

Abstract
Formyl peptide receptors (FPRs) are G protein-coupled receptors (GPCRs) expressed on a variety of cell types.These receptors play an important role in the regulation of inflammatory reactions and sensing cellular damage. They have also been implicated in the pathogenesis of various diseases, including neurodegenerative diseases, cataract formation, and atherogenesis. Thus, FPR ligands, both agonists and antagonists, may represent novel therapeutics for modulating host defense and innate immunity. A variety of molecules have been identified as receptor subtype-selective and mixed FPR agonists with potential therapeutic value during last decade. This review describes our efforts along with recent advances in the identification, optimization, biological evaluation, and structure–activity relationship (SAR) analysis of small molecule non-peptide FPR agonists and antagonists, including chiral molecules. Questions regarding the interaction at the molecular level of benzimidazoles, pyrazolones, pyridazin-3(2H)-ones, N-phenylureas and other derivatives with FPR1 and FPR2 are discussed. Application of computational models for virtual screening and design of FPR ligands is also considered.
Keywords: Agonist, Ca2+ mobilization, chiral recognition, formyl peptide receptor, G protein-coupled receptor, neutrophil, molecular modeling
1. INTRODUCTION
Formyl peptide receptors (FPRs) are G protein-coupled receptors (GPCR) that play an important role in leukocyte activation and chemotaxis [1]. These receptors were originally identified for their ability to bind and be stimulated by N-formyl peptides, which are produced by bacteria but can also be released from damaged mitochondria during tissue injury [2-4]. Phylogenetic analysis has revealed that FPRs belong to a family of chemosensory GPCR [5], and structurally related members of these receptors expressed in the vomeronasal organs of mammals appear to have an olfactory function associated with the identification of pathogenic states [6, 7]. It has been proposed that FPRs act as sensors of pathogen-derived products that recruit leukocytes to sites of infection, where these cells exert antibacterial effector functions and clear cell debris [8, 9]. Indeed, targeted disruption of the fpr1 gene coding for the mouse counterpart of human FPR1 renders mice more susceptible to bacterial infection without other significant phenotypic alterations [10], supporting the role of FPRs in innate host defense based on recognition of bacterial-derived agonists. However, the expression pattern of FPRs in nonphagocytic cells suggests that these receptors also participate in functions other than innate immunity and may represent unique targets for therapeutic drug design [9, 11-13]. For example, it was recently found that aging Fpr1 knockout mice develop spontaneous lens degeneration, suggesting FPR agonists may be useful in drug therapy for cataracts [14].
In humans, there are three FPR isoforms: FPR1 and FPR2 exhibiting 69% amino acid homology to each other, and FPR3 with 56% amino acid sequence identity to FPR1 and 83% to FPR2 [1]. These receptors are expressed on a variety of cell types, including neutrophils, macrophages, T lymphocytes, immature dendritic cells, epithelial cells, hepatocytes, fibroblasts, astrocytes, microvascular endothelial cells, and platelets [1, 15-17]. In mice, the FPR family consists of eight genes, including genes for mFpr1, mFpr2, and mFpr-rs1, which are expressed in leukocytes. Recently, mFpr1/mFpr2 was found to be expressed in dendritic and glial cells [8, 18-20]. Five distinct Fprs, including mFpr-rs1, mFpr-rs3, mFpr-rs4, mFpr-rs6, and mFpr-rs7, are expressed in mouse vomeronasal sensory neurons [5]. Mouse Fpr1 has a 76% sequence identity to human FPR1; however, its putative ligand-binding domains resemble those of human FPR2 rather than FPR1 [8, 21].
Being expressed in the majority of white blood cells, FPRs play an important role in the regulation of inflammatory reactions and cellular dysfunction [15, 22]. A variety of endogenous and pathogen-associated molecules can bind FPR1 and FPR2 with high affinity. Several studies have described intestinal proinflammatory properties for N-formyl peptides (e.g., fMLF), such as in colonic inflammation in vivo [23-25], suggesting a possible role in the etiology of inflammatory bowel disease in humans when bacterial chemotactic peptides breach mucosal defenses. Furthermore, involvement of fMLF in the pathogenesis of pouchitis, ulcerative colitis, and Crohn’s disease is also suggested [26-28]. Fehr et al. [29] reported that fMLF could substitute for the place of the second endotoxin injection in provoking the generalized Shwartzman reaction in rabbits. When injected into rabbits, fMLF causes dose-dependent transient hypotension, as well as neutropenia, thrombocytopenia, and a decrease in systemic vascular resistance [30, 31]. Moreover, fMLF causes a variable degree of bronchoconstriction after inhalation of this peptide by rabbits [32]. Similarly, leucopenia occurs rapidly after inhalation of fMLF by humans and could contribute to bronchial inflammation during bacterial infection [33].
FPR2 behaves as a “pattern recognition” receptor that can be activated by a wide variety of unrelated ligands. These include acute-phase serum amyloid A (SAA), annexin 1, urokinase plasminogen activator receptor (uPAR), the V3 region of HIV-1 envelope glycoprotein gp120, a 42-amino-acid form of β-amyloid, human prion peptide, cathelicidin LL-37, and other molecules (reviewed in [1, 11, 34, 35]). Some of these endogenous FPR2 agonists may play pathophysiological roles. For example, binding of SAA to FPR2 contributed to the destruction of bone and cartilage via the promotion of synoviocyte hyperplasia and angiogenesis in rheumatoid arthritis [36]. Recently, it was proposed that FPR2 could be involved in pathogenic mechanisms of atherogenesis via FPR2-dependent SAA-stimulated foam cell formation [37, 38]. Other examples of FPR involvement in multiple diseases have been recently reviewed [39]. These authors concluded that FPRs form a very attractive family of pharmacological targets and suggested that “the studies of specific agonists and antagonists for FPRs will be of great significance for better understanding of the roles of these receptors in physiology and pathophysiology [39].”
2. THERAPEUTIC EFFICACY OF FPR LIGANDS
Various approaches to activate or inhibit FPR-dependent pathways have been considered for therapeutic development. For example, antagonists that inhibit binding of FPR agonists decrease leukocyte chemotaxis and can potentially diminish release of pro-inflammatory molecules. In addition, some of therapeutic effects of FPR agonists may be related to homo- and/or cross-desensitization mechanisms. Finally, an integral role of FPR agonists in promoting the resolution of inflammatory reactions has been demonstrated [40, 41].
There is evidence that bioactive ligands acting as FPR agonists or antagonists might serve as useful therapeutics in host defense and as immunomodulatory activators to enhance selective innate immune responses in order to reduce detrimental effects associated with inflammation, infectious diseases, and cancer [42-45]. Various endogenous and synthetic peptide FPR agonists have been used to study the potential therapeutic efficacy in models of pathological processes. For example, FPR2 agonist, annexin 1 fragment 2-26 (Anxa12–26), reduced the lipopolysaccharide (LPS)-induced cerebrovascular inflammatory response in a murine model [46], significantly enhanced gastric ulcer healing [47], and prevented reperfusion-induced myocardial dysfunction in rats [48]. In addition, both Anxa12–26 and fMLF induced anti-nociceptive effects in mice [49]. The FPR2 agonist CGEN-855A (21-amino acid peptide) displayed anti-inflammatory activity in a mouse air pouch model and provided protection against ischemia-reperfusion-mediated injury to the myocardium in both murine and rat models [50]. It has also been suggested that FPR agonists could be utilized as anti-HIV therapeutics/immunoregulators to mobilize phagocytic leukocytes in HIV/AIDS patients [51, 52]. One of most potent mixed-type FPR agonists, a D-methionine-containing hexapeptide WKYMVm, potently inhibited HIV-1 Env-mediated fusion and viral infection through heterologous desensitization of C-X-C chemokine receptor 4 (CXCR4) and C-C chemokine receptor 5 (CCR5), suggesting a novel approach to the development of anti-HIV-1 therapeutics [53]. WKYMVm also increased neutrophil bactericidal activity in chemotherapy-treated cancer patients [54], protected the host by enhancing bactericidal activity and inhibiting vital organ inflammation and apoptosis in a sepsis mouse model [55], and enhanced endogenous tumor necrosis factor (TNF)-related apoptosis inducing ligand (TRAIL) gene expression with tumoricidal activity [56]. Furthermore, airway activation of FPRs with WKYMVm effectively prevented the development of allergic inflammation induced by inhalation of LPS-containing allergens. This effect was mediated by the inhibition of dendritic cell infiltration, maturation, and migration, as well as inhibition of both Th1 and Th17 polarization [57]. Based on localization of FPRs in leukocytes and in the central nervous system, these receptors have also emerged as promising therapeutic targets for the treatment of neurodegenerative diseases [58]. In addition, peptide agonists of FPR1 and FPR2 (i.e.,fMLF and MMK-1, respectively) prevented alopecia in neonatal rats induced by the anticancer agent etoposide [59].
The most potent and receptor specific FPR1 antagonists described so far are the fungal hydrophobic cyclic peptides, cyclosporines A and H [60]. Although cyclosporine H attenuated the acute inflammatory response evoked by cigarette smoke [61] and blocked fMLF-induced analgesia [62], in vivo studies of cyclosporines should be interpreted carefully because their main therapeutic effects appear to involve signaling pathways unrelated to FPR1 [63]. Indeed, cyclosporin A, a relatively large molecule (1.2 kDa), inhibited the T-cell receptor signal transduction pathway via the formation of a cyclosporin A-cyclophilin complex, which in turn bound to and inhibited the Ca2+-calmodulin dependent phosphatase calcineurin [64]. Cyclosporin A also inhibited formation and opening of the mitochondrial permeability transition pore [65]. Although cyclosporine H does not bind to immunophilin, this peptide is a potent inhibitor of the Ca2+-calmodulin-dependent phosphorylation of elongation factor 2 (EF-2) [66].
Other known peptide FPR antagonists are Boc-MLF (also termed Boc-1), and Boc-FLFLFL (also termed Boc-2), and there are several reports of in vivo application of Boc-2 [46, 48, 67, 68]. For example, Anxa12–26-induced cardioprotection was abolished by Boc-2 [48]. Similarly, Boc-2 prevented the inhibitory effect of Anxa12–26 on neutrophil extravasation [68]. Recently, tryptophan containing dipeptide derivatives of Boc-2 were reported as FPR1 antagonists [69].
The number of novel synthetic peptide FPR ligands continues to increase, and there are a couple of excellent reviews in the past few years summarizing these molecules [11, 43, 58, 70-73]. However, peptides are difficult to make and administer as therapeutic agents, making small-molecule chemical compounds a better choice for future clinical development. Growing evidence supporting the anti-inflammatory and tissue-protective effects of FPR agonists and antagonists prompted us and other research groups to search for novel small-molecule ligands for these receptors. As result of these efforts, a great number of synthetic non-peptide FPR agonists and antagonists with a wide range of chemical diversity were found using different high-throughput platforms in screening of commercial libraries and/or by structure–activity relationship (SAR)-directed design and synthesis (e.g., [74-87]). These have great potential for development of clinically useful anti-inflammatory and immunomodulatory drugs [85]. The availability of structurally defined non-peptide small-molecule FPR ligands is clearly of substantial benefit in drug development and facilitating SAR analysis to model ligand binding features, which is somewhat difficult with peptide ligands.
In this review, we provide an overview of the current literature, perspectives regarding therapeutic potential of various FPR ligands, details on the discovery and design of novel small-molecule non-peptide FPR agonists and antagonists. Because there have been ongoing efforts in several laboratories to study “FPR-ligand” interaction, questions regarding molecular modeling of FPR recognition are also discussed.
3. SMALL-MOLECULE FPR AGONISTS AND THEIR MODELING
Although using the chemical structure of endogenous ligands is one strategy for design of synthetic GPCR agonists and antagonists, this strategy has not been exploited in the development of non-peptide FPR ligands. Indeed, only lipoxin A4 (LXA4) and bile acids have been reported to date as endogenous non-peptide FPR ligands [88, 89]. Although LXA4 has been described as an anti-inflammatory mediator that exerts its effects through FPR2, the LXA4 target has been a matter of debate, as LXA4 analogues have been reported to be unable to signal through FPR2 [90-93]. In any case, most primary strategies for identifying new FPR agonists have been high-throughput screening (HTS), SAR analysis, and computer-aid drug design.
3.1. Benzimidazole Derivatives
HTS of a chemical library of synthetic small-molecule compounds identified two FPR1-specific agonists 1 and 2, which have a common 2-(benzimidazol-2-ylsulfanyl)-N-phenyl-acetamide scaffold [79]. Both compounds activated chemotaxis in human neutrophils, and SAR analysis of 52 related derivatives revealed 17 additional FPR1-specific agonists and 16 mixed FPR1/FPR2 agonists [79, 82]. Selected FPR agonists 1-9 with the benzimidazole scaffold are shown in (Table 1). It should be noted that all active derivatives contained either a 5-methoxy or 5-ethoxy group in the benzene moiety of the benzimidazole heterocycle, which was an essential feature for activity. Modification of benzene ring A also had effects on activity and receptor specificity, although a wider range of modifications was tolerated in this ring, including such substituents in the para position as nitro (1), ethoxy (2), methoxy (3 and 4), chlorine (5), bromine (6), methylthio (7), and methylcarboxyl (8) (Table 1). However, if the alkoxy chain on benzene ring A in 3 was elongated to four carbons, activity was lost. Introduction of an additional chlorine in compound 5 at the meta position resulted in complete loss of activity. Likewise, substitution of the para methoxy group in compounds 3 and 4 with trifluoromethoxy led to partial or complete loss of FPR1 agonist activity. All tested derivatives where benzene ring A contained fused 1,3-dioxolane or 1,4-dioxane rings were FPR agonists, although only compound 9 was specific for FPR1, and the other compounds with this feature were mixed FPR1/FPR2 agonists [82].
Table 1.
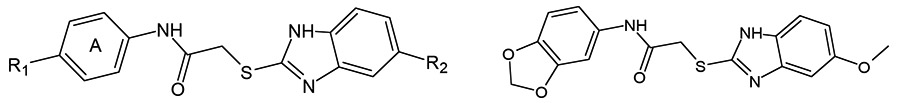 | |||||
|---|---|---|---|---|---|
| # | R1 | R2 | EC50 (μM)a | ||
| FPR1 | FPR2 | PMN | |||
| 1 | NO2 | OCH3 | 3.4 | N.A. | 0.4 |
| 2 | OCH2CH3 | OCH3 | 21.5 | N.A. | 5.8 |
| 3 | OCH3 | OCH2CH3 | 19.1 | N.A. | 0.6 |
| 4 | OCH3 | OCH3 | 1.7 | 5.1 | 0.3 |
| 5 | Cl | OCH3 | 1.6 | 6.0 | 0.7 |
| 6 | Br | OCH3 | 3.3 | N.A. | 1.8 |
| 7 | SCH3 | OCH3 | 2.0 | N.A. | 0.5 |
| 8 | COOCH3 | OCH3 | 1.5 | N.A. | 0.9 |
| 9 | 2.2 | N.A. | 2.6 | ||
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected HL-60 cells or in human polymorphonuclear neutrophils (PMN).N.A., no activity observed
3.1.1. Molecular Docking of Benzimidazole Derivatives
In the absence of an experimentally determined structure of FPR1, homology modeling can provide a rational alternative to a reasonable 3D structure. The FPR1 homology model was created using the crystal structure of bovine rhodopsin, which has a sequence identity of 20% for 348 aligned residues that correspond to the seven transmembrane domains [82, 94]. The position of the ligand binding site in the homology model, based on cross-linking and mutagenesis studies, was found to be located in the upper region of a bundle comprising transmembrane helices 2, 5, 6, and 7 [95, 96] and was confirmed by docking studies with tetrapeptide Ac-QAWF, which is the shortest core structure of the annexin AI-derived peptide [94]. Since pre-docking studies indicated that this region is coincident with the ligand binding site, it was proposed that this feature is relevant to the modeling process [97] and justified use of the rhodopsin-based model as a template for the docking of novel small-molecule FPR1 agonists vs. the other known GPCR crystal structures currently available.
Key sub-areas of the FPR1 binding site include two channels (A and C), two cavities (B and E), and the bottom (D) [82] (see arrows in Fig. (1A). Docking studies [82] showed that, most active FPR1-specific agonists 7 and 8 both had very similar benzimidazole orientations with their methoxy substituents H-bonded to Thr199 in channel A of the FPR1 binding site (Fig. 1B). Compound 8 forms a H-bond of 1.5 kcal/mol with a key residue (Arg205), and this interaction was weaker than the H-bond formed between Arg205 and fMLF. Thirty seven of 43 related alkoxy-substituted benzimidazoles had methoxy or ethoxy oxygen atoms embedded in channel A of the FPR1 binding site or in cavity B, which is located behind the hydrophobic ledge formed by the isobutyl group of Leu198. Thirty-four of these benzimidazole compounds were active FPR1 agonists, demonstrating the importance of the ligand–receptor interactions in regions A and B for FPR1 agonist activity. As examples, positions of alkoxy groups in the binding site are shown in Fig. 1A for FPR1 agonists 1, 7, and 8.
Fig. (1).

Panel A. Poses of agonists 1, 7, and 8 relative to the surface of the FPR1 binding site. Key sub-areas of the FPR1 binding site are indicated with arrows and include two channels (A and C), two cavities (B and E), and the bottom (D), as described previously [82]. Surface coloring was made according to electrostatic properties, whereby negatively and positively charged areas are shown in red and blue, respectively. Panel B. Docking poses of benzimidazoles 7 (purple) and 8 (green) in FPR1. H-bonds are indicated with dashed lines.
3.2. Pyrazolones
Using a Ca2+ flux-based assay in Gα15 and FPR1/FPR2 co-transfected CHO cells for HTS of a small-molecule chemical library, researchers from Amgen identified pyrazolone 10 as a potent FPR2 agonist [98]. Replacement of the iodine atom in position 4 with other halogens resulted in decreased potency, which correlated with the size of the halo-substituent (compounds 11 and 12) (Table 2). However, the analog with an unsubstituted phenyl urea moiety was completely inactive. The 4-methyl and 4-ethyl derivatives were active at FPR2. In contrast, the sterically more demanding 4-isopropyl analog lost activity. A more detailed survey of substituent changes at the phenyl urea moiety demonstrated that the steric and electronic nature of the para-substituent on the phenyl group had important effects on FPR2 agonist activity [98].
Table 2.
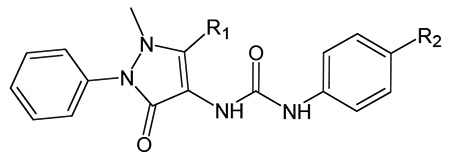 |
||||
|---|---|---|---|---|
| # | R1 | R2 | EC50 (μM)a | |
| FPR1 | FPR2 | |||
| 10 | CH3 | I | N.A. | 0.03 |
| 11 | CH3 | Br | N.A. | 0.09 |
| 12 | CH3 | Cl | N.A. | 0.49 |
| 13 | i-Pr | Cl | N.A. (0.065)b |
0.044 (0.022)b |
| 14 | t-Bu | Cl | N.A. | 0.023 |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected CHO cells;
EC50 values reported [100]. N.A., no activity observed.
Modification of substituents at C(5) of pyrazolone analogs revealed that the potency of compounds with bulkier substituents was consistently enhanced (Me<Et<i-Pr<t-Bu). As examples, structures and activities of compounds 13 and 14 with i-Pr and t-Bu C(5)-substituents are shown in (Table 2). Agonist 13 (designated in most of publications as “Compound 43”) was rediscovered in independent HTS as a mixed FPR1/FPR2 agonist with EC50 ~4 nM at FPR2 [81]. This compound also induced a respiratory burst in mouse and human neutrophils and activated C3 receptor (CR3) mobilization to the surface of human neutrophils [81, 91]. A substitution of the pyrazolone core at N(2) position showed that modifications within this portion of the molecule were relatively well tolerated. Indeed, the pyrimidines and quinolines were roughly equal or more potent than the corresponding phenyl derivatives, while thiazoles showed somewhat reduced activity [98].
Although all 28 pyrazolone-derived FPR2 agonists were reported to have no FPR1 agonist activity up to a concentration of 10 μM [98], Sogawa et al. [99, 100] and Forsman et al. [91] demonstrated that pyrazolone 13 has dual FPR agonist activity in HL-60 and CHO-K1 cells co-transfected with Gα16 and FPR1/FPR2 and suggested that this discrepancy might be due to the different types of G-proteins used in the cell-based screening assays (Gα16 vs. Gα15) [99]. Indeed, compound 13 significantly inhibited the binding of [3H]fMLF and [125I]WKYMVM to FPR1 and FPR2, respectively [100]. Moreover, the FPR1-specific antagonist cyclosporine H completely abolished the neutrophil response to compound 13 [91]. Compound 13 had agonist activity for murine Fpr1 and Fpr2, with EC50 values of 0.35 and 0.89 μM, respectively [100]. Using RBL-2H3 cells expressing mFpr1, mFpr2, and mFpr-rs1, He et al. [21] demonstrated that compound 13 induced Ca2+ mobilization and degranulation via both mFpr1 and mFpr2, but not mFpr-rs1. This agonist induced a rapid extracellular signal-regulated kinase (ERK) phosphorylation in murine peritoneal macrophages [101] and stimulated interleukin (IL)-6 secretion in 50% human blood [76]. Compound 13 also activated chemotactic responses in both human and mouse neutrophils, with a typical bell-shaped dose–response curve, and induced internalization of both FPR1 and FPR2 [81, 99, 100].
The most potent pyrazolone FPR2 agonists 10 and 13 inhibited human neutrophil chemotaxis induced by fMLF and IL-8, probably via homo- and cross-desensitization [98]. Incubation of human neutrophils with compound 13 diminished the expression of C-X-C chemokine receptor types 1/2 (CXCR1/2), complement receptor C5aR, and leukotriene B4 (LTB4) receptor BLT1 [100]. Similarly, pretreatment with compound 13 dose-dependently inhibited Ca2+ flux and the chemotactic response of murine neutrophils stimulated with mouse C5a, LTB4, or keratinocyte chemoattractant (KC/CXCL1), the murine homolog of human IL-8 [100, 102].
Lead pyrazolones also demonstrated anti-inflammatory activity in vivo. Pyrazolone 10 caused reduction of ear edema in mice treated topically or orally prior to induction of edema [98]. Compound 13, administered i.v. prior to IL-1β injection into 6-day-old air pouches, produced 50–75% inhibition of cell recruitment in wild-type mice but were ineffective in Fpr2−/− animals [101]. In addition, compound 13 inhibited neutrophil migration into the airway in LPS-exposed mice, as well as zymosan-induced neutrophil accumulation in the peritoneal cavity, possibly through cross-desensitization [100].
3.3. N-Substituted Benzimidazoles
In the next route of screening by researchers from Amgen, the N-substituted benzimidazole 15 was identified as an FPR2-specific agonist, and subsequent SAR studies identified 15 additional FPR2-specific agonists [76]. The authors found that benzimidazoles with small alkyl substituents, such as ethyl at C(2) (16), exhibited submicromolar FPR2 activity (Table 3), and further modification revealed that contraction of the piperidine ring in 16 to pyrrolidine R-enantiomer 17 resulted in a 20-fold increase in potency. It should be noted that R-enantiomer 17 was significantly more potent than its S-counterpart 18 (Table 3). A brief survey centered around the nature of the C(5)-substituent in the indole fragment revealed that OMe is optimal for FPR2 activity. Indeed, derivatives with F, Cl, Br, O-CF3, and Et substituents at C(5) of the indole ring were less active than OMe in compound 17. The most potent N-substituted benzimidazole 17 inhibited human neutrophil chemotaxis induced by fMLF and IL-8, probably via cross-desensitization. In addition, compound 17, in the presence of IL-1β, dose-dependently stimulated IL-6 secretion in 50% human blood above levels induced by IL-1β alone [76].
Table 3.
Agonist Activity of N-substituted Benzimidazoles 15-18 [76]
| # | Structure | EC50 (μM)a | |
|---|---|---|---|
| FPR1 | FPR2 | ||
| 15 | N.A. | 6.4 | |
| 16 | N.A. | 0.69 | |
| 17 | 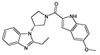 |
N.A. | 0.034 |
| 18 | 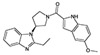 |
N.A. | 3.5 |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2- transfected CHO cells. N.A., no activity observed.
3.4. Pyridazin-3(2H)-ones
In the past six years, our laboratories have been involved in the SAR directed design, synthesis, biological evaluation, and molecular modeling of pyridazin-3(2H)-ones with FPR1/FPR2 agonist activities. These aromatic compounds were screened using Ca2+ flux assay in FPR-transfected human promyelocytic leukemia HL-60 cells. By starting with the known pyrazolone scaffold [98] for FPR agonists (Table 2), we first modified this scaffold but found that related pyridazine derivatives failed to exhibit FPR agonist activity [78]. We next focused on analogues with a substituted benzyl group at position 4 of the pyridazinone moiety and a similar functionalized chain at N-2. This approach led to the identification of compound 19 with medium activity at FPR1/FPR2 (Table 4). Thus this compound was selected as a lead, and extensive SAR studies were performed (Fig. (2).
Table 4.
| # | Structure | EC50 (μM)a | ||
|---|---|---|---|---|
| FPR1 | FPR2 | PMN | ||
| 19 | 3.4 | 3.8 | 2.6 | |
| 20 | 9.7 | 5.4 | 11.3 | |
| 21 | 2.3 | NA | 3.2 | |
| 22 | N.A. | 2.4 | 4.3 | |
| 23 | 6.6 | N.A. | 4.2 | |
| 24 | 9.3 | 2.8 | 4.9 | |
| 25 | 3.0 | 1.0 | 2.3 | |
| 26 | 9.3 | 2.8 | 2.4 | |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected HL-60 cells or in human polymorphonuclear neutrophils (PMN). N.A., no activity observed
Fig. (2).
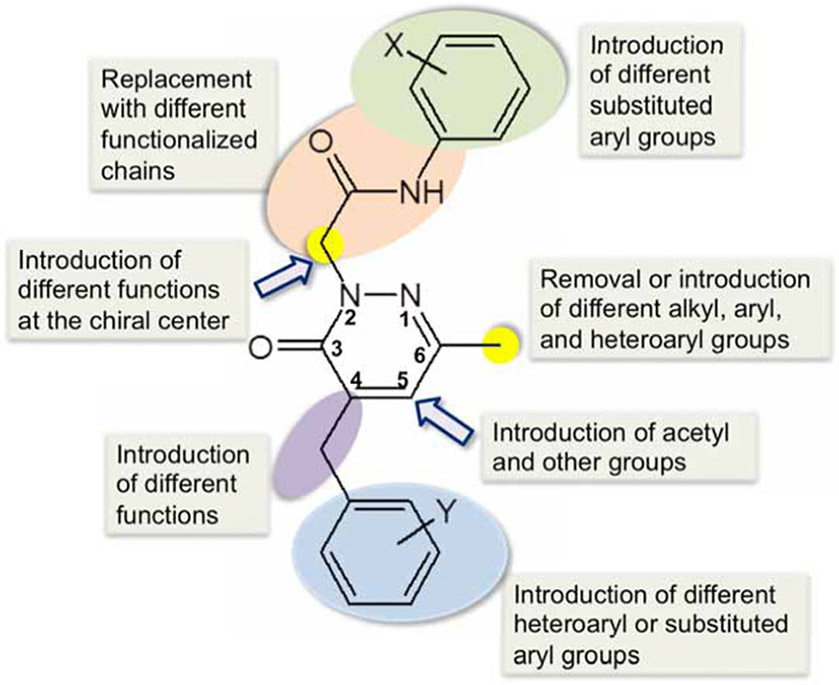
SAR directed design of FPR agonists with pyridazin-3(2H)-one scaffold.
Elongation of the hydrocarbon chain at the acetamide spacer (e.g., compound 20) reduced potency at FPR1/FPR2 (Table 4), and other modifications of the functionalized chain resulted in non-active compounds [78, 103]. However, introduction of different substituted aryl groups at the nitrogen atom of the acetamide fragment was more successful. Among compounds with a halogen-substituted phenyl ring, the 4-Cl and 4-I analogues exhibited the same profile as the 4-Br substituted compound (19), but the 4-F derivative was specific for FPR1. In contrast, moving Br to positions meta or ortho resulted in a complete loss of FPR1/FPR2 activity. The elimination or replacement of 4-Br with substituents having similar steric properties, such as t-Bu, OCF3, and CN also led to loss of FPR agonist activity. The 4-trifluoromethyl, the 4-nitro, 4-methyl, 4-methoxy, and 3,4-dimethoxy analogues had relatively low activity at FPR1/FPR2, whereas the 3,4-methylendioxy derivative 21 was specific for FPR1.
The methyl group at position C-6 of the pyridazinone ring was modified to introduce a cyclohexyl group, a 2-thienyl group, (substituted)aryl groups, and a benzyl group. None of these modifications led to more active FPR1/FPR2 agonist compared to reference compound 19 [104]. SAR analysis based on substituents at position C-4 of the pyridazinone scaffold was also performed [104]. Moving OCH3 from the meta to the para position resulted in compound 22, an FPR2-selective agonist. Substitution of OCH3 in compound 22 with SCH3 resulted loss of activity for all FPRs [103]. Introduction of fluorine in the meta position gave compound 23, which was an FPR1-specific agonist, and a similar selective activity was evident for its chloro-analogue. Replacement of the methoxyphenyl group in compound 19 with 5- and 6-membered heterocycles (e.g., compound 24), including 3-furyl, 3-thienyl, 2-thienyl, and 3-pyridyl groups resulted in mixed FPR1/FPR2 agonists. Introduction of a carbonyl group as spacer in place of CH2 in the benzyl group at position C-4 resulted in compound 25, which is a relatively potent mixed FPR1/FPR2 agonist. Substitution of CH2 with an amidic group was detrimental for FPR1, but not FPR2 activity (compound 26). In addition, compounds 19-26 also activated Ca2+ mobilization and chemotaxis in human neutrophils [78, 104].
Taken together, SAR analysis suggested that the position C-6 of the pyridazinone ring was not readily tolerant to modifications. In contrast, position C-4 was more amenable to chemical manipulation. Indeed, substituted benzyl groups and heterocycles, as well as functionalized spacers (e.g., CO, NH), can be productively introduced at this level, retaining good agonist activity. Regarding the substituted phenyl ring attached to the acetamide fragment, the presence of a lipophilic and/or electronegative substituent, such as F, Br, Cl or CH3, in the para position is an essential requirement for FPR1/FPR2 agonist activity. Likewise, the presence of an acetamide spacer at pyridazine N-2 also plays a critical role in specificity and potency. The optimal length of the spacer is four chemical bonds. The importance of both CO and NH in the spacer seems to indicate that a H-bond donor neighboring a H-bond acceptor system is also an essential requirement for binding at FPRs. This is consistent with the structures of other reported FPR1/FPR2 agonists, where the presence of an urea is a common structural element. Moreover, this H-bond donor/acceptor system must be placed at an appropriate distance from both the aromatic and the heterocyclic scaffold (see below).
3.4.1. Molecular Docking of Pyridazin-3(2H)-ones
Molecular docking of selected pyridazin-3(2H)-ones into the binding sites of FPR1 and FPR2 was performed in order to evaluate how the presence of different substituents and atoms influences the binding modes of the molecules [103, 104]. Molecular skeletons within the binding site of FPR1 were located mainly along cavity B and directed to channels A and C or to cavity E, in accordance with binding modes for 2-arylacetamide pyridazin-3(2H)-ones (Figs. 1A and 3A). Noticeably, bromine-substituted compounds, in their best poses, had bulky and highly hydrophobic P-bromophenyl groups located in the vicinity of channel A and directed outside the binding site (for example, see overlapped docking poses of fMLF and compound 23 in Fig. 3A). Additionally, H-bonding interactions of Thr265 with the pyridazine nitrogen atom are evident in agonists 19 and 25, as are interactions with the carbonyl group of the pyridazinone moiety in compound 24 (Fig. 3B). For compound 25, this carbonyl is also H-bonded to both Thr199 and Asn192. Compound 21, which contains a benzodioxolane moiety instead of a p-bromophenyl group, had the opposite binding mode, with the benzodioxolane protruding into channel C. In its docking pose, this compound formed a H-bond between one of its endocyclic oxygen atoms and the hydroxyl group of Tyr257 (Fig. 3C).
Fig. (3).
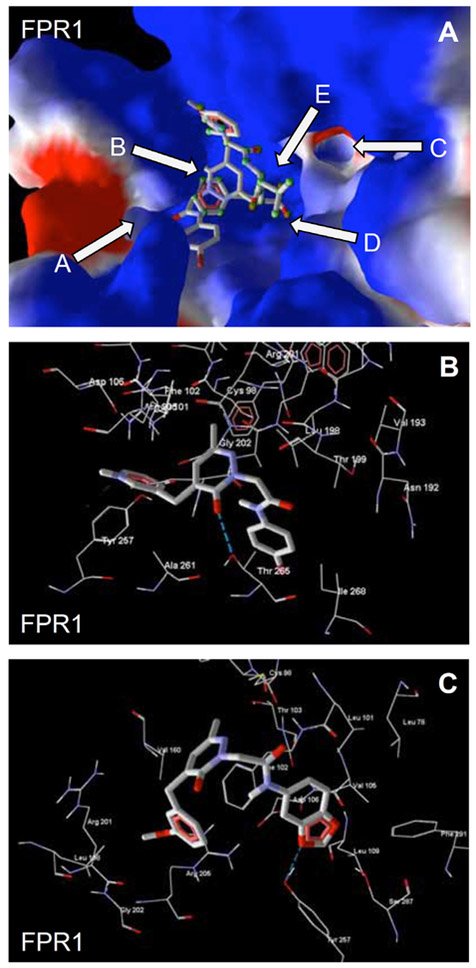
Panel A. Docking poses of fMLF (atoms are highlighted by green spheres) and pyridazin-3(2H)-one 23 (atoms are not highlighted). Key sub-areas of the FPR1 binding site are indicated with arrows as in (Fig. 1B). Panel B. Interactions between Thr265 of FPR1 and molecule 24. H-bond is indicated with a dashed line. Panel C. Specific interaction between Tyr257 of FPR1 and compound 21. H-bond is indicated with a dashed line.
The FPR2 model was constructed by homology modeling using the Phyre2 server. The rhodopsin-based model of FPR2 was selected as the most predictive structure from 18 other homology models based on 8 dissolved crystal structures of GPCRs [105]. Although the CXCR4 structure has a higher sequence identity with FPR2 (28%), this structure has a low resolution (3.2 Å) for the template, as compared to the rhodopsin structure (2.2 Å). The ligand binding site of FPR2 has a non-symmetric dumb-bell shape with two cavities different in size, where the smaller cavity lies deeply in the binding site and is surrounded by residues Val105, Asp106, Leu109, Phe110, Arg201, Trp254, and Gln258 [105]. FPR2-specific peptide agonist WKYMVM in its best docking pose occupies this cavity with an indole moiety near the N-terminus of the peptide. A narrow channel connecting the two cavities is bordered by residues Phe257, Val260, Ala261, Thr177, Phe178, and Phe180. The larger cavity of the docking site opens outside the receptor and has a complex shape.
Docking of FPR2 agonists 19, 22, and 24-26 to FPR2 showed that they adopt conformations that overlap well with the docking pose of WKYMVM. Characteristically, the bromo-substituted phenyl rings of these molecules lie within the smaller cavity found deep within the binding site (Fig. 4A). The narrow channel contains an amide fragment adjacent to the p-bromophenyl groups of the molecules. In addition, the pyridazinone moieties are oriented similarly in the binding site with their carbonyl and methyl groups overlaid in the same positions. Such an orientation is conditioned by the possibility of H-bonding of the nitrogen atom in the pyridazine ring, as well as an oxygen or nitrogen atom in the amide moiety, with the hydroxyl group of Thr177. As an example, specific interactions of agonist 25 with Thr177 and Ala261 are shown in (Fig. 4B).
Fig. (4).
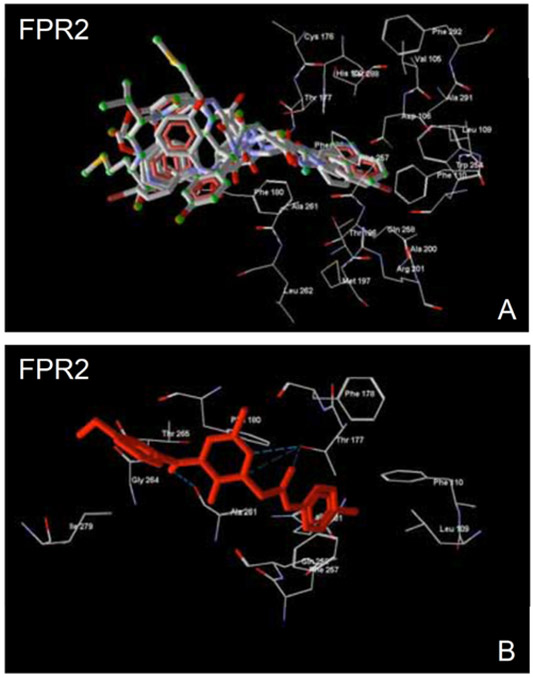
Panel A. Overlapping docking poses of WKYMVM (atoms are indicated with green spheres) and pyridazin-3(2H)-ones 19, 22, and 24-26. FPR2 residues within 6 Å around the smaller cavity are shown. Panel B. Compound 25 docked into the FPR2 binding site. Residues lying within 3.5 Å of the pose are shown. H-bonds are indicated with dashed lines.
As discussed above, Arg205 is one of the key residues important for FPR1 agonist activity [82, 96]. Indeed, active pyridazinone 19 forms a H-bond with Arg205 in FPR1, with total interaction energy of 32.9 kcal/mol with this residue. However, the sulfur-containing analogue with a thio-acetamide fragment had a much weaker non-bonded attractive interaction with Arg205 (8.82 kcal/mol). Another thio-analogue of compound 22 containing a p-methylthio group instead of the p-methoxy substituent was not H-bonded to any FPR1 residue [103]. Docking studies with the FPR2 homology model showed that the binding modes of oxygen-containing derivatives were also quite different from their thio-analogues [103]. The inactive sulfur-containing derivatives contain bulky groups that extend outside the cavity, which caused steric hindrance and prevented effective binding of the ligands with FPR2. Hence, these molecules cannot adopt the more suitable conformations that are possible for the corresponding oxygen-containing derivatives. For example, the best docking pose of the p-methylthio analogue of compound 22 had significant repulsive interactions with Tyr277 and Ile280 of FPR2. Despite H-bonding with Asn171 and Asp173, this docking pose was energetically unfavorable, as compared to that of the active methoxy-derivative 22. For the best pose of compound 22, the closest non-valent contact of 2.48 Å occurs between the methoxy oxygen atom in molecule 22 and Thr168. An analogous pose would be impossible for the corresponding compound with a p-methylthio group, which is more bulky than the methoxy substituent.
3.5. Chiral Pyridazines
Based on the pyridazine-3(2H)-one scaffold, two series of chiral derivatives were synthesized and evaluated [77, 106]. While we found that orientation of the chiral C5-methyl substituent in N-(4-bromophenyl)-2-[5-methyl-6-oxo-3-phenyl-5,6-dihydropyridazin-1(4H)-yl]acetamide and its analogues had little effect on agonist activity, orientation of the chiral center attached to nitrogen atom in position 2 of pyridazinone heterocycle had significant effects [77]. For these compounds, the S-(+) enantiomers lacked FPR agonist activity, whereas the R-(−) counterparts 27-29 had high activity in the micromolar or even nanomolar range (Table 5). In general, elongation of the carbon chain at the chiral center in R-enantiomers increased agonist activity (CH3<C2H5=n-C3H7<n-C4H9=C6H5) at FPR1/FPR2, suggesting the importance of considering this molecular feature in further development of these analogues.
Table 5.
Agonist Activity of Chiral Pyridazin-3(2H)-ones 27-29 [77]
 |
||||
|---|---|---|---|---|
| # | R | EC50 (μM)a | ||
| FPR1 | FPR2 | PMN | ||
| 27 | n-C3H7 | 3.0 | 0.84 | 0.93 |
| 28 | i-C3H7 | 9.4 | 5.4 | 2.3 |
| 29 | n-C4H9 | 0.5 | 0.089 | 0.073 |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2- transfected HL-60 cells or in human polymorphonuclear neutrophils (PMN).
3.6. N-Phenylurea Derivatives
HTS of a large chemolibrary resulted in the identification of FPR2-specific agonist 30 with an N′-phenylurea scaffold [75]. This compound activated reactive oxygen species (ROS) production by murine neutrophils and induced Ca2+ mobilization and chemotaxis in human neutrophils [79]. SAR analysis of related derivatives revealed three additional FPR2-specific agonists 31-33 (Table 6). All active derivatives contained a para methoxy group in benzene ring B, which seems to be an essential feature for activity of these derivatives. However, introduction of additional methoxy groups to ring B resulted in total loss of activity. Most active derivatives contained a halogen atom in the para position of benzene ring A. However, the presence of the halogen atom was not absolutely essential for biological activity, as compound 31 was also an active FPR2 agonist. Moving the halogen atom from the para position to the meta and then ortho positions resulted in decreased or completely lost activity, respectively.
Table 6.
 |
||||
|---|---|---|---|---|
| # | R | EC50 (μM)a | ||
| FPR1 | FPR2 | PMN | ||
| 30 | Cl | N.A. | 0.4 | 0.7 |
| 31 | H | N.A. | 3.0 | 4.8 |
| 32 | F | N.A. | 0.9 | 1.2 |
| 33 | Br | N.A. | 0.1 | 1.2 |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected HL-60 cells or in human polymorphonuclear neutrophils (PMN). N.A., no activity observed.
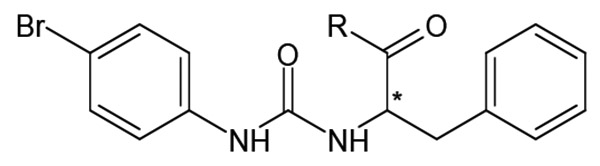
Branching of the hydrocarbon chain in N-phenyl-N’-(2-phenylethyl)urea derivatives resulted in a series of analogs with a chiral sp3 carbon near the carbamide nitrogen atom. The resulting benzyl-containing moiety is structurally related to the phenylalanine derivatives. Our screening demonstrated that nine Phe-based analogs and nine other related analogs were potent FPR agonists [80], and structures of selected compounds 34-47 are shown in (Tables 7 and 8). Although most of the compounds were mixed FPR1/FPR2 agonists, many displayed much higher selectivity for FPR2. It should be noted, that both enantiomers of racemic mixture 35 were reported previously in independent screenings as FPR2 agonists Acadia C5 and Acadia C6 [45], and their reported EC50 at this receptor was ~30-35 nM [81], which is pretty close to the racemic mixture (Table 7). The S-enantiomer has been shown to dose-dependently prevent hyperalgesia induced by carrageenan in rats [45].
Table 7.
Agonist Activity of Chiral N-phenylurea Derivatives 34-38 [80]
 |
|||||
|---|---|---|---|---|---|
| # | R | EC50 (μM)a | |||
| FPR1 | FPR2 | FPR3 | PMN | ||
| 34 | 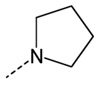 |
4.5 | 0.14 | 11.5 | 3.2 |
| 35 | 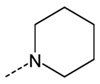 |
1.8 | 0.04 | 6.5 | 1.2 |
| 36 |  |
0.3 | 0.004 | 0.1 | 0.7 |
| 37 |  |
0.08 | 0.007 | 0.5 | 0.5 |
| 38 | 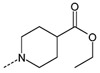 |
N.A. | 0.16 | N.A. | 4.4 |
EC50 values were determined by measurement of Ca2+ flux in FPR1-, FPR2-, and FPR3-transfected
HL-60 cells or in human polymorphonuclear neutrophils (PMN). N.A., no activity observed.
Table 8.
Agonist Activity of Benzodioxole Derivatives of N-phenylureas 39-47 [80]
 |
||||||
|---|---|---|---|---|---|---|
| # | R | EC50 (μM)a | ||||
| FPR1 | FPR2 | FPR3 | PMN | |||
| 39 | H | N.A. | 0.006 | 3.3 | 5.9 | |
| 40 | F | 1.7 | 0.004 | 0.7 | 0.7 | |
| 41 | Cl | 3.7 | 0.002 | 0.2 | 0.06 | |
| 42 | Br | 2.7 | 0.004 | 1.7 | 0.1 | |
| 43 | Me | 5.1 | 0.07 | 10.8 | 4.5 | |
| # | R1 | R2 | EC50 (μM)a | |||
| FPR1 | FPR2 | FPR3 | PMN | |||
| 44 | H | F | N.A. | 0.12 | 1.3 | 1.2 |
| 45 | F | F | 7.5 | 0.02 | 1.2 | 0.14 |
| 46 | Me | F | N.A. | 0.5 | N.A. | 10.1 |
| 47 | Cl | O-Me | 0.11 | 0.0002 | 0.05 | 0.013 |
EC50 values were determined by measurement of Ca2+ flux in FPR1-, FPR2-, and FPR3-transfected
HL-60 cells or in human polymorphonuclear neutrophils (PMN). N.A., no activity observed.
3.7. Chiral 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido] propanamides
Screening of a small library of 32 low-molecular-weight ligands of 24 different GPCRs led to the identification of two bombesin receptor BB1/BB2 antagonists, PD176252 (compound 48) and PD168368 (compound 49), as potent mixed FPR1/FPR2 agonists [80]. Although several potent FPR1/FPR2 agonists were found through secondary screening of Trp-based analogs from commercial chemical libraries, subsequent SAR analysis was completed by synthesis of related enantiomers using stereospecific methods starting from the enantiomerically pure N-Boc-R-Trp or N-Boc-S-Trp [105]. From this approach, several potent FPR agonists, including FPR2-specific agonists 50-53, were identified. Unexpectedly, the majority of compounds found to induce Ca2+ mobilization in FPR-transfected HL-60 cells failed to stimulate this response in human neutrophils. This finding is supported by our previous findings that some chiral FPR agonists and their closely related achiral derivatives are much (up to 600-fold) less active for activation of Ca2+ mobilization in human neutrophils than in the FPR-transfected cells [80]. Pretreatment of human neutrophils with probenecid restored the Ca2+ flux response in neutrophils treated with most of these derivatives. Structures of selected FPR agonists 48-53 are shown in (Table 9). Because substitution of OCH3, CF3, Br, and CH3 in the para position of the phenyl ring with CN or NO2 and removing CH3 at the chiral center of compounds 48 and 49 resulted in loss of ability to activate neutrophil Ca2+ flux without probenecid pretreatment, it appears that the requirement for probenecid is somehow related to compound structure. We suggest that compounds 48 and 49 have more flexible structures than the parent compounds, where an intramolecular H-bond between an amino group and the carbamide carbonyl atom could significantly restrict conformational freedom of these molecules. Thus, increased flexibility of the chiral agonists could be analogous to conformation changes in FPR2 [105]. Alternatively, because OCH3, CF3, Br, and CH3 in the para-position of the phenyl ring of FPR2 agonists are less electronegative than NO2 and CN groups [107], potency of electrostatic and H-bond interactions in FPR2 subpockets could be an important characteristic for activation of G-protein coupling and Ca2+ flux in human neutrophils.
Table 9.
Agonist Activity of Chiral 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamides 48-53 [105]
 | ||||
|---|---|---|---|---|
| # | EC50 (μM)a | |||
| FPR1 | FPR2 | PMN | ||
| w/o Prob. | with Prob. | |||
| 48 | 0.33 | 1.3 | 0.72 | 0.21 |
| 49 | 0.57 | 0.62 | 0.91 | 0.19 |
| 50 | N.A. | 0.023 | N.A. | 0.0032 |
| 51 | N.A. | 0.7 | N.A. | 0.05 |
| 52 | N.A. | 0.36 | N.A. | 0.022 |
| 53 | N.A. | 0.19 | N.A. | 0.46 |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected HL-60 cells or in human polymorphonuclear neutrophils (PMN) analyzed without (w/o) or in the presence of 2.5 mM probenecid (Prob.). N.A., no activity observed.
Analysis of the literature indicates that probenecid is a non-specific inhibitor of multidrug resistance-associated proteins and can have different effects on several other cellular targets. For example, probenecid is able to activate transient receptor potential V2 (TRPV2), Ca2+-permeable nonselective cation channel [108], and transient receptor potential channel subtype A member 1 (TRPA1), another nonselective cation channel [109], and it has recently been reported that probenecid can directly modulate interaction of GPCR and G-proteins [110]. In addition, Prossnitz et al. [111] proposed that primary myeloid cells maintain a subpopulation of FPR in a low-affinity, possibly G protein-free state, which is not a feature of FPR-transfected HL-60 cells. Because allosteric communication between the ligand-binding orthosteric site and the cytoplasmic G-protein-binding surface is a fundamental feature of GPCRs [112], it is possible that certain FPR2 agonists, such as the ureidopropanamides 50-53, could stabilize this receptor in a G-protein-free state, and additional agents (e.g., probenecid) may be required to reactivate G-protein coupling.
3.8. Molecular Modeling of Chiral Recognition at FPR2
To investigate the enantiomer preference observed for FPR2 agonists with different scaffolds and chiral center substituents, we created a pharmacophore model for FPR2 using field point methodology [113-115] based on five potent small-molecule non-peptide FPR2 agonists, including FPR agonist 42 and four chiral molecules (both S- and R- forms) 17, 35, 36, and 49 [80]. The hydrophobic field surface of the 5-molecule FPR2 template consists of three regions (H1, H2, and H3), which correlate with subpockets I, II, and III in the FPR2 binding site, respectively (Fig. 5). FPR2 agonists and their inactive enantiomers were overlaid in the three-subpocket model. The similarity values depend equally on geometric and field similarity of a molecule and template. For most chiral pyridazin-3(2H)-ones [77], the similarity between aligned molecule and the template for R-enantiomers was higher than that of their S-enantiomer counterparts. Moreover, more active enantiomers with a ureidopro-panamide scaffold had better alignments than the corresponding inactive enantiomers [105]. A visual inspection of the molecule overlays on the 5-molecule FPR2 template showed that in most cases the active enantiomers had alignment modes with nonpolar molecular fragments located in subpocket II, while bromo- or nitro-substituted phenyl rings were always positioned in subpocket I. In particular, moieties without polar atoms occupied subpocket II, while polar substructures (methoxy in benzene rings or indole NH groups) were located in subpocket III for these alignments [105].
Fig. (5).

Model of chiral compound docking to FPR2. Geometry of the hydrophobic field surface of the pharmacophore model matches to the binding site geometry of FPR2. An FPR2 agonist can approach the FPR2 binding site from the top (“mouth”) of the cavity, shown by dashed yellow line around the agonist template (hydrophobic regions H2 and H3) and around the cavity mouth, which includes subpockets II and III. Field points are colored as follows: blue, electron-rich (negative): red, electron-deficient (positive): yellow, van der Waals attractive (steric). Hydrophobic region H1 (usually associated with 4-nitrophenyl or 4-bromophenyl groups in FPR2 agonists) should properly fit into subpocket I of the FPR2 ligand-binding site. The cavity of the FPR2 binding site shows the position of side chain tails of compound 53 in subpockets II and III. Surface coloring was made according to electrostatic properties, whereby negatively and positively charged areas are shown in red and blue, respectively. It should be noted, that blue (positively charged) surface areas of the receptor correspond to blue field points obtained with positive probe atom and red (negatively charged) surface areas of the receptor correspond to red field points obtained with negative probe atom. Areas of subpockets are indicated with light-blue arrows. Numeration of subpockets and the hydrophobic surface of the FPR2 pharmacophore model are as reported in [80, 105].
According to docking studies with the FPR2 homology model [105], the pharmacophore subpocket I is surrounded by His102, Val105, Asp106, Leu109, Trp254, Phe257, Ser288, and Phe292 and lies in the smaller cavity. Residues His102, Val105, Asp106, Leu109, and Trp254 were also identified by Fujita et al. [116] as critical for the FPR2 binding site. Residues surrounding subpockets II and III were also identified [105]. The FPR2-specific peptide agonist WKYMVM, in its best docking pose, occupies all three subpockets, with the N-terminal indole moiety located in subpocket I. Subsequent modeling with FPR agonists showed that the geometric configuration of the FPR2 binding site fits well with the shape of the hydrophobic field obtained for the FPR2 agonist pharmacophore model, and extrema of negative and positive fields correspond well to areas of the ligand binding site of the FPR2 homology model (Fig. 5). Thus, this correspondence seems to be important for proper orientation of an agonist molecule for penetration into the binding site.
3.9. 2-(N-Piperazinyl)acetamide Derivatives
Eight FPR2-specific agonists 54-61 with a 2-(N-piperazinyl)acetamide scaffold were discovered while screening a chemical library using the Ca2+ flux assay in FPR1/FPR2 RBL cells as readout (Table 10). All active derivatives contained Br in the para position of benzene ring A, which was required for activity. Furthermore, moving Br from the para position to the ortho or meta positions resulted in loss of activity. Finally, replacement of para Br in ring A with a variety of other substituents resulted in loss of activity [79]. It is noteworthy that related piperazines were reported as selective agonists of the dopamine D4 receptor, which is also a GPCR [117]. Whether our FPR2 agonists are also D4 receptor agonists has not been evaluated.
Table 10.
Agonist Activity of 2-(N-piperazinyl)acetamides 54-61 [79]
 | |||||||
|---|---|---|---|---|---|---|---|
| # | R1 | R2 | R3 | R4 | EC50 (μM)a | ||
| FPR1 | FPR2 | PMN | |||||
| 54 | H | H | F | H | N.A. | 19.3 | 10.5 |
| 55 | H | Cl | H | H | N.A. | 15.1 | 16.4 |
| 56 | H | H | H | H | N.A. | 8.7 | 2.3 |
| 57 | H | H | Cl | H | N.A. | 2.1 | 2.0 |
| 58 | H | Cl | CH3 | H | N.A. | 4.3 | 1.0 |
| 59 | CH3 | Cl | H | H | N.A. | 4.1 | 1.8 |
| 60 | CH3 | H | H | Cl | N.A. | 9.4 | 0.4 |
| 61 | H | OCH3 | H | H | N.A. | 11.0 | 7.9 |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected RBL cells or in human polymorphonuclear neutrophils (PMN). N.A., no activity observed.
3.10. Quinazolinones
Quinazolinone 62 (designated in most of publications as Quin-C1) was described as one of first small-molecule non-peptide FPR agonists [74]. This compound selectively activated FPR2 vs. FPR1 (EC50 ~1.4 μM), induced ROS production, degranulation, and chemotaxis in human neutrophils and stimulated internalization of FPR2 and ERK phosphorylation [74, 81]. Compound 62 activated both mFpr1/mFpr2 [21] and demonstrated anti-inflammatory properties in a mouse model of lung injury [118]. Furthermore, HTS and SAR-directed design and synthesis identified analogs 63-66 [79, 119], including FPR2-specific agonists containing methyl (63), nitro (64), and 3,4-methylenedioxy (66) groups in the para-position of benzene ring A (Table 11). Substitution with bulky groups, such as isobutoxy or butoxy, at this position resulted in loss of agonist activity, and activity was also lost when the nitro group in compound 64 was changed to amino or dimethylamino groups. Elimination of the methoxy group in compound 62 or introduction of an additional methoxy group in the meta position of 62 also resulted in inactive compounds [119]. Our recent screening identified related quinazolinone 65 as a potent FPR agonist [79].
Table 11.
 | |||||
|---|---|---|---|---|---|
| # | R1 | R2 | EC50 (μM) | ||
| FPR1 | FPR2 | PMN | |||
| 62 | OCH3 | butoxy | N.A. | 1.4 | N.D. |
| 63 | CH3 | butoxy | N.D. | ≥1.4 | N.D. |
| 64 | NO2 | butoxy | N.D. | >1.4 | N.D. |
| 65 | OCH3 | NO2 | N.A. | 0.3 | 1.4 |
| 66 | N.D. | >1.4 | N.D. | ||
N.D., EC50was not reported. N.A., no activity observed.
3.11. Miscellaneous FPR Agonists
Structures of FPR agonists with varying scaffolds but with no clear SAR emerging from evaluation of their analogs are discussed here (Table 12). Five acetohydrazides, including chiral compound 67, dibenzoylhydrazine 68, and thienylpentanamide 69 were identified as FPR2-specific agonists in library screening [79]. Derivatives of 2-(indol-3-yl)-1,3-thiazolidin-4-one 70, N,N′-diphenylurea 71, and (3-thienyl)methylene acetylhydrazide 72 were found to be mixed FPR1/FPR2 agonists, and compounds 68-72 activated chemotaxis in human neutrophils [79]. Two other phenyl-substituted ureas 73 and 74 have been reported as potent FPR2-specific agonists [81]. Compounds 75 and 76 [81] are unique from other described FPR agonists because these compounds have carboxyl/hydroxyl groups. It should be noted that compounds 75 and 76 activated intracellular Ca2+ mobilization in CHO cells expressing FPR1 as well as FPR2, but the EC50 for FPR1 agonist activity was not reported [81]. Compound 77, with a unique 2-(4-phenyl-5-((phenylamino) methyl)-1,2,4-triazol-3-ylthio)acetamide scaffold, and its closely related derivatives were patented as the first small-molecule non-peptide FPR2 agonists, but their activities (EC50 values) at this receptor were not reported [45]. Compound 77 was further rediscovered in independent screening as a potent FPR2-specific agonist with nanomolar activity [81]. Compounds 78 [81] and 79 [120] each have identical groups at the sp3 carbon atom, resembling structures of other FPR agonists 39-43 [80]. It should be noted, that the low activity FPR1 agonist 79 was found using computer-aided prioritization and subsequent physical screening [120].
Table 12.
Agonist Activity of Miscellaneous Derivatives 67-84[45, 75, 79, 81,116,121,123,124]
 | |||||||
|---|---|---|---|---|---|---|---|
| # | EC50 (μM) | # | EC50 (μM)a | ||||
| FPR1 | FPR2 | PMN | FPR1 | FPR2 | PMN | ||
| 67 | N.A. | 3.9 | 1.1 | 77 | N.D. | 0.045 | N.D. |
| 68 | N.A. | 12.6 | 7.0 | 78 | N.D. | 0.2 | N.D. |
| 69 | N.A. | 9.7 | 28.8 | 79 | ~20.0 | N.D. | N.D. |
| 70 | N.A. | 10.8 | 8.2 | 80 | 6.6 | N.A. | 0.63 |
| 71 | 18.6 | 11.1 | 1.9 | 81 | N.A. | 19.1 | 0.3 |
| 72 | 19.7 | 15.0 | 23.6 | 82 | N.A. | 18.3 | 0.6 |
| 73 | N.D.b | 0.04 | N.D. | 83 | 0.13 | N.A. | N.D. |
| 74 | N.D. | 0.1 | N.D. | 84 | N.A. | 0.14 | N.D. |
| 75 | N.D. | 0.006 | N.D. | 85 | N.A. | ~1.0 | N.D. |
| 76 | N.D. | 0.006 | N.D. | 86 | N.A. | N.D. | N.D. |
EC50 values were determined by measurement of Ca2+ flux in FPR1- and FPR2-transfected RBL cells (compounds 67-72 and 80-82), CHO cells (compounds 73-78), FPR1-transfected U937 cells (compound 79), or HEK-293 cells (compounds 83-84).
N.D., no data or EC50 was not reported. N.A., no activity observed.
Compounds 80-82 containing an arylcarboxylic acid hydrazide core structure potently induced TNF production in macrophages, presumably via FPR activation [121, 122]. Compound 80 is an FPR1-specific agonist, whereas compounds 81 and 82 are low activity FPR2-specific agonists (Table 12). Compounds 80-82 stimulated intracellular Ca2+ mobilization, chemotaxis, and ROS production in murine and human neutrophils [75, 121]. Currently, compound 80 is sold by Tocris Bioscience as an FPR1 agonist designated as FPRA14.
Recently, polyphenylure 83 and pyrrolidine bis-diketopiperazine 84 were found to be specific high-affinity FPR1 and FPR2 agonists, respectively, using mixture-based combinatorial libraries [123]. However, activity of these potent agonists in neutrophils was not evaluated.
Two non-peptide calpain inhibitors that are α-mercaptoacrylic acid derivatives, PD150606 (compound 85) and PD151746 (compound 86), were reported to induce Ca2+ mobilization in FPR2-transfected HEK-293 cells [116, 124]. These compounds have atypical chemical structures, as compared with most reported FPR agonists. Indeed, a visual inspection of molecules 1-84 shows that a typical FPR agonist contains at least two heterocycles separated by a chemical linker with >2 bonds (see also [79]). We evaluated ability of the calpain inhibitors 85 and 86 to stimulate Ca2+ mobilization in FPR1/FPR2 transfected HL-60 cells, but both compounds were inactive in our hands at concentrations up to 30 μM (unpublished data). The explanation for this discrepancy might be due to the different types of G-proteins used in the cell-based screening assays. Although docking simulations demonstrated several low-energy poses of compound 85 in the orthosteric binding site of FPR2 [116], pepducin-like allosteric interaction of these calpain inhibitors with allosteric sites, including the FPR2-G-protein interface, or even direct effects on G-proteins could not be excluded.
Two methionine-derived benzimidazoles were recently identified as neutrophil agonists in a Ca2+ mobilization assay by Unitt et al. [84] during HTS and subsequent SAR analysis [84]. However, FPR subtype specificity for these compounds was not reported.
4. SMALL-MOLECULE FPR ANTAGONISTS AND THEIR MODELING
4.1. Diamides
Using HTS for compound inhibition of Alexa647-labeled fMLFK-specific binding to HEL-293 cells expressing FPR1 and Gα16 and subsequent analysis for inhibition of fMLF-stimulated intracellular Ca2+ mobilization in human neutrophils, Unitt et al. [84] identified diamide 87, which antagonized FPR1 function in human neutrophils (Table 13). The most potent diamide analog identified (compound 88) possessed a more lipophilic substitent R, as compared to compounds 89-92, which contain 4-chlorophenyl, benzyl, cyclohexyl, and 1-methylimidazolyl substituents, respectively. For the benzoyl moiety, an ortho hydroxy group was essential to maintain FPR1 antagonist activity, and replacing the hydroxyl in compound 89 with OCH3 or CH3 led to inactive compounds. In addition, the nature of the antagonism exhibited by these diamides (i.e., competitive vs. non-competitive) was not evaluated. Initial SAR for the diamides and several of their analogs suggested that in order to generate a good FPR1 antagonist profile, compounds would require a number of parameters that would be challenging to successfully modify in latter phases of drug development. As result, further work, including testing enantiomers, was not undertaken [84].
Table 13.
FPR1 Antagonist Activity of Diamides 87-92 [84]
| # | R | IC50 (μM)a |
|---|---|---|
| 87 |  |
0.3 |
| 88 |  |
0.08 |
| 89 | 0.8 | |
| 90 |  |
1.3 |
| 91 |  |
0.8 |
| 92 |  |
1.6 |
Inhibition of fMLF-stimulated intracellular Ca2+ flux in human neutrophils; IC50 values were recalculated from reported pIC50 values.
4.2. Methionine-derived Benzimidazoles
Another FPR1 antagonist discovered by Unitt et al. [84] was the methionine-derived benzimidazole 93 (Table 14). Compound 93 demonstrated low antagonism at rat Fpr1 (IC50 = 10 μM), but was inactive at mouse Fpr1. Modification of the benzofuran group in 93, including removal of the ethoxy group (compound 94) or fused benzene ring (compound 95), reduced activity. Compound 96, with a cyclohexyl moiety, was more active in comparison with compound 97, which contained a phenyl substituent. Derivative 98 with a 3-chloro-2-thienyl group was very close in activity to cyclohexyl-containing compound 96. Modification of the benzimidazole group by introduction of a benzyl group reduced activity (compound 99), although subsequent introduction of a cyano group in the para position of the benzyl moiety increased antagonist activity (compound 100). Incorporation of heteroatoms into the phenyl ring of the benzimidazole had variable effects, depending on the substitution pattern. Purine (compound 101) and 4-azabenzimidazole (compound 102) derivatives were less active than reference compound 93. Moreover, substitution of the benzimidazole group in compound 93 with 5-azabenzimidazole led to the appearance of FPR agonist activity, although FPR subtype specificity for this compound was not reported. S-enantiomer 103 with an ethylthiomethyl group was equiactive to compound 93, although its R-counterpart 104 had a lower antagonistic activity.
Table 14.
FPR1 Antagonist Activity of Methionine-derived Benzimidazoles 93-104 [84]
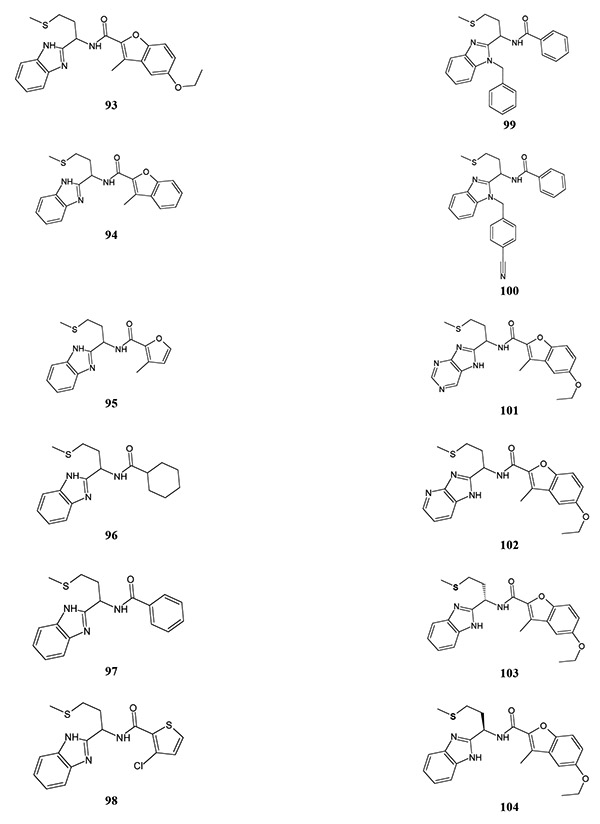 | |||
|---|---|---|---|
| # | IC50(μM)a | # | IC50 (μM) |
| 93 | 0.5 | 99 | 2.5 |
| 94 | 4.0 | 100 | 0.32 |
| 95 | 4.0 | 101 | 5.0 |
| 96 | 2.5 | 102 | 3.2 |
| 97 | 5.0 | 103 | 0.63 |
| 98 | 2.0 | 104 | <25 |
Inhibition of fMLF-stimulated intracellular Ca2+ flux in human neutrophils; IC50 values were recalculated from reported pIC50 values.
4.3. Pyrazole-4-carboxamides
A series of pyrazole-based FPR1 antagonists have been identified from HTS and subsequent optimization [86, 87] (Table 15). Compound 105 was first identified by HTS [87], although attempts to obtain more potent antagonists by modification or replacement of the sulphonamide and amide motif were unsuccessful, suggesting that the amidic group is necessary for FPR1 agonist activity. However, conformational effects of these changes cannot be excluded. Introduction of a methyl substituent at the 3 position of the pyrazole scaffold resulted in a modest increase in potency (compound 105 vs. 106), and modification of the carboxamide moiety led to S-enantiomer 107, which was preferentially active over the R-counterpart 108 by ~6-fold. Introduction of strongly electron-withdrawing fluorine and cyano groups into both phenyl rings of the S-enantiomer led to compound 109, which possessed a substantially higher antagonist activity than the initial pyrazole 107. During subsequent optimization, the alkyl pyrazole substituent was varied in compound 107, with the most potent analogs 110-112 bearing cyclohexyl, tetrahydrothiopyran-4-yl, and 4-methoxycyclohexyl groups, respectively [86]. It is interesting to note that the trans-diastereoisomer of compound 112 was completely inactive, highlighting specificity of the interaction of the methoxy motif with FPR1. Compound 112 has good pharmacokinetic properties and is the most potent FPR1 antagonist among the known synthetic small-molecule FPR antagonists. Note, however, that the nature of the antagonist activity (i.e., competitive vs. non-competitive) was not evaluated for these methionine-derived benzimidazoles and pyrazole-4-carboxamides, so allosteric inhibition cannot be excluded.
Table 15.
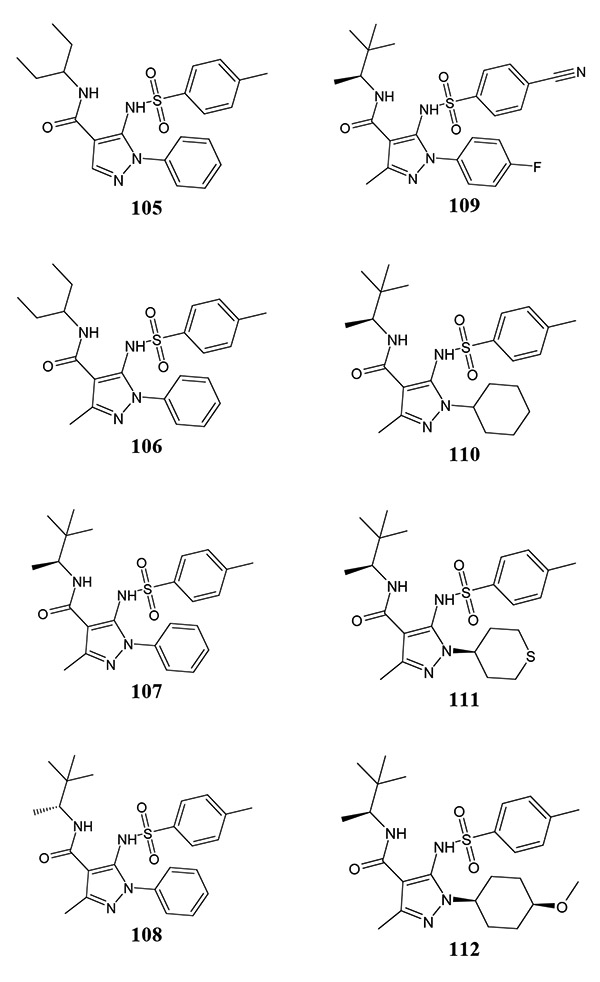 | |||
|---|---|---|---|
| # | IC50 (μM)a | # | IC50 (μM) |
| 105 | 2.0 | 109 | 0.025 |
| 106 | 0.5 | 110 | 0.032 |
| 107 | 0.08 | 111 | 0.04 |
| 108 | 0.5 | 112 | 0.004 |
Inhibition of fMLF-stimulated intracellular Ca2+ flux in human neutrophils or HEK cells expressing recombinant human FPR1; IC50 values were recalculated from reported pIC50 values.
4.4. Miscellaneous FPR Antagonists
Sulfinpyrazone 113 (Table 16) and its related derivative 1,2-diphenyl-4-(3-(1 -naphthyl)-propyl)-3,5-pyrazolidinedione (DNP) were reported as the first competitive FPR1 antagonists [125, 126]. The low affinity of sulfinpyrazone at FPR1 (Ki=14 μM) was later supported by Young et al. [127]. It should be noted that sulfinpyrazone is a non-steroidal antiinflammatory drug (NSAID) that alters platelet function by inhibiting cyclooxygenase activity [128]. In addition, this drug acts as a general inhibitor of organic anion transporters [129]. Other NSAIDs, including diclofenac, piroxicam (compound 114), and its close analog tenoxicam were found to be low activity FPR1 antagonists [130, 131]. Losartan, a selective antagonist of AT1 receptor activation by angiotensin II and sulfasalazine, a drug useful in the therapy of inflammatory bowel disease [132], also decreased the binding of [3H]fMLF to neutrophils [133]. Recently, propofol, a widely used i.v. anesthetic drug, was reported to exhibit FPR1 antagonist activity [134]. Propofol and its halogenated analogs are also high-affinity inhibitors of voltage-operated sodium channels [135]. Because NSAIDs, losartan, sulfasalazine, and propofol exhibit various pharmacological properties, these drugs are not suitable for in vivo studies designed to probe the physiological role of FPR.
Table 16.
Antagonist Activity of Miscellaneous Derivatives at FPR1 (Compounds 113-126, and 132) and FPR2 (Compounds 127-131) [83, 120, 123, 127]
 | |||||
|---|---|---|---|---|---|
| # | Ki,(μM) | # | Ki,(μM) | # | Ki,(μM) |
| 113 | 14.0 | 120 | 12.0a | 127 | 0.27 |
| 114 | 18.0 | 121 | 2.0a | 128 | 1.8 |
| 115 | 4.0a | 122 | 4.0 | 129 | 3.2 |
| 116 | 7.0a | 123 | 0.095 | 130 | 6.7b |
| 117 | 4.0a | 124 | 0.55 | 131 | 0.001a |
| 118 | 11.0a | 125 | 0.75 | 132 | 0.1 |
| 119 | 3.0a | 126 | 0.002a | ||
Inhibition of fluorescent ligand (WKYMVm-FITC) binding in U937 (FPR1) or RBL (FPR2) cells were used.
Binding ability (IC50) was found in competition assay [125I]-WKYMVm with membranes prepared from RBL-FPR2 cells.
Several new scaffolds for competitive FPR1 antagonists were discovered by Edwards et al. [120] using a flow cytometry-based competition assay (Table 16). These scaffolds included phenyl-1,4,6,7,8,9-hexahydro-5H-pyrazolo[3,4- b]quinolin-5-one (compound 115), 5-oxo-4-phenyl-1,4,5,6, 7,8-hexahydroquinoline (compound 116), (Z)-5-(2-oxoindolin-3-ylidene)-2-thioxothiazolidin-4-one (compound 117), [(3-cyclohexylpropanoyl)amino]-4-thiophene-3-carboxylate (compound 118), benzenesulfonamide (compound 119), 4-anilino-1,2,3,4-tetrahydroquinoline (compound 120), 4H-1-benzopyran-4-one (compound 121), and 4-benzoyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one (compound 122). Although seven compounds with a 2,2'-(phenylmethanediyl)bis(3-hydroxy-5,5-dimethylcyclohex-2-en-1-one) scaffold were reported as relatively low activity FPR1 antagonists with Ki values in the 1 to 32 μM range [120], these compounds were also identified to be partial agonists, with most the potent being compound 79 (Table 12). Thus, it appears that these partial agonists may still be able to desensitize cells but are probably not receptor antagonists, because they can directly induce a Ca2+ flux.
Further HTS efforts supported two of the chemotypes described above as FPR1 antagonists, including 4H-1- benzopyran-4-one and 4-benzoyl-3-hydroxy-5-phenyl-1H pyrrol-2(5H)-one [83], with compounds 123 and 124 as examples, respectively. Although nine FPR1-specific antagonists with the same 4H-1-benzopyran-4-one scaffold were reported in both publications [83, 120], SAR analysis of the substituents attached to the 4H-pyran-4-one and benzyl rings or via the carboxyl bridge is limited, and further evaluation will be necessary. Another FPR1 antagonist identified in the screening by Young et al. [83] is pyrazolo[1,5-a]pyrimidin- 7(1H)-one 125. Pyrrolidine bis-diketopiperazine 126 was recently described as potent and selective FPR1 antagonist by screening 37 different combinatorial libraries and totaling more than five million small molecules [123, 136].
Using of a flow-cytometry-based competition assay in “duplex” format, Edwards et al. [137] and Strouse et al. [138] identified three FPR2-specific antagonists with an 2-phenylimidazo[1,2-a]pyrimidine scaffold, the most potent being compound 127. Other less potent FPR2 antagonists include anthrone derivative 128 and 2-(piperazin-1-yl)pyrimidine 129. It should be noted, that the assay detected active structures, which blocked the binding of a fluorescent ligand to FPR on the surface of intact cells, thus detecting both agonists and antagonists [127, 139]. However, testing of possible agonist effects was not reported by Young et al. [83]. To evaluate if these antagonists could have agonist effects and whether receptor desensitization could explain the reported antagonism, we evaluated their activity in HL-60 cells transfected with FPR1/FPR2. However, no direct agonist effects were found for the most potent compounds 123 and 127 in the Ca2+ flux assay (unpublished), indicating they are likely true competitive antagonists.
Quinazolinone 130 with a hydroxyl group in the para position of benzene ring A was described as an FPR2 antagonist (see Tables 11 and 16). This compound did not activate Ca2+ flux in FPR2 transfected cells, but inhibited WKYMVm-stimulated Ca2+ flux, chemotaxis, and ERK1/ ERK2 phosphorylation in FPR2 transfected RBL cells. Moreover, compound 130 inhibited arachidonic acid-induced ear edema [119].
Several pyrrolidine bis-diketopiperazine-based FPR2 antagonists, including compound 131, were recently identified by screening combinatorial libraries [123, 136]. Similar to quinazolinone derivatives 62/130, modification of the propyl functionality of FPR2 agonist 84 (see Table 12) to an isopropyl yielded FPR2 antagonist 131, which is the most potent non-peptide FPR2 antagonist identified to date.
Compound 132 (BVT173187; 3,5-dichloro-N-(2-chloro-5-methyl-phenyl)-2-hydroxy-benzamide) was reported as an inhibitor of FPR1-dependent signals, but without effect on an agonist binding [85]. Although this compound did not inhibit FPR2-dependent responses, neutrophil responses to C5a and IL-8 were attenuated by this molecule, suggesting a broader specificity and/or interaction with various intracellular domains [85]. Indeed, the chemical scaffold of this compound has similarity with other bioactive molecules, including a potent NF-κB inhibitor IMD-0354 (N-(3,5-bistrifluoromethyl-phenyl)-5-chloro-2-hydroxy-benzamide) [140], naphtol AS-E and its derivatives, which are inhibitors of cyclic AMP-response element binding (CREB)-mediated gene transcription [141], and m-3M3FBS, which is a putative phospholipase C activator [142], as well as an inhibitor of Kv7 (KCNQ) voltage-activated potassium current [143].
Bile acids, such as deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA), were reported to antagonize FPR1 at high concentrations (>100 μM) [88, 89]. It should be noted that bile acids have multiple physiological functions, including activation of the farnesoid X receptor (FXR) [144] and Takeda G-protein-coupled receptor (TGR5), a G protein-coupled bile acid receptor (GPBAR1) [145]. Both FXR and TGR5 demonstrate pleiotropic functions, including immune modulation.
4.5. Molecular Modeling of FPR Antagonists and their Receptor Binding
Docking studies of low-activity non-peptide FPR1 antagonists, including sulfasalazine, losartan, phenylbutazone, sulpninpyrazone, several FPR1 ligands, and bile acids DCA and CDCA have been performed [120, 146]. These studies suggest that FPR1 antagonists could block the access of agonists to the receptor by binding at the outer side of the transmembrane helices, between loops E2 and E3 [146]. The authors concluded that while the interaction with Tyr257 is crucial for anchoring the carboxylic group of the bile acids to the receptor, H-bonding with Thr177 and Thr265 is important to determine the potency of bile acids.
Both research groups that described docking studies of FPR1 antagonists also reported pharmacophore models for these antagonists. The pharmacophore model developed by Edwards et al. [120] was based on best docking poses of four FPR1 antagonists, including cyclosporin H, phenylbutazone, and 2 peptide FPR1 antagonists [147, 148], into a rhodopsin-based FPR1 homology structure. This pharmacophore model has three points: two acceptors for H-bonding and one hydrophobic point. Using this model, structures of several FPR1 ligands were prioritized, including FPR1 agonist 79. Moreover, our further pharmacophore modeling also showed that this model is not specific for FPR1 antagonists, because FPR1 agonists (e.g., compound 80) also fit this model [75, 121]. Ferrari et al. [146] described two pharmacophore models for FPR1 antagonists that contain either two hydrophobic centers, one H-bonding donor site, and one H-bond acceptor/negative site (model I) or two H-bond acceptor sites and three hydrophobic centers (model II).
The present overview of FPR ligands can provide some general observations concerning differences between structures of agonists and antagonists. Indeed, many FPR1 antagonists investigated contain OH groups, which can serve as H-bond donors and/or acceptors upon binding to the receptor. This feature is much more characteristic for antagonists than of agonists. For example, FPR1 antagonists 112, 113, 115, 116, 118, 120, 122, 128, all diamides (Table 13), and FPR2 antagonist 127 contain hydroxyl substituents, while there are no OH-containing FPR1 agonists and only a couple OH-containing FPR1/FPR2 agonists reported (compounds 75, and 76). It should be noted that substitution of methoxy, methyl, or nitro groups with a hydroxyl group in FPR1 agonists 62-64 converted them to FPR1 antagonists (e.g., compound 130) [119]. The presence of OH groups in the structures of known FPR1 antagonists is in agreement with models of Ferrari et al. [146], which contain pharmacophore points with H-bond donor and acceptor nature. To date, there are no reports describing modeling of FPR2 antagonists. Because several potent FPR1/FPR2 antagonists were recently described, further molecular modeling should be conducted for determination of peculiarities of their ligand- receptor interactions.
5. COMPUTATIONAL MODELS FOR VIRTUAL SCREENING AND DESIGN OF FPR LIGANDS
Computational drug discovery is an effective strategy for accelerating and economizing the drug discovery and development process. Over the past decades, computational drug discovery methods, such as virtual screening, pharmacophore modeling, and molecular docking have been greatly improved [149]. Since the 3D crystal structures of FPRs have not yet been determined experimentally, ligand-based virtual screening represents an alternative approach for developing new structurally diverse FPR ligands. Among ligand-based virtual screening technologies, pharmacophore modeling and (quantitative) structure-activity relationship [(Q)SAR] analysis are methods for estimating properties of a chemical from its molecular structure. These methods have been used extensively in drug discovery research (reviewed in [150]). The pharmacophore and (Q)SAR functions can be useful for predicting the biological properties of virtual hits or untested compounds obtained from ligand-based virtual screening.
5.1. (Q)SAR Models
While a variety of molecular parameters can be used in the computational methods for (Q)SAR analysis [151], some of these parameters are complex physicochemical or geometrical 3D descriptors whose calculation is associated with difficulties resulting from molecular flexibility. Conversely, topological 2D descriptors, obtainable from the structural formula of a compound are very attractive because of their simplicity. A reasonable compromise between ease of interpretation and ease of computation was reported by Carhart et al. [152], who introduced atom pair descriptors as features of the environments of all atoms in the 2D representation of a chemical structure. This methodology has been widely used in the context of fragment-based similarity searches, ligand-based in silico drug screening, and database mining [153, 154].
Recently we applied atom pair descriptors to represent selected set of FPR1/FPR2 agonists and obtained SAR rules, which were based on six atom pair descriptors [155]. Using binary classification tree methodology, we found that FPR1 agonists in the series investigated could be characterized by simultaneously satisfying certain conditions, as described [155]. Good quality and high predictive ability of the SAR model, as well as simplicity and rapidity of calculations associated with the binary tree algorithm, suggest promise in using the classification tree approach for large database mining and virtual screening of FPR agonists. This application of simple classification rules is especially attractive with the appearance of very large compound databases. However, virtual screening with highly diverse databases should be preceded by development of more sophisticated SAR rules based on large training sets consisting of compounds with various chemical types.
Within a series of molecules with a common scaffold, deriving QSAR models is useful for effective drug design. So far, QSAR models have not been published for FPR agonists or antagonists, and further efforts are necessary to develop such models.
5.2. FPR Pharmacophore Models
A pharmacophore is a model which represents the key physicochemical interactions that mediate biological activity [156]. A pharmacophore model can be used for ligand-based virtual screening and to characterize molecular features of ligands and their structural requirements for biological interaction [157]. Moreover, during virtual screening based on GPCR structure, the data set could be filtered with a simple pharmacophore model representative of the desired receptor ligands in order to exclude compounds considered to be unable to bind [158].
Several pharmacophore models of FPR1/FPR2 ligands have been reported to date [75, 79, 120, 121, 146]. We built a pharmacophore model of FPR1 agonists based on field point methodology developed by Cheeseright et al. [113-115]. This approach allowed us to compare diverse molecules in terms of their field similarity and create an alignment of their bioactive conformations [159]. Although the results of our modeling [79] are in agreement with the three-centered pharmacophore model obtained by Edwards et al. [120], our model also elucidates features not evident in the Edwards model. For example, the compact group of red spheres in the lower part of (Fig. 6) is considered a potentially important characteristic of the template, likely representing interaction with electronegative features of the ligand-binding site. Investigation of a wider series of highly active FPR agonists in the future will generate more information about the significance of these field points.
Fig. (6).
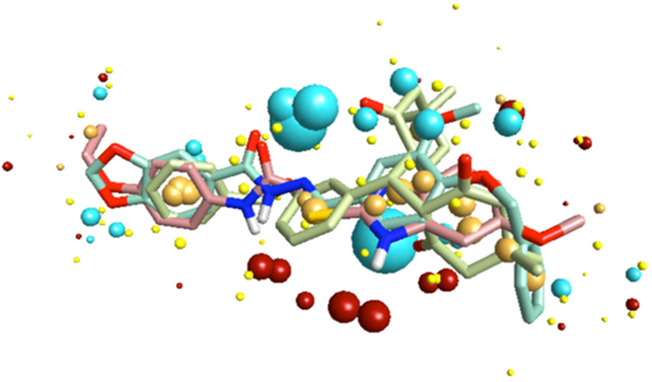
Multi-molecule template for FPR1 developed from compounds 2, 79, and 81.
Another useful approach to the construction of pharmacophore models consists of applying comparative molecular field analysis (CoMFA) [160]. comparative molecular similarity indices analysis (CoMSIA) [161], and related ligand-based methodologies. This approach allows one to obtain a QSAR model along with spatial arrangement of pharmacologically important areas or points. As an example, QSAR based on CoMFA, CoMSIA, and molecular descriptors was built for ligands of P2Y1, an ADP-activated GPCR [162]. Hence, further is needed to derive such models within series of structurally related FPR agonists or antagonists.
5.3. Homology Models and Molecular Docking
Molecular docking is becoming a routine technique during the last decade because of the great success in establishing 3D structures of ligand-receptor complexes and in homology modeling. Although docking is hardly applicable now to large database mining because of high computational demands, its use for small data sets of FPR agonists and other compounds is very helpful for understanding peculiarities of interaction between ligands and receptors. Successful homology models of GPCRs can provide a structural basis for understanding ligand binding and receptor activation of GPCRs, and have been used for determining and comparing specificity profiles of related receptors, guiding drug design, virtual screening, and enriching the rate of ligand hits relative to a random collection of small-molecule compounds [163-166]. Moreover, induced fit docking (IFD) recently was introduced as a method that combines flexible ligand docking with receptor structure prediction and side chain refinement and could be considered as an approach for ligand-based protein structure optimization and homology modeling of GPCR [158, 167]. For drug design, it is important to understand the extent to which crystal structures or homology models can be used to best predict the binding mode of compounds. Thus, the set of active FPR ligands described in this review could be useful for selection of the best FPR homology model and/or for ligand-based FPR structure optimization.
For the identification of ligands based on novel chemotypes, a proven technique is virtual screening through molecular docking of chemically diverse libraries that bind to various GPCRs [168]. Although several homology models of FPR1/FPR2 have been reported to date [82, 105, 116, 120, 146, 169], most of them were used to explain the binding of FPR1/FPR2 agonists. One exception is the use of a combination of the FPR1 homology model and its pharmacophore model [120]. The combined model includes two electron acceptors, one hydrophobic point, and 23 sterically forbidden zones (i.e., exclusion spheres). This computational model was used for in silico screening of ~480,000 drug-like small molecules and improved the physical screening hit rate by 12-fold (1.2% vs. 0.1% hit-rate in a random compound collection).
6. CONCLUSION AND PERSPECTIVES
Although FPRs have great potential as pharmacologic targets for treatment of a variety of inflammatory diseases [9, 12, 13], a key question in the field is whether FPR agonists or antagonists have the most therapeutic potential. An additional question is whether FPR agonists/antagonists could represent more specific and/or effective therapeutics than currently available treatments for these processes and pathologies. Indeed, studies indicate that FPRs have a number of functions outside of the immune system and are involved in a variety of signaling systems. Thus, the off-target effects of FPR ligands in vivo may be unpredictable. Clearly, further studies are required to understand the complex role of FPRs in vivo and how they might serve as therapeutic targets. Although detailed pharmacological characterization of rodent FPRs is clearly interesting as a model for human diseases and determination of the biological roles of FPRs, rodent FPRs are distinct from their human counterparts and many agonists of human FPRs do not activate murine FPRs. Thus, the selection of appropriate in vivo models is also important to consider.
Current small-molecule FPR ligands have been discovered as result of HTS efforts and/or synthesis of new structures based on discovered hits and subsequent SAR-directed drug design. Because a key question in drug discovery is how to best prioritize and select hits for further evaluation from the many weakly active compounds, SAR information is the next step in attempts to estimate the chemical optimization potential of hits [170]. Note, however, that SAR analysis of small-molecule ligands for murine counterparts of the human FPRs has not been performed and is clearly necessary if these compounds are being developed as therapeutics in murine models.
Despite the increased number of FPR agonists discovered in recent years, pharmacokinetic analysis has only been reported for one FPR2 agonist (compound 13) [98] and all 3 known series of FPR1 antagonists [84, 86, 87]. This may reflect relatively weak interest from pharmaceutical companies for FPRs as perspective therapeutic targets. Indeed, only three companies have reported interest in development of FPR ligands, including Acadia Pharmaceuticals, Inc. (FPR2 agonists) [45], Amgen, Inc. (FPR2 agonists) [76, 98], and AstraZeneca, R&D, Charnwood (FPR1 antagonists) [84, 86, 87]. Furthermore, there are still no therapeutic drugs targeting FPRs in preclinical or clinical trials. Another possibility to apply such non-peptidic FPR ligands in clinical medicine is in the development of imaging agents. Indeed, several imaging conjugates based on the peptide FPR1 antagonist cFLFLFK have been reported, including a 64Cu conjugate for a positron emission tomography (PET) [171], a 99mTc conjugate for a single photon emission computed tomography (SPECT) [172], a cyanine-7 conjugate for noninvasive near infrared fluorescence imaging [173], and a Gd(III) contrast agent for magnetic resonance imaging (MRI) [174]. Thus, imaging agents based on small-molecule FPR agonists could be desirable agents imaging neutrophils at sites of inflammation.
ACKNOWLEDGEMENTS
This work was supported in part by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number GM103500, an equipment grant from the M. J. Murdock Charitable Trust, and the Montana State University Agricultural Experimental Station.
ABBREVIATIONS
- Anxa12–26
Annexin 1 fragment 2-26
- CoMFA
Comparative molecular field analysis
- CXCR
C-X-C chemokine receptor
- DCA
Deoxycholic acid
- FPR
Formyl peptide receptor
- GPCR
G protein-coupled receptor
- HIV
Human immunodeficiency virus
- HTS
High-throughput screening
- IL
Interleukin
- LPS
Lipopolysaccharide
- LTB
Leukotriene B4
- LXA4
Lipoxin A4
- NSAID
Non-steroidal anti-inflammatory drug
- QSAR
Quantitative structure-activity relationship
- SAA
Serum amyloid A
- SAR
Structure–activity relationship
- TNF
Tumor necrosis factor
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
REFERENCES
- [1].Ye RD; Boulay F; Wang JM; Dahlgren C; Gerard C; Parmentier M; Serhan CN; Murphy PM International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the Formyl Peptide Receptor (FPR) Family. Pharmacol. Rev, 2009, 61, 119–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schiffmann E; Corcoran BA; Wahl SM N-formylmethionyl peptides as chemoattractants for leucocytes. Proc. Natl. Acad. Sci.USA, 1975, 72, 1059–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Aswanikumar S; Corcoran B; Schiffman E; Day AR; Freer RJ; Showell HJ; Becker EL; Pert CB Demonstration of a receptor on rabbit neutrophils for chemotactic peptides. Biochem. Biophys. Res. Commun, 1977, 74, 810–817. [DOI] [PubMed] [Google Scholar]
- [4].Carp H Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med, 1982, 155, 264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rivière S; Challet L; Fluegge D; Spehr M; Rodriguez I Formyl peptide receptor-like proteins are a novel family of vomeronasal chemosensors. Nature, 2009, 459, 574–577. [DOI] [PubMed] [Google Scholar]
- [6].Liberles SD; Horowitz LF; Kuang D; Contos JJ; Wilson KL; Siltberg-Liberles J; Liberles DA; Buck LB Formyl peptide receptors are candidate chemosensory receptors in the vomeronasal organ. Proc. Natl. Acad. Sci. USA, 2009, 106, 9842–9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bufe B; Schumann T; Zufall F Formyl peptide receptors from immune and vomeronasal system exhibit distinct agonist properties. J. Biol. Chem, 2012, 287, 33644–33655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gao JL; Murphy PM Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J. Biol. Chem, 1993, 268, 25395–25401. [PubMed] [Google Scholar]
- [9].Liu M; Zhao J; Chen K; Bian X; Wang C; Shi Y; Wang JM G protein-coupled receptor FPR1 as a pharmacologic target in inflammation and human glioblastoma. Int. Immunopharmacol, 2012, 14, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gao JL; Lee EJ; Murphy PM Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J. Exp. Med, 1999, 189, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cattaneo F; Parisi M; Ammendola R Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Molec. Sci, 2013, 14, 7193–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Iribarren P; Zhou Y; Hu J; Le Y; Wang JM Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol. Res, 2005, 31, 165–176. [DOI] [PubMed] [Google Scholar]
- [13].Cui Y; Le Y; Yazawa H; Gong W; Wang JM Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer's disease. J. Leukoc. Biol, 2002, 72, 628–635. [PubMed] [Google Scholar]
- [14].Schneider EH; Weaver JD; Gaur SS; Tripathi BK; Jesaitis AJ; Zelenka PS; Gao JL; Murphy PM The leukocyte chemotactic receptor FPR1 is functionally expressed on human lens epithelial cells. J. Biol. Chem, 2012, 287, 40779–40792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Migeotte I; Communi D; Parmentier M Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev., 2006, 17, 501–519. [DOI] [PubMed] [Google Scholar]
- [16].Czapiga M; Gao JL; Kirk A; Lekstrom-Himes J Human platelets exhibit chemotaxis using functional N-formyl peptide receptors. Exp. Hematol, 2005, 33, 73–84. [DOI] [PubMed] [Google Scholar]
- [17].McCoy R; Haviland DL; Molmenti EP; Ziambaras T; Wetsel RA; Perlmutter DH N-formylpeptide and complement C5a receptors are expressed in liver cells and mediate hepatic acute phase gene regulation. J. Exp. Med, 1995, 182, 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schreiner J; Kretschmer D; Klenk J; Otto M; Buhring HJ; Stevanovic S; Wang JM; Beer-Hammer S; Peschel A; Autenrieth SE Staphylococcus aureus phenol-soluble modulin peptides modulate dendritic cell functions and increase in vitro priming of regulatory t cells. J. Immunol, 2013, 190, 3417–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang ZG; Ye RD Characterization of two new members of the formyl peptide receptor gene family from 129S6 mice. Gene, 2002, 299, 57–63. [DOI] [PubMed] [Google Scholar]
- [20].Slowik A; Merres J; Elfgen A; Jansen S; Mohr F; Wruck CJ; Pufe T; Brandenburg LO Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)--and amyloid beta 1-42-induced signal transduction in glial cells. Mol. Neurodegener, 2012, 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].He HQ; Liao D; Wang ZG; Wang ZL; Zhou HC; Wang MW; Ye RD Functional characterization of three mouse formyl peptide receptors. Mol. Pharmacol, 2013, 83, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gavins FN Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol. Sci, 2010, 31, 266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chester JF; Ross JS; Malt RA; Weitzman SA Acute colitis produced by chemotactic peptides in rats and mice. Am J Pathol, 1985, 121, 284–290. [PMC free article] [PubMed] [Google Scholar]
- [24].LeDuc LE; Nast CC, Chemotactic peptide-induced acute colitis in rabbits. Gastroenterology, 1990, 98, 929–935. [DOI] [PubMed] [Google Scholar]
- [25].Colucci M; Mastriota M; Maione F; Di Giannuario A; Mascolo N; Palmery M; Severini C; Perretti M; Pieretti S Guinea pig ileum motility stimulation elicited by N-formyl-Met-Leu-Phe (fMLF) involves neurotransmitters and prostanoids. Peptides, 2011, 32, 266–271. [DOI] [PubMed] [Google Scholar]
- [26].Anton PA; Targan SR; Shanahan F Increased neutrophil receptors for and response to the proinflammatory bacterial peptide formyl-methionyl-leucyl-phenylalanine in Crohn's disease. Gastroenterology, 1989, 97, 20–28. [DOI] [PubMed] [Google Scholar]
- [27].Ledesma de Paolo MI; Celener Gravelle P; De Paula JA; Panzita MT; Bandi JC; Bustos Fernandez L Stimulation of inflammatory mediators secretion by chemotactic peptides in rat colitis model. Acta Gastroenterol. Latinoam, 1996, 26, 23–30. [PubMed] [Google Scholar]
- [28].Nast CC; LeDuc LE Chemotactic peptides. Mechanisms, functions, and possible role in inflammatory bowel disease. Dig. Dis. Sci, 1988, 33, 50S–57S. [DOI] [PubMed] [Google Scholar]
- [29].Fehr J; Dahinden C; Russi R Formylated chemotactic peptides can mimic the secondary, provoking endotoxin injection in the generalized Shwartzman reaction. J. Infect. Dis, 1984, 150, 160–161. [DOI] [PubMed] [Google Scholar]
- [30].Jonsson M; Tzanela M; Kolbeck RC; McCormick JR Hemodynamic and metabolic effects of intravenous formyl-methionyl-leucyl-phenylalanine (FMLP) in rabbits. In Vivo, 1997, 11, 133–139. [PubMed] [Google Scholar]
- [31].Tzanela M; Orfanos S; McCormick JR Endotoxin augments hemodynamic and metabolic effects of formyl-methionyl-leucyl-phenylalanine (FMLP) in rabbits. In Vivo, 2007, 21, 81–87. [PubMed] [Google Scholar]
- [32].Berend N; Armour CL; Black JL Formyl-methionyl-leucyl-phenylalanine causes bronchoconstriction in rabbits. Agents Actions, 1986, 17, 466–471. [DOI] [PubMed] [Google Scholar]
- [33].Peters MJ; Breslin AB; Kemp AS; Chu J; Berend N Haematological effects of inhalation of N-formyl-methionyl-leucyl-phenylalanine in man. Thorax, 1992, 47, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Le Y; Murphy PM; Wang JM Formyl-peptide receptors revisited. Trends Immunol., 2002, 23, 541–548. [DOI] [PubMed] [Google Scholar]
- [35].Iaccio A; Angiolillo A; Ammendola R Intracellular signaling triggered by formyl-peptide receptors in nonphagocytic cells. Curr. Signal Transduct. Ther, 2008, 3, 88–96. [Google Scholar]
- [36].Lee MS; Yoo SA; Cho CS; Suh PG; Kim WU; Ryu SH Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J. Immunol, 2006, 177, 5585–5594. [DOI] [PubMed] [Google Scholar]
- [37].Wan W; Gao JL Leukocyte chemoattractant receptor FPR2 may accelerate atherogenesis. Med. Hypotheses, 2012, 79, 101–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee HY; Kim SD; Baek SH; Choi JH; Bae YS Role of formyl peptide receptor 2 on the serum amyloid A-induced macrophage foam cell formation. Biochem. Biophys. Res. Commun, 2013, 433, 255–259. [DOI] [PubMed] [Google Scholar]
- [39].Li Y; Ye D Molecular biology for formyl peptide receptors in human diseases. J. Mol. Med, 2013, 91(7),781–789. [DOI] [PubMed] [Google Scholar]
- [40].Maderna P; Cottell DC; Toivonen T; Dufton N; Dalli J; Perretti M; Godson C FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J., 2010, 24, 4240–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maderna P; Godson C Phagocytosis of apoptotic cells and the resolution of inflammation. Biochim. Biophys. Acta, 2003, 1639, 141–151. [DOI] [PubMed] [Google Scholar]
- [42].Zhang L; Falla TJ Host defense peptides for use as potential therapeutics. Curr. Opin. Investig. Drugs, 2009, 10, 164–171. [PubMed] [Google Scholar]
- [43].Dufton N; Perretti M Therapeutic anti-inflammatory potential of formyl-peptide receptor agonists. Pharmacol. Ther, 2010, 127, 175–188. [DOI] [PubMed] [Google Scholar]
- [44].McDonald B; Pittman K; Menezes GB; Hirota SA; Slaba I; Waterhouse CC; Beck PL; Muruve DA; Kubes P Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science, 2010, 330, 362–366. [DOI] [PubMed] [Google Scholar]
- [45].Nash N, Scully A, Gardell L, Olsson R, Gustafsson M Use of the lipoxin receptor, FPRL1, as a tool for identifying compounds effective in the treatment of pain and inflammation. Patent US 60/592,926, 2004.
- [46].Gavins FN; Hughes EL; Buss NA; Holloway PM; Getting SJ; Buckingham JC Leukocyte recruitment in the brain in sepsis: involvement of the annexin 1-FPR2/ALX anti-inflammatory system. FASEB J., 2012, 26, 4977–4989. [DOI] [PubMed] [Google Scholar]
- [47].Martin GR; Perretti M; Flower RJ; Wallace JL Annexin-1 modulates repair of gastric mucosal injury. Am. J. Physiol. Gastrointest. Liver Physiol, 2008, 294, G764–G769. [DOI] [PubMed] [Google Scholar]
- [48].Qin C; Buxton KD; Pepe S; Cao AH; Venardos K; Love JE; Kaye DM; Yang YH; Morand EF; Ritchie RH Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2-26). Br. J. Pharmacol, 2013, 168, 238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pieretti S; Di Giannuario A; De Felice M; Perretti M; Cirino G, Stimulus-dependent specificity for annexin 1 inhibition of the inflammatory nociceptive response: the involvement of the receptor for formylated peptides. Pain, 2004, 109, 52–63. [DOI] [PubMed] [Google Scholar]
- [50].Hecht I; Rong J; Sampaio AL; Hermesh C; Rutledge C; Shemesh R; Toporik A; Beiman M; Dassa L; Niv H; Cojocaru G; Zauberman A; Rotman G; Perretti M; Vinten-Johansen J; Cohen Y A novel peptide agonist of formyl-peptide receptor-like 1 (ALX) displays anti-inflammatory and cardioprotective effects. J. Pharmacol. Exp. Ther, 2009, 328, 426–434. [DOI] [PubMed] [Google Scholar]
- [51].Su SB; Gong WH; Gao JL; Shen WP; Grimm MC; Deng X; Murphy PM; Oppenheim JJ; Wang JM T20/DP178, an ectodomain peptide of human immunodeficiency virus type 1 gp41, is an activator of human phagocyte N-formyl peptide receptor. Blood, 1999, 93, 3885–3892. [PubMed] [Google Scholar]
- [52].Le Y; Oppenheim JJ; Wang JM Pleiotropic roles of formyl peptide receptors. Cytokine Growth Factor Rev., 2001, 12, 91–105. [DOI] [PubMed] [Google Scholar]
- [53].Li BQ; Wetzel MA; Mikovits JA; Henderson EE; Rogers TJ; Gong W; Le Y; Ruscetti FW; Wang JM The synthetic peptide WKYMVm attenuates the function of the chemokine receptors CCR5 and CXCR4 through activation of formyl peptide receptor-like 1. Blood, 2001, 97, 2941–2947. [DOI] [PubMed] [Google Scholar]
- [54].Kim H; Park JH; Lee EH; Kim MJ; Park SK; Heo SK; Kim BS; Min YJ Granulocyte function is stimulated by a novel hexapeptide, WKYMVm, in chemotherapy-treated cancer patients. Exp. Hematol, 2006, 34, 407–413. [DOI] [PubMed] [Google Scholar]
- [55].Kim SD; Kim YK; Lee HY; Kim YS; Jeon SG; Baek SH; Song DK; Ryu SH; Bae YS The agonists of formyl peptide receptors prevent development of severe sepsis after microbial infection. J. Immunol, 2010, 185, 4302–4310. [DOI] [PubMed] [Google Scholar]
- [56].Lin C; Wei W; Zhang J; Liu S; Liu Y; Zheng D Formyl peptide receptor-like 1 mediated endogenous TRAIL gene expression with tumoricidal activity. Mol. Cancer Ther, 2007, 6, 2618–2625. [DOI] [PubMed] [Google Scholar]
- [57].Tae YM; Park HT; Moon HG; Kim YS; Jeon SG; Roh TY; Bae YS; Gho YS; Ryu SH; Kwon HS; Kim YK Airway activation of formyl peptide receptors inhibits Th1 and Th17 cell responses via inhibition of mediator release from immune and inflammatory cells and maturation of dendritic cells. J. Immunol, 2012, 188, 1799–1808. [DOI] [PubMed] [Google Scholar]
- [58].Mollica A; Stefanucci A; Costante R; Pinnen F Role of formyl peptide receptors (FPR) in abnormal inflammation responses involved in neurodegenerative diseases. Antiinflamm. Antiallergy Agents Med. Chem, 2012, 11, 20–36. [DOI] [PubMed] [Google Scholar]
- [59].Tsuruki T; Takahata K; Yoshikawa M Mechanism of the protective effect of intraperitoneally administered agonists for formyl peptide receptors against chemotherapy-induced alopecia.Biosci. Biotechnol. Biochem, 2007, 71, 1198–1202. [DOI] [PubMed] [Google Scholar]
- [60].Stenfeldt AL; Karlsson J; Wennerås C; Bylund J; Fu H; Dahlgren C Cyclosporin H, Boc-MLF and Boc-FLFLF are antagonists that preferentially inhibit activity triggered through the formyl peptide receptor. Inflammation, 2007, 30, 224–229. [DOI] [PubMed] [Google Scholar]
- [61].Cardini S; Dalli J; Fineschi S; Perretti M; Lungarella G; Lucattelli M Genetic ablation of the fpr1 gene confers protection from smoking-induced lung emphysema in mice. Am. J. Respir. Cell Mol. Biol, 2012, 47, 332–339. [DOI] [PubMed] [Google Scholar]
- [62].Rittner HL; Hackel D; Voigt P; Mousa S; Stolz A; Labuz D; Schafer M; Schaefer M; Stein C; Brack A Mycobacteria attenuate nociceptive responses by formyl peptide receptor triggered opioid peptide release from neutrophils. PLoS Pathog., 2009, 5, e1000362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhou C; Zhou Y; Wang J; Feng Y; Wang H; Xue J; Chen Y; Ye RD; Wang MW V101L of human formyl peptide receptor 1 (FPR1) increases receptor affinity and augments the antagonism mediated by cyclosporins. Biochem. J, 2013, 451, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Fruman DA; Klee CB; Bierer BE; Burakoff SJ Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc. Natl. Acad. Sci. USA, 1992, 89, 3686–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Broekemeier KM; Pfeiffer DR Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry, 1995, 34, 16440–16449. [DOI] [PubMed] [Google Scholar]
- [66].Gschwendt M; Kittstein W; Marks F The weak immunosuppressant cyclosporine D as well as the immunologically inactive cyclosporine H are potent inhibitors in vivo of phorbol ester TPA-induced biological effects in mouse skin and of Ca2+/calmodulin dependent EF-2 phosphorylation in vitro. Biochem. Biophys. Res. Commun, 1988, 150, 545–551. [DOI] [PubMed] [Google Scholar]
- [67].da Silva NG; Sampaio SC; Goncalves LR Inhibitory effect of Crotalus durissus terrificus venom on chronic edema induced by injection of bacillus Calmette-Guerin into the footpad of mice. Toxicon, 2013, 63, 98–103. [DOI] [PubMed] [Google Scholar]
- [68].Gastardelo TS; Damazo AS; Dalli J; Flower RJ; Perretti M; Oliani SM Functional and ultrastructural analysis of annexin A1 and its receptor in extravasating neutrophils during acute inflammation. Am. J. Pathol, 2009, 174, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hwang TL; Hung CH; Hsu CY; Huang YT; Tsai YC; Hsieh PW Design and synthesis of tryptophan containing dipeptide derivatives as formyl peptide receptor 1 antagonist. Org. Biomol. Chem, 2013, 11, 3742–3755. [DOI] [PubMed] [Google Scholar]
- [70].Cavicchioni G; Fraulini A; Falzarano S; Spisani S Structure-activity relationship of for-L-Met L-Leu-L-Phe-OMe analogues in human neutrophils. Bioorg. Chem, 2006, 34, 298–318. [DOI] [PubMed] [Google Scholar]
- [71].Selvatici R; Falzarano S; Mollica A; Spisani S Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur. J. Pharmacol, 2006, 534, 1–11. [DOI] [PubMed] [Google Scholar]
- [72].Selvatici R; Siniscalchi A; Spisani S Effects of Synthetic peptides on the inflammatory response and their therapeutic potential. Mini Rev. Med. Chem, 2012, 13, 553–564. [DOI] [PubMed] [Google Scholar]
- [73].Dalpiaz A; Scatturin A Peptide derivatives as agonists or antagonists of formylpeptide receptors: analysis of their effects on neutrophils. Mini Rev. Med. Chem, 2003, 3, 167–173. [DOI] [PubMed] [Google Scholar]
- [74].Nanamori M; Cheng X; Mei J; Sang H; Xuan Y; Zhou C; Wang MW; Ye RD A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol. Pharmacol, 2004, 66, 1213–1222. [DOI] [PubMed] [Google Scholar]
- [75].Schepetkin IA; Kirpotina LN; Khlebnikov AI; Quinn MT High-throughput screening for small-molecule activators of neutrophils: Identification of novel N-formyl peptide receptor agonists. Mol. Pharmacol, 2007, 71, 1061–1074. [DOI] [PubMed] [Google Scholar]
- [76].Frohn M; Xu H; Zou X; Chang C; McElvaine M; Plant MH; Wong M; Tagari P; Hungate R; Bürli RW New 'chemical probes' to examine the role of the hFPRL1 (or ALXR) receptor in inflammation. Bioorg. Med. Chem, 2007, 17, 6633–6637. [DOI] [PubMed] [Google Scholar]
- [77].Cilibrizzi A; Schepetkin IA; Bartolucci G; Crocetti L; Dal P,V; Giovannoni MP; Graziano A; Kirpotina LN; Quinn MT; Vergelli C Synthesis, enantioresolution, and activity profile of chiral 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones as potent N-formyl peptide receptor agonists. Bioorg. Med. Chem, 2012, 20, 3781–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Cilibrizzi A; Quinn MT; Kirpotina LN; Schepetkin IA; Holderness J; Ye RD; Rabiet MJ; Biancalani C; Cesari N; Graziano A; Vergelli C; Pieretti S; Dal P,V; Giovannoni MP 6-Methyl-2,4-disubstituted pyridazin-3(2H)-ones: A novel class of small-molecule agonists for formyl peptide receptors. J. Med. Chem, 2009, 52, 5054–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kirpotina LN; Khlebnikov AI; Schepetkin IA; Ye RD; Rabiet MJ; Jutila MA; Quinn MT Identification of novel small-molecule agonists for human formyl peptide receptors and pharmacophore models of their recognition. Mol. Pharmacol, 2010, 77, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Schepetkin IA; Kirpotina LN; Khlebnikov AI; Jutila MA; Quinn MT Gastrin-releasing peptide/neuromedin B receptor antagonists PD176252, PD168368, and related analogs are potent agonists of human formyl-peptide receptors. Mol. Pharmacol, 2011, 79, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Forsman H; Kalderen C; Nordin A; Nordling E; Jensen AJ; Dahlgren C Stable formyl peptide receptor agonists that activate the neutrophil NADPH-oxidase identified through screening of a compound library. Biochem. Pharmacol, 2011, 81, 402–411. [DOI] [PubMed] [Google Scholar]
- [82].Khlebnikov AI; Schepetkin IA; Kirpotina LN; Brive L; Dahlgren C; Jutila MA; Quinn MT Molecular docking of 2-(benzimidazol-2-ylthio)-N-phenylacetamide-derived small-molecule agonists of human formyl peptide receptor 1. J. Mol. Model, 2012, 18, 2831–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Young SM; Bologa CM; Fara D; Bryant BK; Strouse JJ; Arterburn JB; Ye RD; Oprea TI; Prossnitz ER; Sklar LA; Edwards BS Duplex high-throughput flow cytometry screen identifies two novel formylpeptide receptor family probes. Cytometry A, 2009, 75, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Unitt J; Fagura M; Phillips T; King S; Perry M; Morley A; MacDonald C; Weaver R; Christie J; Barber S; Mohammed R; Paul M; Cook A; Baxter A Discovery of small molecule human FPR1 receptor antagonists. Bioorg. Med. Chem. Lett, 2011, 21, 2991–2997. [DOI] [PubMed] [Google Scholar]
- [85].Cevik-Aras H; Kalderen C; Jenmalm JA; Oprea T; Dahlgren C; Forsman H A non-peptide receptor inhibitor with selectivity for one of the neutrophil formyl peptide receptors, FPR 1. Biochem. Pharmacol, 2012, 83, 1655–1662. [DOI] [PubMed] [Google Scholar]
- [86].Morley AD; King S; Roberts B; Lever S; Teobald B; Fisher A; Cook T; Parker B; Wenlock M; Phillips C; Grime K Lead optimisation of pyrazoles as novel FPR1 antagonists. Bioorg. Med. Chem. Lett, 2012, 22, 532–536. [DOI] [PubMed] [Google Scholar]
- [87].Morley AD; Cook A; King S; Roberts B; Lever S; Weaver R; Macdonald C; Unitt J; Fagura M; Phillips T; Lewis R; Wenlock M Discovery of pyrazoles as novel FPR1 antagonists. Bioorg. Med. Chem. Lett, 2011, 21, 6456–6460. [DOI] [PubMed] [Google Scholar]
- [88].Chen X; Yang D; Shen W; Dong HF; Wang JM; Oppenheim JJ; Howard MZ Characterization of chenodeoxycholic acid as an endogenous antagonist of the G-coupled formyl peptide receptors. Inflamm. Res, 2000, 49, 744–755. [DOI] [PubMed] [Google Scholar]
- [89].Chen X; Mellon RD; Yang L; Dong H; Oppenheim JJ; Howard OM Regulatory effects of deoxycholic acid, a component of the anti-inflammatory traditional Chinese medicine Niuhuang, on human leukocyte response to chemoattractants. Biochem. Pharmacol, 2002, 63, 533–541. [DOI] [PubMed] [Google Scholar]
- [90].Hanson J; Ferreiros N; Pirotte B; Geisslinger G; Offermanns S Heterologously expressed formyl peptide receptor 2 (FPR2/ALX) does not respond to lipoxin A. Biochem. Pharmacol, 2013, 85, 1795–1802. [DOI] [PubMed] [Google Scholar]
- [91].Forsman H; Onnheim K; Andreasson E; Dahlgren C What formyl peptide receptors, if any, are triggered by compound 43 and lipoxin A4? Scand. J. Immunol, 2011, 74, 227–234. [DOI] [PubMed] [Google Scholar]
- [92].Forsman H; Dahlgren C Lipoxin A(4) metabolites/analogues from two commercial sources have no effects on TNF-alphamediated priming or activation through the neutrophil formyl peptide receptors. Scand. J. Immunol, 2009, 70, 396–402. [DOI] [PubMed] [Google Scholar]
- [93].Su SB; Gao J; Gong W; Dunlop NM; Murphy PM; Oppenheim JJ; Wang JM T21/DP107, A synthetic leucine zipper-like domain of the HIV-1 envelope gp41, attracts and activates human phagocytes by using G-protein-coupled formyl peptide receptors. J. Immunol, 1999, 162, 5924–5930. [PubMed] [Google Scholar]
- [94].Movitz C; Brive L; Hellstrand K; Rabiet MJ; Dahlgren C The annexin I sequence gln(9)-ala(10)-trp(11)-phe(12) is a core structure for interaction with the formyl peptide receptor 1. J.Biol.Chem, 2010, 285, 14338–14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Miettinen HM; Mills JS; Gripentrog JM; Dratz EA; Granger BL; Jesaitis AJ The ligand binding site of the formyl peptide receptor maps in the transmembrane region. J. Immunol, 1997, 159, 4045–4054. [PubMed] [Google Scholar]
- [96].Mills JS; Miettinen HM; Cummings D; Jesaitis AJ Characterization of the binding site on the formyl peptide receptor using three receptor mutants and analogs of Met-Leu-Phe and Met-Met-Trp-Leu-Leu. J. Biol. Chem, 2000, 275, 39012–39017. [DOI] [PubMed] [Google Scholar]
- [97].Kufareva I; Rueda M; Katritch V; Stevens RC; Abagyan R Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure, 2011, 19, 1108–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bürli RW; Xu H; Zou X; Muller K; Golden J; Frohn M; Adlam M; Plant MH; Wong M; McElvain M; Regal K; Viswanadhan VN; Tagari P; Hungate R Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg. Med. Chem. Lett, 2006, 16, 3713–3718. [DOI] [PubMed] [Google Scholar]
- [99].Sogawa Y; Shimizugawa A; Ohyama T; Maeda H; Hirahara K The pyrazolone originally reported to be a formyl peptide receptor (FPR) 2/ALX-selective agonist is instead an FPR1 and FPR2/ALX dual agonist. J. Pharmacol. Sci, 2009, 111, 317–321. [DOI] [PubMed] [Google Scholar]
- [100].Sogawa Y; Ohyama T; Maeda H; Hirahara K Inhibition of neutrophil migration in mice by mouse formyl peptide receptors 1 and 2 dual agonist: indication of cross-desensitization in vivo. Immunology, 2011, 132, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Dufton N; Hannon R; Brancaleone V; Dalli J; Patel HB; Gray M; D'Acquisto F; Buckingham JC; Perretti M; Flower RJ Anti-inflammatory role of the murine formyl-peptide receptor 2: ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol, 2010, 184, 2611–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sogawa Y; Ohyama T; Maeda H; Hirahara K Formyl peptide receptor 1 and 2 dual agonist inhibits human neutrophil chemotaxis by the induction of chemoattractant receptor cross-desensitization. J. Pharmacol. Sci, 2011, 115, 63–68. [DOI] [PubMed] [Google Scholar]
- [103].Crocetti L; Vergelli C; Cilibrizzi A; Graziano A; Khlebnikov AI; Kirpotina LN; Schepetkin IA; Quinn MT; Giovannoni MP Synthesis and pharmacological evaluation of new pyridazin-based thioderivatives as FPR agonists. Drug Dev. Res, 2013, 74, 259–271. [Google Scholar]
- [104].Giovannoni MP; Schepetkin IA; Cilibrizzi A; Crocetti L; Khlebnikov AI; Dahlgren C; Graziano A; Dal Piaz V; Kirpotina LN; Zerbinati S; Vergelli C; Quinn MT Further studies on 2-arylacetamide pyridazin-3(2H)-ones: Design, synthesis and evaluation of 4,6-disubstituted analogues as formyl peptide receptors (FPRs) agonists. Eur. J. Med. Chem, 2013, 64, 512–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Schepetkin IA; Kirpotina LN; Khlebnikov AI; Leopoldo M; Lucente E; Lacivita E; De Giorgio P; Quinn MT 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: Molecular modeling of chiral recognition by FPR2. Biochem. Pharmacol, 2013, 85, 404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Cilibrizzi A; Crocetti L; Giovannoni MP; Graziano A; Vergelli C; Bartolucci G; Soldani G; Quinn MT; Schepetkin IA; Faggi C Synthesis, HPLC enantioresolution, and X-ray analysis of a new series of C5-methyl pyridazines as N-formyl peptide receptor (FPR) agonists. Chirality, 2013, 25, 400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Marriott S; Reynolds WF; Taft RW; Topsom RD Substituent electronegativity parameters. J. Org. Chem, 1984, 49, 959–965. [Google Scholar]
- [108].Bang S; Kim KY; Yoo S; Lee SH; Hwang SW Transient receptor potential V2 expressed in sensory neurons is activated by probenecid. Neurosci. Lett, 2007, 425, 120–125. [DOI] [PubMed] [Google Scholar]
- [109].McClenaghan C; Zeng F; Verkuyl JM TRPA1 agonist activity of probenecid desensitizes channel responses: consequences for screening. Assay Drug Dev. Technol, 2012, 10, 533–541. [DOI] [PubMed] [Google Scholar]
- [110].Greene TA; Alarcon S; Thomas A; Berdougo E; Doranz BJ; Breslin PA; Rucker JB Probenecid inhibits the human bitter taste receptor TAS2R16 and suppresses bitter perception of salicin. PLoS One, 2011, 6, e20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Prossnitz ER; Quehenberger O; Cochrane CG; Ye RD Signal transducing properties of the N-formyl peptide receptor expressed in undifferentiated HL60 cells. J. Immunol, 1993, 151, 5704–5715. [PubMed] [Google Scholar]
- [112].Rosenbaum DM; Zhang C; Lyons JA; Holl R; Aragao D; Arlow DH; Rasmussen SG; Choi HJ; Devree BT; Sunahara RK; Chae PS; Gellman SH; Dror RO; Shaw DE; Weis WI; Caffrey M; Gmeiner P; Kobilka BK Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature, 2011, 469, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Cheeseright T; Mackey M; Rose S; Vinter A Molecular field extrema as descriptors of biological activity: definition and validation. J. Chem. Inf. Model, 2006, 46, 665–676. [DOI] [PubMed] [Google Scholar]
- [114].Cheeseright T; Mackey M; Rose S; Vinter A Molecular field technology applied to virtual screening and finding the bioactive conformation. Expert Opin. Drug Discov, 2007, 2, 131–144. [DOI] [PubMed] [Google Scholar]
- [115].Cheeseright TJ; Mackey MD; Scoffin RA High content pharmacophores from molecular fields: a biologically relevant method for comparing and understanding ligands. Curr. Comput. Aided Drug Des, 2011, 7, 190–205. [DOI] [PubMed] [Google Scholar]
- [116].Fujita H; Kato T; Watanabe N; Takahashi T; Kitagawa S Stimulation of human formyl peptide receptors by calpain inhibitors: homology modeling of receptors and ligand docking simulation. Arch. Biochem. Biophys, 2011, 516, 121–127. [DOI] [PubMed] [Google Scholar]
- [117].Matulenko MA; Hakeem AA; Kolasa T; Nakane M; Terranova MA; Uchic ME; Miller LN; Chang R; Donnelly-Roberts DL; Namovic MT; Moreland RB; Brioni JD; Stewart AO Synthesis and functional activity of (2-aryl-1-piperazinyl)-N-(3-methylphenyl)acetamides: selective dopamine D4 receptor agonists. Bioorg. Med. Chem, 2004, 12, 3471–3483. [DOI] [PubMed] [Google Scholar]
- [118].He M; Cheng N; Gao WW; Zhang M; Zhang YY; Ye RD; Wang MW Characterization of Quin-C1 for its anti-inflammatory property in a mouse model of bleomycin-induced lung injury. Acta Pharmacol. Sin, 2011, 32, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zhou C; Zhang S; Nanamori M; Zhang Y; Liu Q; Li N; Sun M; Tian J; Ye PP; Cheng N; Ye RD; Wang MW Pharmacological characterization of a novel nonpeptide antagonist for formyl peptide receptor-like 1. Mol. Pharmacol, 2007, 72, 976–983. [DOI] [PubMed] [Google Scholar]
- [120].Edwards BS; Bologa C; Young SM; Balakin KV; Prossnitz ER; Savchuck NP; Sklar LA; Oprea TI Integration of virtual screening with high-throughput flow cytometry to identify novel small molecule formylpeptide receptor antagonists. Mol. Pharmacol, 2005, 68, 1301–1310. [DOI] [PubMed] [Google Scholar]
- [121].Schepetkin IA; Kirpotina LN; Tian J; Khlebnikov AI; Ye RD; Quinn MT Identification of novel formyl peptide receptor-like 1 agonists that induce macrophage tumor necrosis factor α production. Mol. Pharmacol, 2008, 74, 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Khlebnikov AI; Schepetkin IA; Kirpotina LN; Quinn MT Computational structure-activity relationship analysis of non-peptide inducers of macrophage tumor necrosis factor-alpha production. Bioorg. Med. Chem, 2008, 16, 9302–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Pinilla C; Edwards BS; Appel JR; Yates-Gibbins T; Giulianotti MA; Medina-Franco JL; Young SM; Santos RG; Sklar LA; Houghten RA Selective agonists and antagonists of formylpeptide receptors: duplex flow cytometry and mixture-based positional scanning libraries. Mol. Pharmacol, 2013, 84, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Fujita H; Kato T; Watanabe N; Takahashi T; Kitagawa S Calpain inhibitors stimulate phagocyte functions via activation of human formyl peptide receptors. Arch. Biochem. Biophys, 2011, 513, 51–60. [DOI] [PubMed] [Google Scholar]
- [125].Dahinden C, J. F Receptor-directed inhibition of chemotactic factor-induced neutrophil hyperactivity by pyrazolon derivatives. Definition of a chemotactic peptide antagonist. J. Clin. Invest, 1980, 66, 884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Levesque L; Gaudreault RC; Marceau F The interaction of 3,5-pyrazolidinedione drugs with receptors for f-Met-Leu-Phe on human neutrophil leukocytes: a study of the structure-activity relationship. Can. J. Physiol. Pharmacol, 1991, 69, 419–425. [DOI] [PubMed] [Google Scholar]
- [127].Young SM; Bologa C; Prossnitz ER; Oprea TI; Sklar LA; Edwards BS High-throughput screening with HyperCyt flow cytometry to detect small molecule formylpeptide receptor ligands. J. Biomol. Screen, 2005, 10, 374–382. [DOI] [PubMed] [Google Scholar]
- [128].Del Maschio A; Livio M; Cerletti C; De Gaetano G Inhibition of human platelet cyclo-oxygenase activity by sulfinpyrazone and three of its metabolites. Eur. J. Pharmacol, 1984, 101, 209–214. [DOI] [PubMed] [Google Scholar]
- [129].Baus E; Urbain J; Leo O; Andris F Flow cytometric measurement of calcium influx in murine T cell hybrids using Fluo-3 and an organic-anion transport inhibitor. J. Immunol. Methods, 1994, 173, 41–47. [DOI] [PubMed] [Google Scholar]
- [130].Stenfeldt AL; Karlsson J; Wenneras C; Bylund J; Fu H; Dahlgren C The non-steroidal anti-inflammatory drug piroxicam blocks ligand binding to the formyl peptide receptor but not the formyl peptide receptor like 1. Biochem. Pharmacol, 2007, 74, 1050–1056. [DOI] [PubMed] [Google Scholar]
- [131].Colli S; Colombo S; Tremoli E; Stragliotto E; Nicosia S Effects of tenoxicam on superoxide anion formation, beta-glucuronidase release and fMLP binding in human neutrophils: comparison with other NSAIDs. Pharmacol. Res, 1991, 23, 367–379. [DOI] [PubMed] [Google Scholar]
- [132].Stenson WF; Mehta J; Spilberg I Sulfasalazine inhibition of binding of N-formyl-methionyl-leucyl-phenylalanine (FMLP) to its receptor on human neutrophils. Biochem. Pharmacol, 1984, 33, 407–412. [DOI] [PubMed] [Google Scholar]
- [133].Raiden S; Giordano M; Andonegui G; Trevani AS; Lopez DH; Nahmod V; Geffner JR Losartan, a selective inhibitor of subtype AT1 receptors for angiotensin II, inhibits the binding of N-formylmethionyl-leucyl-phenylalanine to neutrophil receptors. J. Pharmacol. Exp. Ther, 1997, 281, 624–628. [PubMed] [Google Scholar]
- [134].Yang SC; Chung PJ; Ho CM; Kuo CY; Hung MF; Huang YT; Chang WY; Chang YW; Chan KH; Hwang TL Propofol inhibits superoxide production, elastase release, and chemotaxis in formyl peptide-activated human neutrophils by blocking formyl peptide receptor 1. J. Immunol, 2013, 190, 6511–6519. [DOI] [PubMed] [Google Scholar]
- [135].Haeseler G; Karst M; Foadi N; Gudehus S; Roeder A; Hecker H; Dengler R; Leuwer M High-affinity blockade of voltage-operated skeletal muscle and neuronal sodium channels by halogenated propofol analogues. Br. J. Pharmacol, 2008, 155, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Medina-Franco JL; Edwards BS; Pinilla C; Appel JR; Giulianotti MA; Santos RG; Yongye AB; Sklar LA; Houghten RA Rapid scanning structure-activity relationships in combinatorial data sets: identification of activity switches. J. Chem. Inf. Model, 2013, 53, 1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Edwards BS; Young SM; Ivnitsky-Steele I; Ye RD; Prossnitz ER; Sklar LA High-content screening: flow cytometry analysis. Methods Mol. Biol, 2009, 486, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Strouse JJ; Young SM; Mitchell HD; Ye RD; Prossnitz ER; Sklar LA; Edwards BS A novel fluorescent cross-reactive formylpeptide receptor/formylpeptide receptor-like 1 hexapeptide ligand. Cytometry A, 2009, 75, 264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Edwards BS; Young SM; Oprea TI; Bologa CG; Prossnitz ER; Sklar LA Biomolecular screening of formylpeptide receptor ligands with a sensitive, quantitative, high-throughput flow cytometry platform. Nat. Protoc, 2006, 1, 59–66. [DOI] [PubMed] [Google Scholar]
- [140].Sugita A; Ogawa H; Azuma M; Muto S; Honjo A; Yanagawa H; Nishioka Y; Tani K; Itai A; Sone S Antiallergic and anti-inflammatory effects of a novel I kappaB kinase beta inhibitor, IMD-0354, in a mouse model of allergic inflammation. Int. Arch. Allergy Immunol, 2009, 148, 186–198. [DOI] [PubMed] [Google Scholar]
- [141].Li BX; Yamanaka K; Xiao X Structure-activity relationship studies of naphthol AS-E and its derivatives as anticancer agents by inhibiting CREB-mediated gene transcription. Bioorg. Med. Chem, 2012, 20, 6811–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Bae YS; Lee TG; Park JC; Hur JH; Kim Y; Heo K; Kwak JY; Suh PG; Ryu SH Identification of a compound that directly stimulates phospholipase C activity. Mol. Pharmacol, 2003, 63, 1043–1050. [DOI] [PubMed] [Google Scholar]
- [143].Horowitz LF; Hirdes W; Suh BC; Hilgemann DW; Mackie K; Hille B Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J. Gen. Physiol, 2005, 126, 243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Makishima M; Okamoto AY; Repa JJ; Tu H; Learned RM; Luk A; Hull MV; Lustig KD; Mangelsdorf DJ; Shan B Identification of a nuclear receptor for bile acids. Science, 1999, 284, 1362–1365. [DOI] [PubMed] [Google Scholar]
- [145].Kawamata Y; Fujii R; Hosoya M; Harada M; Yoshida H; Miwa M; Fukusumi S; Habata Y; Itoh T; Shintani Y; Hinuma S; Fujisawa Y; Fujino M A G protein-coupled receptor responsive to bile acids. J. Biol. Chem, 2003, 278, 9435–9440. [DOI] [PubMed] [Google Scholar]
- [146].Ferrari C; Macchiarulo A; Costantino G; Pellicciari R Pharmacophore model for bile acids recognition by the FPR receptor. J. Comput. Aided Mol. Des, 2006, 20, 295–303. [DOI] [PubMed] [Google Scholar]
- [147].Dalpiaz A; Ferretti ME; Vertuani G; Traniello S; Scatturin A; Spisani S C- and N-terminal residue effect on peptide derivatives' antagonism toward the formyl-peptide receptor. Eur. J. Pharmacol, 2002, 436, 187–196. [DOI] [PubMed] [Google Scholar]
- [148].Derian CK; Solomon HF; Higgins JD 3rd; Beblavy MJ; Santulli RJ; Bridger GJ; Pike MC; Kroon DJ; Fischman AJ Selective inhibition of N-formylpeptide-induced neutrophil activation by carbamate-modified peptide analogues. Biochemistry, 1996, 35, 1265–1269. [DOI] [PubMed] [Google Scholar]
- [149].Ou-Yang SS; Lu JY; Kong XQ; Liang ZJ; Luo C; Jiang H Computational drug discovery. Acta Pharmacol. Sin, 2012, 33, 1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Dong X; Ebalunode JO; Yang SY; Zheng W Receptor-based pharmacophore and pharmacophore key descriptors for virtual screening and QSAR modeling. Curr. Comput. Aided Drug Des, 2011, 7, 181–189. [DOI] [PubMed] [Google Scholar]
- [151].Gute BD; Basak SC Optimal neighbor selection in molecular similarity: comparison of arbitrary versus tailored prediction spaces. SAR QSAR. Environ. Res, 2006, 17, 37–51. [DOI] [PubMed] [Google Scholar]
- [152].Carhart RE; Smith DH; Venkataraghavan R Atom pairs as molecular features in structure activity studies - Definition and applications. J. Chem. Inf. Comput. Sci, 1985, 25, 64–73. [Google Scholar]
- [153].Khlebnikov AI; Schepetkin IA; Quinn MT Structure-activity relationship analysis of N-benzoylpyrazoles for elastase inhibitory activity: A simplified approach using atom pair descriptors. Bioorg. Med. Chem, 2008, 16, 2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Chen YF; Hsu KC; Lin PT; Hsu DF; Kristal BS; Yang JM LigSeeSVM: ligand-based virtual screening using support vector machines and data fusion. Int. J. Comput. Biol. Drug Des, 2011, 4, 274–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Khlebnikov AI; Schepetkin IA; Quinn MT Computational structure-activity relationship analysis of small-molecule agonists for human formyl peptide receptors. Eur. J. Med. Chem, 2010, 45, 5406–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Luu TT; Malcolm N; Nadassy K Pharmacophore modeling methods in focused library selection - applications in the context of a new classification scheme. Comb. Chem. High Throughput. Screen, 2011, 14, 488–499. [DOI] [PubMed] [Google Scholar]
- [157].Ashton M; Charlton MH; Schwarz MK; Thomas RJ; Whittaker M The selection and design of GPCR ligands: from concept to the clinic. Comb. Chem. High Throughput Screen, 2004, 7, 441–452. [DOI] [PubMed] [Google Scholar]
- [158].Kolaczkowski M; Bucki A; Feder M; Pawlowski M Ligand-optimized homology models of D1 and D2 dopamine receptors: Application for virtual screening. J. Chem. Inf. Model, 2013, 53, 638–648. [DOI] [PubMed] [Google Scholar]
- [159].Low CM; Vinter JG Rationalizing the activities of diverse cholecystokinin 2 receptor antagonists using molecular field points. J Med. Chem, 2008, 51, 565–573. [DOI] [PubMed] [Google Scholar]
- [160].Cramer RD; Patterson DE; Bunce JD Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc, 1988, 110, 5959–5967. [DOI] [PubMed] [Google Scholar]
- [161].Klebe G; Abraham U; Mietzner T Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem, 1994, 37, 4130–4146. [DOI] [PubMed] [Google Scholar]
- [162].Costanzi S; Tikhonova IG; Ohno M; Roh EJ; Joshi BV; Colson AO; Houston D; Maddileti S; Harden TK; Jacobson KA P2Y1 antagonists: combining receptor-based modeling and QSAR for a quantitative prediction of the biological activity based on consensus scoring. J. Med. Chem, 2007, 50, 3229–3241. [DOI] [PubMed] [Google Scholar]
- [163].Senderowitz H; Marantz Y G Protein-Coupled Receptors: target-based in silico screening. Curr. Pharm. Des, 2009, 15, 4049–4068. [DOI] [PubMed] [Google Scholar]
- [164].de Graaf C; Rognan D Customizing G Protein-coupled receptor models for structure-based virtual screening. Curr. Pharm. Des, 2009, 15, 4026–4048. [DOI] [PubMed] [Google Scholar]
- [165].Levit A; Barak D; Behrens M; Meyerhof W; Niv MY Homology model-assisted elucidation of binding sites in GPCRs. Methods Mol. Biol, 2012, 914, 179–205. [DOI] [PubMed] [Google Scholar]
- [166].Anighoro A; Rastelli G Enrichment factor analyses on G-protein coupled receptors with known crystal structure. J. Chem. Inf. Model, 2013, 53, 431–440. [DOI] [PubMed] [Google Scholar]
- [167].Phatak SS; Gatica EA; Cavasotto CN Ligand-steered modeling and docking: A benchmarking study in class A G protein-coupled receptors. J. Chem. Inf. Model, 2010, 50, 2119–2128. [DOI] [PubMed] [Google Scholar]
- [168].Costanzi S Modeling G protein-coupled receptors and their interactions with ligands. Curr. Opin. Struct. Biol, 2013, 23, 185–190. [DOI] [PubMed] [Google Scholar]
- [169].Yuan S; Ghoshdastider U; Trzaskowski B; Latek D; Debinski A; Pulawski W; Wu R; Gerke V; Filipek S The role of water in activation mechanism of human N-formyl peptide receptor 1 (FPR1) based on molecular dynamics simulations. PLoS One, 2012, 7, e47114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Wawer M; Bajorath J Extraction of structure-activity relationship information from high-throughput screening data. Curr. Med. Chem, 2009, 16, 4049–4057. [DOI] [PubMed] [Google Scholar]
- [171].Locke LW; Chordia MD; Zhang Y; Kundu B; Kennedy D; Landseadel J; Xiao L; Fairchild KD; Berr SS; Linden J; Pan D A novel neutrophil-specific PET imaging agent: cFLFLFKPEG-64Cu. J. Nucl. Med, 2009, 50, 790–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Zhang Y; Xiao L; Chordia MD; Locke LW; Williams MB; Berr SS; Pan D Neutrophil targeting heterobivalent SPECT imaging probe: cFLFLF-PEG-TKPPR-99m Tc. Bioconjug. Chem, 2010, 21, 1788–1793. [DOI] [PubMed] [Google Scholar]
- [173].Xiao L; Zhang Y; Liu Z; Yang M; Pu L; Pan D Synthesis of the Cyanine 7 labeled neutrophil-specific agents for noninvasive near infrared fluorescence imaging. Bioorg. Med. Chem. Lett, 2010, 20, 3515–3517. [DOI] [PubMed] [Google Scholar]
- [174].Stasiuk GJ; Smith H; Wylezinska-Arridge M; Tremoleda JL; Trigg W; Luthra SK; Morisson Iveson V; Gavins FN; Long NJ Gd3+ cFLFLFK conjugate for MRI: a targeted contrast agent for FPR1 in inflammation. Chem. Commun, 2013, 49, 564–566 [DOI] [PubMed] [Google Scholar]
- [175].Sadiq SK; Guixa-Gonzalez R; Dainese E; Pastor M; De Fabritiis G; Selent J Molecular modeling and simulation of membrane lipid-mediated effects on GPCRs. Curr. Med. Chem, 2013, 20(1), 22–38. [PubMed] [Google Scholar]