

Abstract
A short, enantioselective synthesis of (–)-nodulisporic acid C is described. The route features two highly diastereoselective polycyclizations en route to the terpenoid core and the indenopyran fragment and a highly convergent assembly of a challenging indole moiety. Application of this chemistry allows for a 12-step synthesis of the target indoloterpenoid from commercially available material.
Nodulisporic acids (e.g., 1, 2, and 3;Figure 1) comprise a group of secondary metabolites isolated from the endophytic fungus Hypoxylon pulicicidum (previously known as Nodulisporium sp.) that exhibit potent insecticidal activities.1–6 These biological effects arise from the specific activation of a subset of ligand-gated chloride ion channels found in arthropods, but not in mammals, resulting in a potentially useful selectivity profile.7 For example, the flagship congener, nodulisporic acid A (1), exhibits high efficacy against fleas while lacking overt toxicity in dogs.5,8 The producing organism has proven difficult to culture.9,10 Nevertheless, detailed investigations of nodulisporic acids as a starting point for the development of new antiflea medications for companion animals have been performed, resulting in identification of promising lead compounds.11 As a part of broader efforts toward the paxilline indoloterpenoids,12–14 multiple inquiries from organic chemists15 recently culminated in the syntheses of nodulisporic acids B (2), C (3), and D.16,17 In addition to the common indoloterpenoid core, these natural products contain an indenopyran motif found in other members of the paxilline family, such as the janthitrems and shearinines.18,19 Here we demonstrate a 12-step asymmetric synthesis of (–)-nodulisporic acid C (3) enabled by the development and implementation of new polycyclization-based strategies and a highly convergent assembly of a challenging indole moiety.
Figure 1.
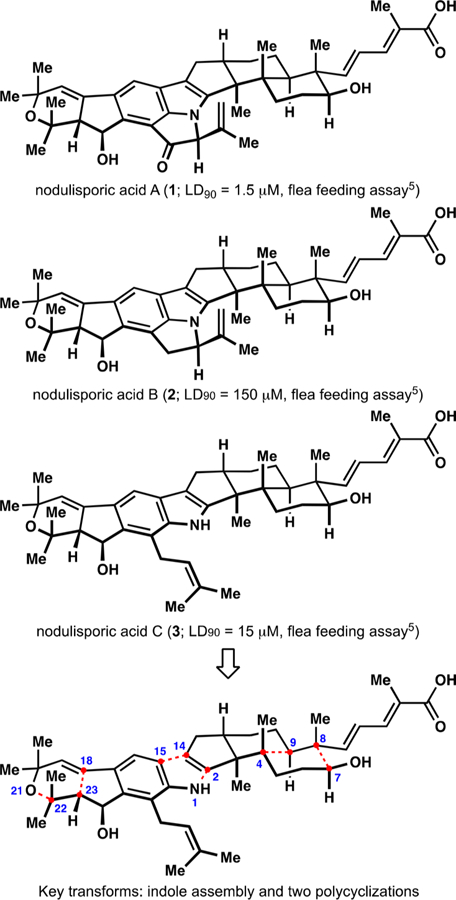
Representative nodulisporic acids and our analysis of nodulisporic acid C.
The key transforms in our retrosynthetic analysis of nodulisporic acid C (3) included a late-stage indole assembly to form the C14–C15 and N1–C2 bonds and two polycyclization events to access the terpenoid and indenopyran motifs. We envisioned a radical-polar crossover cascade initiated by a chemoselective20,21 hydrogen atom transfer (HAT)22–24 to construct the C4–C9 and C8–C7 bonds of the decalin fragment and a gold-catalyzed cycloisomerization25 to construct the C18–C23 and O21–C22 bonds of the dihydropyran fragment. We previously discovered a diastereoselective polycyclization related to the proposed HAT-initiated cascade, which relied on a transient lactolization event to control the relative orientation of the C3–C4 vicinal quaternary stereocenters.13 Implementation of this approach was expected to require multiple manipulations to accommodate both the assembly of the reactant and the late-stage installation of the indole moiety; therefore, an alternative solution was sought. The outcome of the polycyclization en route to the indenopyran motif was difficult to predict at the outset of our work.26,27
Our synthesis began with a copper-catalyzed asymmetric conjugate addition of alkylmagnesium bromide 5 to commercially available cyclopentenone 4 (Scheme 1). Extensive screening identified JosiPhos derivative SL-J015–1 as a suitable ligand for production of silyl enol ether 6 in excellent yield and with synthetically useful levels of enantioenrichment.28 Alkenylation of enoxysilane 6 with pentynol derivative 7 was best performed in the presence of substoichiometric amounts of indium(III) bromide and acidic workup secured access to product 8.13,29 Cyclopentanone 8 was converted to the corresponding protected cyanohydrin, which underwent Sharpless allylic oxidation30 and subsequent double oxidation of the resulting diol to deliver dialdehyde 9.
Scheme 1.
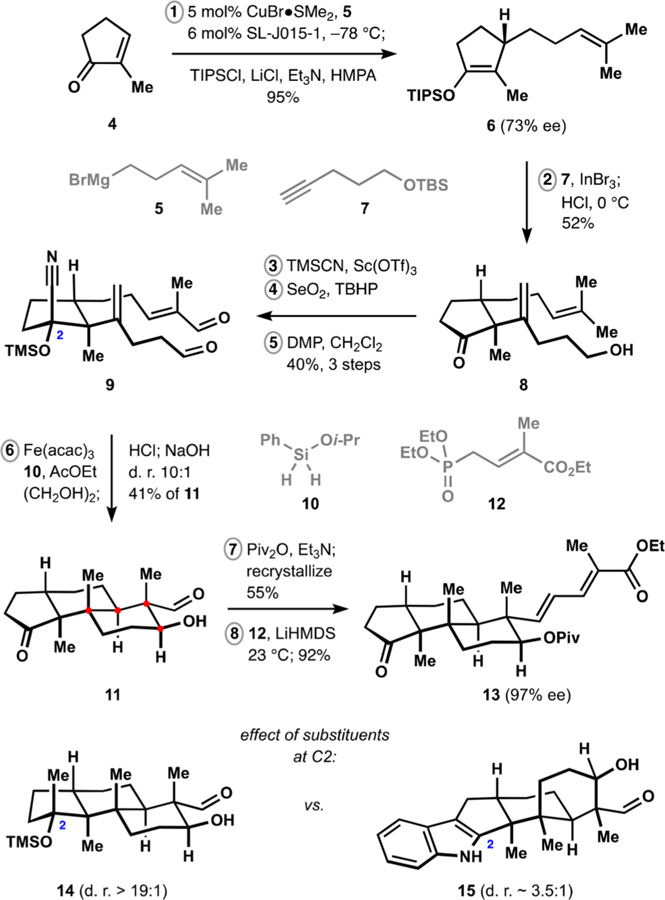
Synthesis of the Terpenoid Fragment
Subjection of intermediate 9 to the conditions of ironmediated HAT20,31 resulted in highly diastereoselective formation of the trans-decalin fragment and subsequent workup secured direct access to the cyclopentanone functionality of the desired tricyclic product 11. The pronounced preference for the trans-decalin motif represents an unusual case of efficient differentiation between methyl and linear alkyl substituents and is in stark contrast to the lack of diastereoselectivity observed with the corresponding unprotected cyclopentanone derivative.13 Presence of the pseudoaxial substituent at C2 proved crucial for obtaining high levels of diastereocontrol. Thus, only the trans-decalin is produced during the polycyclization to corresponding tert-alkyl ether 14 while low selectivity was previously observed in the case of the related sec-alkyl ether, which lacked the methyl substituent at C2.13 Furthermore, undesired cis-decalin 15 is favored during the polycyclization of the corresponding dialdehyde, where C2 is sp2 hybridized and the indole N–H group presumably exhibits unfavorable steric interactions en route to the trans-decalin product.
Protection of alcohol 11 delivered the corresponding pivalate, which could be obtained in a highly enantioenriched form (97% ee) upon recrystallization from hexanes. The pivalate was subjected to Horner–Wadsworth–Emmons olefination with phosphonate 12,32 providing access to ketone 13 in eight steps from commercially available material.
With the decalin-containing fragment of nodulisporic acid C in hand, we proceeded to assemble the indenopyran motif of the target natural product. We began with a three-step synthesis of arene 17 (Scheme 2), which included iodination of commercially available aniline 16, formylation of the intermediate iodide, and nitration of the resulting arene. Addition of diorganozinc 18 to aldehyde 17 was accomplished in the presence of catalytic amounts of aminoalcohol 19 and efficiently delivered benzyl alcohol 20 with high enantioselectivity.33 Sonogashira cross-coupling34 of dibromide 20 with alkyne 21 proceeded chemoselectively and the intermediate diol was protected to yield cycloisomerization precursor 22.
Scheme 2.
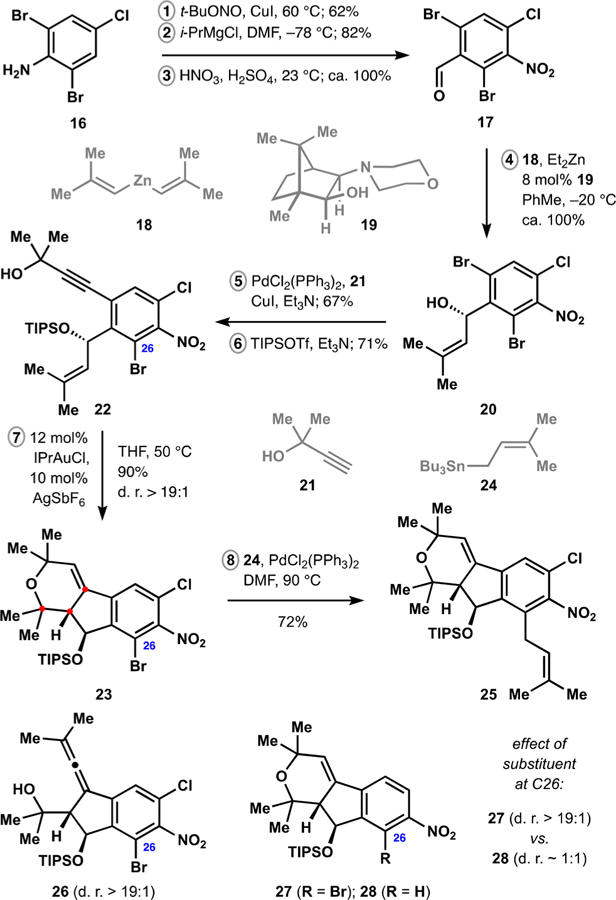
Synthesis of the Indenopyran Fragment
We discovered that treatment of enyne 22 with catalytic amounts of a gold(I) complex in the presence of a halide scavenger resulted in highly efficient and stereoselective formation of dihydropyran 23. The process likely involves the initial nucleophilic attack on the alkyne-gold complex by the isobutenyl fragment and subsequent trapping of the resulting tertiary carbocation with the pendant hydroxyl group. This proposal is supported by the observation of allene 26, which likely results from elimination of an alkenylgold intermediate26 and accumulates in the reaction mixture under anhydrous conditions. Neither dihydropyran 23 nor allene 26 is observed upon treatment of alkyne 22 with acid, but these conditions are competent in converting intermediate 26 to the desired tricyclic product. The presence of the substituent at C26 proved to be crucial for obtaining high levels of stereocontrol during the reaction. Thus, products 23 and 27 are formed as single diastereomers in contrast to dihydropyran 28. Allylic strain between the benzyl ether and the ortho substituent is expected to force rotation of the silyl group35 toward the reaction center in the transition state of the initial cyclization, which can lead to the dramatic enhancement of the diastereoselectivity en route to products 23 and 27. This proposal is consistent with the observed restricted rotation of the siloxy-bearing fragment in the ortho-substituted substrates.
Installation of the prenyl group into the aromatic moiety was best accomplished after the cycloisomerization. Thus, Stille cross-coupling36 of bromide 23 with stannane 24 delivered chloroarene 25 in good yield, completing assembly of the indenopyran motif in eight steps from commercially available starting material and setting the stage for the fragment union.
After significant experimentation, we discovered that a trialkyltin enolate of ketone 13 could undergo a palladiumcatalyzed arylation with nearly stoichiometric quantities of chloroarene 25 (Scheme 3). A RuPhos-based palladium precatalyst37 provided optimal performance and the arylketone was produced in a diastereoselective manner.38 The stereochemical outcome of the reaction proved crucial for the successful completion of the synthesis. Treatment of arylation product 30 with zinc in the presence of acetic acid resulted in efficient formation of the desired indole under mild conditions. In this setting, diastereomer 29 returned the corresponding aminoketone and attempted cyclodehydration under forcing conditions led to elimination of the benzyl ether moiety. The sensitive nature of the indenopyran fragment was previously highlighted in the isolation and synthetic literature.3,17 The facility of conversion of ketone 30 to the desired indole is in stark contrast to the previous reports of cyclodehydrations of relevant aminoketones39 and can be instructive during synthetic planning toward the other paxilline indoloterpenoids. Final manipulations in our synthesis of (–)-nodulisporic acid C (3) included deprotection of the silyl ether and saponification of the intermediate diester, which delivered the natural product in 12 steps from commercially available material (longest linear sequence).
Scheme 3.
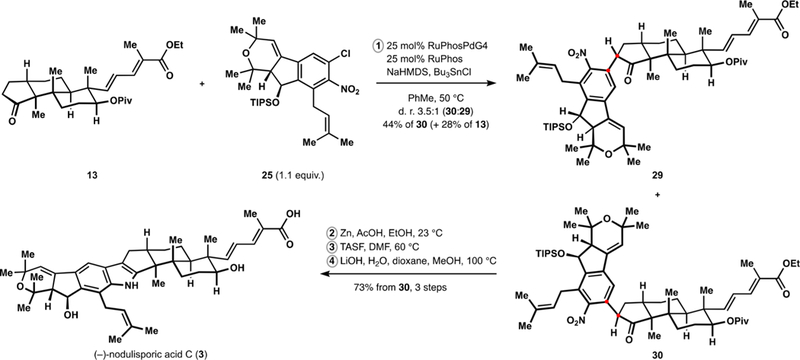
Synthesis of (–)-Nodulisporic acid C (3)
In summary, we disclose an enantioselective synthesis of (–)-nodulisporic acid C that proceeds in a highly convergent manner. The relative brevity of our route was enabled by the development of two polycyclizations to rapidly assemble the decalin and indenopyran motifs and a ketone arylation protocol to unite the two complex fragments. In these processes, seemingly minor structural modifications within the crowded steric environments have a significant influence on the reactivity and have allowed for complete stereocontrol over the polycyclization events and facile construction of a sensitive indole fragment. The preponderance of these and related structural motifs among the paxilline indoloterpenoids suggests that lessons learned during our synthesis will find application in the assembly of other members of this fascinating family and simplify production of unnatural analogs for biological studies.
ACKNOWLEDGMENTS
Financial support from the National Institutes of Health (R01GM121678), the University of California Irvine, the Hellman Foundation, the National Science Foundation (DGE-1321846 to N.A.G.), and the Natural Sciences and Engineering Research Council of Canada (PGSD3-487506-2016 to D.J.S.) is gratefully acknowledged. We thank Dr. David George for preparation of compound 15 and Dr. Joseph Ziller and Austin Ryan for X-ray crystallographic analysis. We also thank Professors Larry Overman, Chris Vanderwal, and Scott Rychnovsky for providing routine access to their instrumentation and helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Experimental procedures and spectroscopic data for new compounds (PDF)
- CIF file for compound 15 (CIF)
- CIF file for compound 23 (CIF)
- CIF file for compound 26 (CIF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Ondeyka JG; Helms GL; Hensens OD; Goetz MA; Zink DL; Tsipouras A; Shoop WL; Slayton L; Dombrowski AW; Polishook JD; Ostlind DA; Tsou NN; Ball RG; Singh SB J. Am. Chem. Soc 1997, 119, 8809–8816. [Google Scholar]
- (2).Hensens OD; Ondeyka JD; Dombrowski AW; Ostlind DA; Zink DL Tetrahedron Lett 1999, 40, 5455–5458. [Google Scholar]
- (3).Ondeyka JG; Dahl-Roshak AM; Tkacz JS; Zink DL; Zakson-Aiken M; Shoop WL; Goetz MA; Singh SB Bioorg. Med. Chem. Lett 2002, 12, 2941–2944. [DOI] [PubMed] [Google Scholar]
- (4).Ondeyka JG; Byrne K; Vesey D; Zink DL; Shoop WL; Goetz MA; Singh SB J. Nat. Prod 2003, 66, 121–124. [DOI] [PubMed] [Google Scholar]
- (5).Singh SB; Ondeyka JG; Jayasuriya H; Zink DL; Ha SN; Dahl-Roshak A; Greene J; Kim JA; Smith MM; Shoop WL; Tkacz JS J. Nat. Prod 2004, 67, 1496–1506. [DOI] [PubMed] [Google Scholar]
- (6).Bills GF; González-Menéndez V; Martín J; Platas G; Fournier J; Peršoh D; Stadler M PLoS One 2012, 7, No. e46687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ludmerer SW; Warren VA; Williams BS; Zheng Y; Hunt DC; Ayer MB; Wallace MA; Chaudhary AG; Egan MA; Meinke PT; Dean DC; Garcia ML; Cully DF; Smith MM Biochemistry 2002, 41, 6548–6560. [DOI] [PubMed] [Google Scholar]
- (8).Shoop WL; Gregory LM; Zakson-Aiken M; Michael BF; Haines HW; Ondeyka JG; Meinke PT; Schmatz DM J. Parasitol 2001, 87, 419–423. [DOI] [PubMed] [Google Scholar]
- (9).Byrne KM; Smith SK; Ondeyka JG J. Am. Chem. Soc 2002, 124, 7055–7060. [DOI] [PubMed] [Google Scholar]
- (10).Van de Bittner KC; Nicholson MJ; Bustamante LY; Kessans SA; Ram A; van Dolleweerd CJ; Scott B; Parker EJ J. Am. Chem. Soc 2018, 140, 582–585. [DOI] [PubMed] [Google Scholar]
- (11).Meinke PT; Colletti SL; Fisher MH; Wyvratt MJ; Shih TL; Ayer MB; Li C; Lim J; Ok D; Salva S; Warmke LM; Zakson M; Michael BF; deMontigny P; Ostlind DA; Fink D; Drag M; Schmatz DM; Shoop WL J. Med. Chem 2009, 52, 3505–3515. [DOI] [PubMed] [Google Scholar]
- (12).(a) Smith AB III; Mewshaw RJ Am. Chem. Soc 1985, 107, 1769–1771. [Google Scholar]; (b) Mewshaw R; Taylor MD; Smith AB III J. Org. Chem 1989, 54, 3449–3462. [Google Scholar]; (c) Smith AB III; Sunazuka T; Leenay TL; Kingery-Wood JJ Am. Chem. Soc 1990, 112, 8197– 8198. [Google Scholar]; (d) Smith AB III; Kingery-Wood J; Leenay TL; Nolen EG; Sunazuka TJ Am. Chem. Soc 1992, 114, 1438–1449. [Google Scholar]; (e) Smith AB III; Kanoh N; Ishiyama H; Hartz RA J. Am. Chem. Soc 2000, 122, 11254–11255. [Google Scholar]; (f) Smith AB III; Kanoh N; Ishiyama H; Minakawa N; Rainier JD; Hartz RA; Cho YS; Cui H; Moser WM J. Am. Chem. Soc 2003, 125, 8228–8237. [DOI] [PubMed] [Google Scholar]; (g) Smith AB III; Cui H Org. Lett 2003, 5, 587–590. [DOI] [PubMed] [Google Scholar]; (h) Smith AB III; Cui H Helv. Chim. Acta 2003, 86, 3908–3938. [Google Scholar]; (i) Enomoto M; Morita A; Kuwahara S Angew. Chem., Int. Ed 2012, 51, 12833–12836. [DOI] [PubMed] [Google Scholar]; (j) Teranishi T; Murokawa T; Enomoto M; Kuwahara S Biosci., Biotechnol., Biochem 2015, 79, 11–15. [DOI] [PubMed] [Google Scholar]; (k) Sharpe RJ; Johnson JS J. Am. Chem. Soc 2015, 137, 4968–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Sharpe RJ; Johnson JS J. Org. Chem 2015, 80, 9740–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).George DT; Kuenstner EJ; Pronin SV J. Am. Chem. Soc 2015, 137, 15410–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For recent reviews, see: (a) Corsello MA; Kim J; Garg NK Chem. Sci 2017, 8, 5836–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zou Y; Smith AB III. J. Antibiot 2018, 71, 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Magnus P; Mansley TE Tetrahedron Lett 1999, 40, 6909–6912. [Google Scholar]; (b) Smith AB III; Ishiyama H; Cho YS; Ohmoto K Org. Lett 2001, 3, 3967–3970. [DOI] [PubMed] [Google Scholar]; (c) Smith AB III; Cho YS; Ishiyama H Org. Lett 2001, 3, 3971–3974. [DOI] [PubMed] [Google Scholar]; (d) Smith AB III; Davulcu AH; Kürti L Org. Lett 2006, 8, 1665–1668. [DOI] [PubMed] [Google Scholar]; (e) Smith AB III; Davulcu AH; Kürti L Org. Lett 2006, 8, 1669–1672. [DOI] [PubMed] [Google Scholar]; (f) Smith AB III; Kürti L; Davulcu AH Org. Lett 2006, 8, 2167–2170. [DOI] [PubMed] [Google Scholar]; (g) Smith AB III; Kürti L; Davulcu AH; Cho YS Org. Process Res. Dev 2007, 11, 19–24. [Google Scholar]; (h) Smith AB III; Davulcu AH; Cho YS; Ohmoto K; Kürti L; Ishiyama HJ Org. Chem 2007, 72, 4596–4610. [DOI] [PubMed] [Google Scholar]; (i) Smith AB III; Kürti L; Davulcu AH; Cho YS; Ohmoto KJ Org. Chem 2007, 72, 4611–4620. [DOI] [PubMed] [Google Scholar]
- (16).Zou Y; Melvin JE; Gonzales SS; Spafford MJ; Smith AB III. J. Am. Chem. Soc 2015, 137, 7095–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zou Y; Li X; Yang Y; Berritt S; Melvin J; Gonzales S; Spafford M; Smith AB III. J. Am. Chem. Soc 2018, 140, 9502– 9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) de Jesus AE; Steyn PS; van Heerden FR; Vleggaar RJ Chem. Soc., Perkin Trans. 1 1984, 0, 697–701. [Google Scholar]; (b) Wilkins AL; Miles CO; Ede RM; Gallagher RT; Munday SC J. Agric. Food Chem 1992, 40, 1307–1309. [Google Scholar]
- (19).(a) Belofsky GN; Gloer JB; Wicklow DT; Dowd PF Tetrahedron 1995, 51, 3959–3968. [Google Scholar]; (b) Xu M; Gessner G; Groth I; Lange C; Christner A; Bruhn T; Deng Z; Li X; Heinemann SH; Grabley S; Bringmann G; Sattler I; Lin W Tetrahedron 2007, 63, 435–444. [Google Scholar]
- (20).(a) Lo JC; Yabe Y; Baran PS J. Am. Chem. Soc 2014, 136, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC; Gui J; Yabe Y; Pan C-M; Baran PS Nature 2014, 516, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lo JC; Kim D; Pan C-M; Edwards JT; Yabe Y; Gui J; Qin T; Gutiérrez S; Giacoboni J; Smith MW; Holland PL; Baran PS J. Am. Chem. Soc 2017, 139, 2484–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yan M; Lo JC; Edwards JT; Baran PS J. Am. Chem. Soc 2016, 138, 12692–12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).:Mukaiyama T; Yamada T Bull. Chem. Soc. Jpn 1995, 68, 17.and references therein.For a recent review, see: (b) Crossley SWM; Martinez RM; Obradors C; Shenvi RA Chem. Rev 2016, 116, 8912–9000.Shenvi and Herzon propose HAT as the initial step in their related hydrogenations: (c) Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA J. Am. Chem. Soc 2014, 136, 1300.(d) King SM; Ma X; Herzon SB J. Am. Chem. Soc 2014, 136, 6884.For similar mechanistic considerations, see: (e) Eisenberg DC; Norton JR Isr. J. Chem 1991, 31, 55–66.
- (23).For relevant aldol reactions, see: Isayama S; Mukaiyama T Chem. Lett 1989, 18, 2005–2008. [Google Scholar]
- (24).For recent examples of HAT-initiated cyclizations in synthesis, see: (a) Deng H; Cao W; Liu R; Zhang Y; Liu B Angew. Chem., Int. Ed 2017, 56, 5849–5852. [DOI] [PubMed] [Google Scholar]; (b) Lu Z; Zhang X; Guo Z; Chen Y; Mu T; Li AJ Am. Chem. Soc 2018, 140, 9211–9218. [DOI] [PubMed] [Google Scholar]; It is plausible that the peroxyradical cyclization en route to (+)-cardamom peroxide is initiated via HAT: Hu X; Maimone TJ J. Am. Chem. Soc 2014, 136, 5287–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).For recent examples of gold-catalyzed cascades in synthesis, see: (a) Yue G; Zhang Y; Fang L; Li C.-c.; Luo T; Yang Z Angew. Chem., Int. Ed 2014, 53, 1837–1840. [DOI] [PubMed] [Google Scholar]; (b) McGee P; Bétournay G; Barabé F; Barriault L Angew. Chem., Int. Ed 2017, 56, 6280–6283. [DOI] [PubMed] [Google Scholar]; (c) Ferrer S; Echavarren AM Org. Lett 2018, 20, 5784–5788. [DOI] [PubMed] [Google Scholar]
- (26).Harrak Y; Simonneau A; Malacria M; Gandon V; Fensterbank L Chem. Commun 2010, 46, 865–867. [DOI] [PubMed] [Google Scholar]
- (27).Sanjuan AM; Martínez A; García-García P; Fernandeź-Rodríguez MA; Sanz R Beilstein J. Org. Chem 2013, 9, 2242–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).For relevant asymmetric cuprate additions, see: Calvo BC; Madduri AVR; Harutyunyan SR; Minnaard AJ Adv. Synth. Catal 2014, 356, 2061–2069. [Google Scholar]
- (29).In this setting, the corresponding catalytic alkenylation proved less efficient: Holmbo SD; Godfrey NA; Hirner JJ; Pronin SV J. Am. Chem. Soc 2016, 138, 12316–12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Umbreit MA; Sharpless KB J. Am. Chem. Soc 1977, 99,5526. [Google Scholar]
- (31).Obradors C; Martinez RM; Shenvi RA J. Am. Chem. Soc 2016, 138, 4962–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32). Prepared in two steps from ethyl tiglate as described in SI.
- (33).(a) Nugent WA Chem. Commun 1999, 35, 1369–1370. [Google Scholar]; (b) Lurain AE; Maestri A; Kelly AR; Carroll PJ; Walsh PJ J. Am. Chem. Soc 2004, 126, 13608–13609. [DOI] [PubMed] [Google Scholar]
- (34).Sonogashira KJ Organomet. Chem 2002, 653, 46–49. and references therein. [Google Scholar]
- (35).Wilcox CS; Babston RE J. Org. Chem 1984, 49, 1451–1453. [Google Scholar]
- (36).Stille JK Pure Appl. Chem 1985, 57, 1771–1780. and references therein. [Google Scholar]
- (37).Bruno NC; Niljianskul N; Buchwald SL J. Org. Chem 2014, 79, 4161–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38). Relative configuration of the newly formed stereocenter was established by NOE experiments. See SI for details.
- (39). See references 12a–c, k, l.