and eluents were loaded immediately on SDS-PAGE. Then Western blotting was performed to monitor interactions. Cross-linking IP was performed according to the protocol mentioned in the Abcam website.
Colony formation assay
Colony formation assay was performed as described previously (42). NS and FBXO31KD MCF7 cells (0.4 × 106) were seeded in a 35-mm dish 1 day prior to the replication stress induction. Replicative stress was induced in NS and FBXO31KD cells by treating the cells with 0.25 mm hydroxyurea for 24 h followed by releasing the cells for 24 h. This cycle was repeated three times, and then the cells were allowed to grow for 10 days to form colonies. Finally, the colonies were stained with crystal violet (Sigma, HT90132). The experiment was repeated three times.
Cycloheximide pulse–chase experiment
Freshly prepared cycloheximide (Sigma, C1988) solution (40 μg/ml) was added to the growing cells, and then cells were collected at specified time points. Cells were harvested, prepared whole-cell lysate, and performed Western blotting as described above.
Polyubiquitination assay
FLAG–cyclin A was co-transfected into cells along with either HA–ubiquitin or myc-FBXO31 or in a combination of both. Cells were collected after 36 h of transfection. Then, the cells were lysed and subjected to IP as described above. For Lys-48–linked polyubiquitination, NS and FBXO31KD cells were lysed and subjected to immunoprecipitation for cyclin A. For the cell cycle–dependent polyubiquitination experiment, NS and FBXO31KD cells were transfected with FLAG–cyclin A in combination with either with His–Lys-11-only ubiquitin or His–Lys-48-only ubiquitin plasmid for 36 h. After 24 h of transfection, cells were grown in the presence of 125 nm nocodazole for 16 h and then the cells were released and grown in the absence of nocodazole for the indicated time periods. MG132 was added 2 h prior to harvesting of cells at the indicated time points, and immunoprecipitation was performed followed by Western blotting to monitor polyubiquitination.
Chromatin and nonchromatin fractionation
Cells were resuspended in ice-cold buffer A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 340 mm sucrose, and 10% glycerol and before using the following were added: 0.1% Triton X-100, 1 mm DTT, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture) and were rotated in a cold room for 8 min and then centrifuged at 1300 × g for 5 min at 4 °C (43). The supernatant was removed and labeled as soluble cytoplasmic fraction. The soluble cytoplasmic fraction was clarified by centrifuging at 21,000 × g for 10 min at 4 °C. The nuclear pellet was washed with buffer A (without Triton X-100) by pipetting up and down and then was centrifuged at 350 × g for 10 min at 4 °C. The washed nuclear pellet was resuspended in buffer B (3 mm EDTA, 0.2 mm EGTA, 1 mm DTT, 1 mm phenylmethylsulfonyl fluoride, protease inhibitor mixture) and rotated for 30 min at 4 °C and then centrifuged at 1700 × g for 10 min at 4 °C. The supernatant was removed and labeled as a soluble nuclear fraction. The pellet or insoluble/chromatin fraction was washed with 500 μl of buffer B and centrifuged at 1700 × g for 10 min at 4 °C. The chromatin pellet was resuspended in 200 μl of 1× SDS-PAGE sample buffer and sonicated to reduce the viscosity. Mixed cytoplasmic and soluble nuclear fraction and labeled as nonchromatin fraction.
Immunofluorescence
Cells were grown on poly-l-lysine (Sigma, P4707)-coated coverslips, fixed in 4% paraformaldehyde for 20 min at 25 °C, permeabilized in 0.1% Triton X-100 for 15 min, and blocked by incubating with 2% BSA in PBS for 0.5 h. After blocking, the cells were incubated with primary antibody for 2 h at 25 °C. Cells were then incubated with fluorochrome-conjugated secondary antibody for 1 h (Life Technologies, Inc.) and finally with Hoechst-33258 (Sigma, 94403). After each step, the cells were washed three times in PBS. Next, the stained cells were mounted in mounting media; the circumference of the coverslips was sealed with nail polish; the slides were observed under an epifluorescence microscope (Olympus 1X71 microscope), and data were analyzed using Q-capture pro software. For confocal microscopy studies, Leica SP5 II confocal microscope was used, and data were analyzed using Leica AIM 4.2 software.
Antibodies used
The following antibodies were used: FBXO31 (Sigma, F4431), cyclin A (Santa Cruz Biotechnology, sc-751), tubulin (Sigma, T5168), p-histone 3-S10 (ab47297), MCM4 (Cell Signaling, 12973), histone (Santa Cruz Biotechnology, sc-10809), Myc (Roche Applied Science, 11667203001), FLAG (Sigma, 1804), Cullin 1 (sc-17775), HA (Santa Cruz Biotechnology, sc-805), Lys-48 ubiquitin (Cell Signaling 8081), CDH1 (Santa Cruz Biotechnology, sc-166714), CDC20 (Santa Cruz Biotechnology, sc-13162), CDK1 (Santa Cruz Biotechnology, sc-54), CDK2 (sc-6248), His (Santa Cruz Biotechnology, sc-8036), Lamin B1 (Abcam, ab16048), PCNA (Santa Cruz Biotechnology, sc-25280), and pγ-H2AX S-139 (Abcam ab22551).
Plasmids used in the study
HA–ubiquitin was procured from Addgene-18712. Scrambled shRNA and shRNA against FBXO31 were the kind gifts from Prof. Michael R. Green (University of Massachusetts Medical School). myc-FBXO31 and myc-ΔF-FBXO31 were the kind gifts from Dr. David F. Callen (the University of Adelaide, and Hanson Institute, Australia). Lys-11–only ubiquitin and Lys-48–only ubiquitin mutants were the kind gifts from Dr. Wuhan Xiao (Chinese Academy of Science, China). GST–cyclin A was the kind gift from Prof. Jonathon Pines (The Institute of Cancer Research, London, UK), which was subcloned in p3xFLAG-myc-CMV vector (Sigma, E7283) at HindIII and EcoRI restriction sites using the cloning primer, the details of which are mentioned in Table S3. D297A, D298A, and L299A mutants of FBXO31 were generated using the standard site-directed mutagenesis technique. WT FBXO31, F-box–deleted FBXO31, and mutant FBXO31 were cloned in pCMV-Myc-N vector using BglII restriction sites (Clontech) and in pEGFP-C1 vector (Addgene) using BglII restriction sites. F-box–deleted FBXO31 was cloned using the same primers as WT FBXO31 but the DNA template used was F-box–deleted FBXO31 construct obtained from Dr. David Callen. The primer sequences used in this study are listed in Table S3.
Real-time RT-PCR
Total RNA was isolated using TRIzol reagent according to the manufacturer's protocol (Invitrogen, 15596018). 1 μg of purified RNA (for 20-μl reaction) was used to synthesize cDNA using PrimeScript first strand cDNA synthesis kit (Takara, 6110A). Real-time PCR was performed using the SYBR Green reagent (Takara, RR82WR). The primers used are described in Table S3.
Author contributions
P. D., M. R. W., S. Chatterjee, and M. K. S. conceptualization; P. D. and M. K. S. data curation; P. D., S. I., and S. Choppara validation; P. D., S. I., S. Choppara, P. S., Anil Kumar, and Avinash Kumar investigation; P. D., S. I., S. Choppara, P. S., Anil Kumar, Avinash Kumar, and M. K. S. methodology; P. D. and S. I. writing-original draft; P. D., S. I., P. S., S. Chatterjee, and M. K. S. formal analysis; M. R. W. and M. K. S. supervision; M. K. S. funding acquisition; M. K. S. project administration; M. K. S. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. M. R. Green (University of Massachusetts Medical School) for providing cell lines and lentiviral shRNAs; Prof. J. Pines (The Institute of Cancer Research, London, United Kingdom) for the GST–cyclin A construct; Dr. David F. Callen (the University of Adelaide and Hanson Institute, Australia) for providing FBXO31 clones, and Dr. Wuhan Xiao (Chinese Academy of Science, China) for providing His–Lys-11-only and His–Lys-48-only constructs and clones. We also thank Dr. Arnab Ray Chaudhuri (Erasmus MC, Netherlands) and Dr. Vasudevan Seshadri (National Centre for Cell Science, Pune, India) for useful discussions and Ganesh Kumar Barik for proofreading the paper.
This work was supported by the University Grants Commission fellowship (to P. D.), DBT fellowship (to S. I.), CSIR fellowship (to S. Ch., P. S., and A. K.), and in part by National Centre for Cell Science and Department of Biotechnology, Government of India, Grant BT/PR6690/GBD/27/475/2012. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7 and Tables S1–S3.
- MCM
- minichromosome maintenance complex
- DMEM
- Dulbecco's modified Eagle's medium
- PCNA
- proliferating cell nuclear antigen
- NS
- nonspecific
- HU
- hydroxyurea
- CDK
- cyclin-dependent kinase
- APC/C
- anaphase-promoting complex
- IP
- immunoprecipitation
- Ni-NTA
- nickel-nitrilotriacetic acid
- MD
- molecular dynamics
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation.
References
- 1. Kumar R., Neilsen P. M., Crawford J., McKirdy R., Lee J., Powell J. A., Saif Z., Martin J. M., Lombaerts M., Cornelisse C. J., Cleton-Jansen A. M., and Callen D. F. (2005) FBXO31 is the chromosome 16q24.3 senescence gene, a candidate breast tumor suppressor, and a component of an SCF complex. Cancer Res. 65, 11304–11313 10.1158/0008-5472.CAN-05-0936 [DOI] [PubMed] [Google Scholar]
- 2. Tan Y., Liu D., Gong J., Liu J., and Huo J. (2018) The role of F-box only protein 31 in cancer (review). Oncol. Lett. 15, 4047–4052 10.3892/ol.2018.7816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Santra M. K., Wajapeyee N., and Green M. R. (2009) F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459, 722–725 10.1038/nature08011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malonia S. K., Dutta P., Santra M. K., and Green M. R. (2015) F-box protein FBXO31 directs degradation of MDM2 to facilitate p53-mediated growth arrest following genotoxic stress. Proc. Natl. Acad. Sci. U.S.A. 112, 8632–8637 10.1073/pnas.1510929112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choppara S., Malonia S. K., Sankaran G., Green M. R., and Santra M. K. (2018) Degradation of FBXO31 by APC/C is regulated by AKT- and ATM-mediated phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 115, 998–1003 10.1073/pnas.1705954115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choppara S., Ganga S., Manne R., Dutta P., Singh S., and Santra M. K. (2018) The SCFFBXO46 ubiquitin ligase complex mediates degradation of the tumor suppressor FBXO31 and thereby prevents premature cellular senescence. J. Biol. Chem. 293, 16291–16306 10.1074/jbc.RA118.005354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Johansson P., Jeffery J., Al-Ejeh F., Schulz R. B., Callen D. F., Kumar R., and Khanna K. K. (2014) SCF-FBXO31 E3 ligase targets DNA replication factor Cdt1 for proteolysis in the G2 phase of cell cycle to prevent Re-replication. J. Biol. Chem. 289, 18514–18525 10.1074/jbc.M114.559930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeffery J. M., Kalimutho M., Johansson P., Cardenas D. G., Kumar R., and Khanna K. K. (2017) FBXO31 protects against genomic instability by capping FOXM1 levels at the G2/M transition. Oncogene 36, 1012–1022 10.1038/onc.2016.268 [DOI] [PubMed] [Google Scholar]
- 9. Mankouri H. W., Huttner D., and Hickson I. D. (2013) How unfinished business from S-phase affects mitosis and beyond. EMBO J. 32, 2661–2671 10.1038/emboj.2013.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambert S., and Carr A. M. (2013) Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma 122, 33–45 10.1007/s00412-013-0398-9 [DOI] [PubMed] [Google Scholar]
- 11. Symeonidou I. E., Kotsantis P., Roukos V., Rapsomaniki M. A., Grecco H. E., Bastiaens P., Taraviras S., and Lygerou Z. (2013) Multi-step loading of human minichromosome maintenance proteins in live human cells. J. Biol. Chem. 288, 35852–35867 10.1074/jbc.M113.474825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nishitani H., and Lygerou Z. (2002) Control of DNA replication licensing in a cell cycle. Genes Cells 7, 523–534 10.1046/j.1365-2443.2002.00544.x [DOI] [PubMed] [Google Scholar]
- 13. Greil C., Krohs J., Schnerch D., Follo M., Felthaus J., Engelhardt M., and Wäsch R. (2016) The role of APC/CCdh1 in replication stress and origin of genomic instability. Oncogene 35, 3062–3070 10.1038/onc.2015.367 [DOI] [PubMed] [Google Scholar]
- 14. Ibarra A., Schwob E., and Méndez J. (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 105, 8956–8961 10.1073/pnas.0803978105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uzbekov R. E. (2004) Analysis of the cell cycle and a method employing synchronized cells for study of protein expression at various stages of the cell cycle. Biochemistry 69, 485–496 [DOI] [PubMed] [Google Scholar]
- 16. Podhorecka M., Skladanowski A., and Bozko P. (2010) H2AX phosphorylation: its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 920161 10.4061/2010/920161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pagano M., Pepperkok R., Verde F., Ansorge W., and Draetta G. (1992) Cyclin A is required at two points in the human cell cycle. EMBO J. 11, 961–971 10.1002/j.1460-2075.1992.tb05135.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. den Elzen N., and Pines J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol. 153, 121–136 10.1083/jcb.153.1.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coverley D., Laman H., and Laskey R. A. (2002) Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol. 4, 523–528 10.1038/ncb813 [DOI] [PubMed] [Google Scholar]
- 20. Furuno N., den Elzen N., and Pines J. (1999) Human cyclin A is required for mitosis until mid-prophase. J. Cell Biol. 147, 295–306 10.1083/jcb.147.2.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pines J., and Hunter T. (1991) Human cyclin-A and cyclin-B1 are differentially located in the cell and undergo cell-cycle dependent nuclear transport. J. Cell Biol. 115, 1–17 10.1083/jcb.115.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glotzer M., Murray A. W., and Kirschner M. W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 10.1038/349132a0 [DOI] [PubMed] [Google Scholar]
- 23. Bai C., Sen P., Hofmann K., Ma L., Goebl M., Harper J. W., and Elledge S. J. (1996) SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86, 263–274 10.1016/S0092-8674(00)80098-7 [DOI] [PubMed] [Google Scholar]
- 24. Schulman B. A., Carrano A. C., Jeffrey P. D., Bowen Z., Kinnucan E. R., Finnin M. S., Elledge S. J., Harper J. W., Pagano M., and Pavletich N. P. (2000) Insights into SCF ubiquitin ligases from the structure of the Skp1–Skp2 complex. Nature 408, 381–386 10.1038/35042620 [DOI] [PubMed] [Google Scholar]
- 25. Kipreos E. T., and Pagano M. (2000) The F-box protein family. Genome Biol. 1, REVIEWS3002 10.1186/gb-2000-1-5-reviews3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosenberg A. R., Zindy F., Le Deist F., Mouly H., Métézeau P., Bréchot C., and Lamas E. (1995) Overexpression of human cyclin A advances entry into S phase. Oncogene 10, 1501–1509 [PubMed] [Google Scholar]
- 27. Resnitzky D., Hengst L., and Reed S. I. (1995) Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol. Cell. Biol. 15, 4347–4352 10.1128/MCB.15.8.4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chibazakura T., Kamachi K., Ohara M., Tane S., Yoshikawa H., and Roberts J. M. (2011) Cyclin A promotes S-phase entry via interaction with the replication licensing factor Mcm7. Mol. Cell. Biol. 31, 248–255 10.1128/MCB.00630-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Williamson A., Wickliffe K. E., Mellone B. G., Song L., Karpen G. H., and Rape M. (2009) Identification of a physiological E2 module for the human anaphase-promoting complex. Proc. Natl. Acad. Sci. U.S.A. 106, 18213–18218 10.1073/pnas.0907887106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsumoto M. L., Wickliffe K. E., Dong K. C., Yu C., Bosanac I., Bustos D., Phu L., Kirkpatrick D. S., Hymowitz S. G., Rape M., Kelley R. F., and Dixit V. M. (2010) K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell 39, 477–484 10.1016/j.molcel.2010.07.001 [DOI] [PubMed] [Google Scholar]
- 31. Wheeler L. W., Lents N. H., and Baldassare J. J. (2008) Cyclin A-CDK activity during G1 phase impairs MCM chromatin loading and inhibits DNA synthesis in mammalian cells. Cell Cycle 7, 2179–2188 10.4161/cc.7.14.6270 [DOI] [PubMed] [Google Scholar]
- 32. Diffley J. F. (2004) Regulation of early events in chromosome replication. Curr. Biol. 14, R778–786 10.1016/j.cub.2004.09.019 [DOI] [PubMed] [Google Scholar]
- 33. Labib K., Tercero J. A., and Diffley J. F. (2000) Uninterrupted MCM2–7 function required for DNA replication fork progression. Science 288, 1643–1647 10.1126/science.288.5471.1643 [DOI] [PubMed] [Google Scholar]
- 34. Essers J., Theil A. F., Baldeyron C., van Cappellen W. A., Houtsmuller A. B., Kanaar R., and Vermeulen W. (2005) Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell. Biol. 25, 9350–9359 10.1128/MCB.25.21.9350-9359.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boehm E. M., Gildenberg M. S., and Washington M. T. (2016) The many roles of PCNA in eukaryotic DNA replication. Enzymes 39, 231–254 10.1016/bs.enz.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schönenberger F., Deutzmann A., Ferrando-May E., and Merhof D. (2015) Discrimination of cell cycle phases in PCNA-immunolabeled cells. BMC Bioinformatics 16, 180 10.1186/s12859-015-0618-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fung T. K., Yam C. H., and Poon R. Y. (2005) The N-terminal regulatory domain of cyclin A contains redundant ubiquitination targeting sequences and acceptor sites. Cell Cycle 4, 1411–1420 10.4161/cc.4.10.2046 [DOI] [PubMed] [Google Scholar]
- 38. Li Y., Jin K., Bunker E., Zhang X., Luo X., Liu X., and Hao B. (2018) Structural basis of the phosphorylation-independent recognition of cyclin D1 by the SCF FBXO31 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 115, 319–324 10.1073/pnas.1708677115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kabeche L., and Compton D. A. (2013) Cyclin A regulates kinetochore microtubules to promote faithful chromosome segregation. Nature 502, 110–113 10.1038/nature12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laoukili J., Alvarez M., Meijer L. A., Stahl M., Mohammed S., Kleij L., Heck A. J., and Medema R. H. (2008) Activation of FoxM1 during G2 requires Cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell. Biol. 28, 3076–3087 10.1128/MCB.01710-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 42. Rosenblum M. L., Knebel K. D., Wheeler K. T., Barker M., and Wilson C. B. (1975) Development of an in vitro colony formation assay for the evaluation of in vivo chemotherapy of a rat brain tumor. In Vitro 11, 264–273 10.1007/BF02615637 [DOI] [PubMed] [Google Scholar]
- 43. Méndez J., and Stillman B. (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol. 20, 8602–8612 10.1128/MCB.20.22.8602-8612.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.