

Abstract
A catalytic formal ene reaction between ketone-derived silyl enol ethers and terminal alkynes is described. This transformation is uniquely capable of bimolecular assembly of 2-siloxy-1,4-dienes and can be used to access β,γ-unsaturated ketones containing quaternary carbons in the α-position.
Intermolecular alkenylation of enolates or their surrogates offers direct and convenient access to β,γ-unsaturated ketones, versatile building blocks that contain two orthogonal functionalization sites. Over 30 years ago, Migita reported pioneering examples of such sp2–sp3 coupling processes, which relied on a Pd(0)-catalyzed alkenylation of in situ-generated tri-n-butyltin enolates with substituted vinyl bromides.1 Subsequent works from other laboratories further demonstrated the power of Pd(0)- and Ni(0)-catalyzed alkenylations in the synthesis of β,γ-unsaturated ketones,2–8 although these reactions are typically limited to aryl alkyl or alkenyl alkyl ketones. Intermolecular alkenylations of dialkyl ketones bearing two unactivated enolizable positions are much less represented.9,10 In this context, selective formation of quaternary centers poses a particular challenge, and only isolated examples can be found in the literature.7,11 It is noteworthy that analogous intramolecular alkenylations are much more common and have found a number of applications in synthesis, likely owing to high levels of regiocontrol enforced by kinetic selectivity during cyclization.12 Here we demonstrate a new catalytic formal ene reaction between silyl enol ethers and terminal alkynes that is uniquely capable of bimolecular assembly of 2-siloxy-1,4-dienes and can be used to access β,γ-unsaturated ketones containing quaternary carbons in the α-position.
During our work on the synthesis of paxilline indole diterpenes, we identified an indium(III) bromide-mediated alkenylation of a silyl enol ether with a terminal alkyne as suitable means for assembly of the requisite β,γ-unsaturated ketone.13 We became intrigued by the idea of performing intermolecular alkenylations in a catalytic fashion with retention of the silyl enol ether functionality in the product.14 Electrophilic activation of alkynes toward nucleophilic attack by silyl enol ethers has found extensive application in α alkenylation of ketones. In his pioneering work, Conia described the first, to our knowledge, examples of such reactivity.15 A number of reports of related Al(III)-mediated16 and transition-metal-catalyzed17–22 intramolecular alkenylations of ketone-derived enoxysilanes followed in subsequent years, including some asymmetric variants.23 However, relevant intermolecular alkenylations of silyl enol ethers find very little precedent,13,24,25 and the only available catalytic intermolecular process requires the use of 1,3-dicarbonyls as activated precursors26 (Scheme 1). In the latter example, facile enolization of the substrates enables the ene-like reaction with alkynes. A similar process between ketone-derived silyl enol ethers and alkynes is considerably more challenging, as it requires a net proton transfer from the allylic position of the enoxysilane to the former alkyne fragment that has never been observed in a bimolecular setting.27 Furthermore, the propensity of silyl enol ethers derived from unsymmetrical dialkyl ketones to isomerize under acidic conditions28,29 poses a potential issue of site selectivity. Finally, the product of the alkenylation, a 2-siloxy-1,4-diene, would itself represent a substrate for the reaction, posing an inherent issue of chemoselectivity. In particular, application of such a reaction to the selective construction of quaternary centers would require preferential alkenylation of fully substituted enoxysilane starting materials in the presence of less substituted enoxysilane products.
Scheme 1.
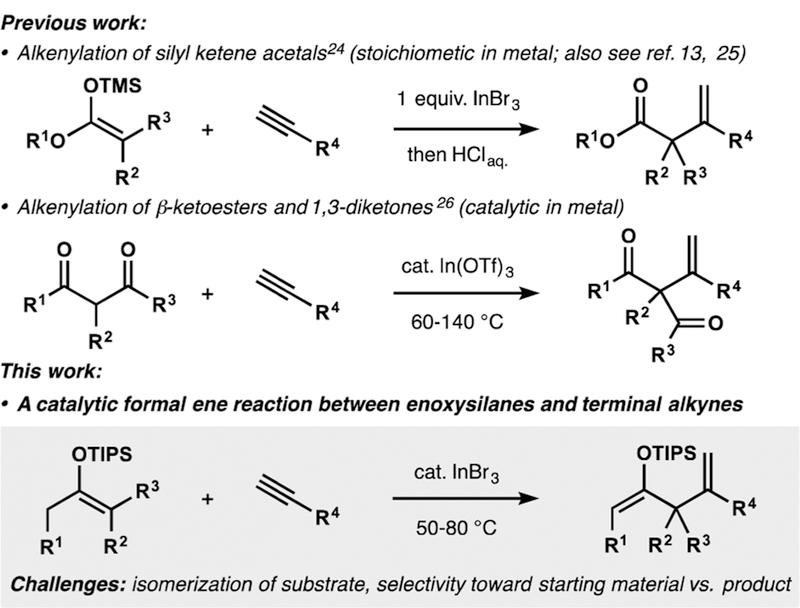
Comparison of Relevant Intermolecular Alkenylations with Current Work
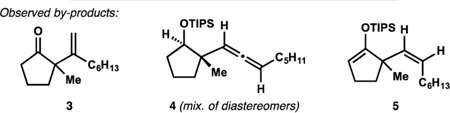
With these considerations in mind, we set out to investigate the catalytic formal ene reaction between silyl enol ether 1 and 1-octyne (Table 1; see Supporting Information (SI) for additional details). Gold(I) complexes19 proved to be poor catalysts for the desired transformation with the best-performing combination of a NHC-derived precatalyst and a halidescavenging additive providing only small amounts of diene 2 and the hydrolyzed product 3 (entry 1). Application of zinc(II) bromide22 was unsuccessful (entry 2). Conditions similar to those previously employed by Nakamura26 afforded only traces of enoxysilane 2 (entry 3). However, changing the counter-anion in the indium(III)-based catalyst produced instructive trends. While chloride (entry 4) offered only marginal improvement over triflate, bromide (entry 5) allowed for the efficient alkenylation of substrate 1, and only small amounts of ketone 3, allene 4,30 and regioisomer 5 were observed. Application of indium(III) iodide and introduction of halide-scavenging additives were less efficient (entries 6 and 7). Catalytic alkenylation in the presence of indium(III) bromide could be performed at temperatures as low as 50 °C, which decreased the content of allene 4 and regioisomer 5 (entry 8). Attempted alkenylation in the presence of catalytic amounts of hydrogen bromide (generated in situ, entry 9) as well as triflic acid (entry 10) did not lead to product formation, confirming the catalytic role of the metal derivative. The robust TIPS-enol ethers31 performed better than more labile TMS and TBS derivatives. Interestingly, attempted alkenylation of isomeric silyl enol ether 6 afforded siloxydiene 2 as a major product, suggesting isomerization of 6 to 1 under the reaction conditions.
Table 1.
Development of the Catalytic Formal Ene Reaction
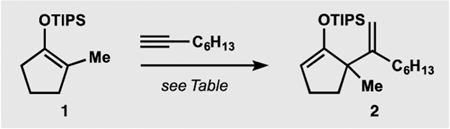 | |||||
 | |||||
| Entry | Conditionsa | 2(%)b | 3(%)b | 4(%)b,c | 5(%)b |
| 1 | 5 mol%IPrAuCI,5 mol% NaBArF, 65℃ | 11 | 8 | 10 | <1 |
| 2 | 10 mol% ZnBr2, 65℃ | 4 | 5 | 1 | <1 |
| 3 | 5 mol% In(OTf)3, 65℃ | <1 | 1 | <1 | <1 |
| 4 | 5 mol% InCI3, 65℃ | 5 | 9 | <1 | <1 |
| 5 | 5 mol% InBr3, 65℃ | 72 | 5 | 6 | 2 |
| 6 | 5 mol% InI3, 65℃ | 36 | 6 | 7 | <1 |
| 7 | 5 mol% InBr3, 5 mol% NaBArF,65℃ | 40 | 11 | 3 | <1 |
| 8d | 5 mol% InBr3,50℃ | 65 | 9 | 3 | <1 |
| 9 | 5 mol% MeOH, 8 mol% TMSBr, 65℃ | <1 | <1 | <1 | <1 |
| 10 | 5 mol% TfOH, 65℃ | <1 | <1 | <1 | <1 |
| Attempted alkenylation of silyl enol ether 6: | |||||
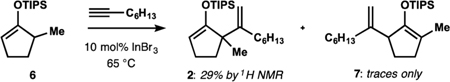 | |||||
0.25–0.5 mmol of 1, 1.5 equiv of alkyne, 0.25–0.5 mL of (CH2Cl)2, 20–28 h.
Based on internal standard and determined by 1H NMR.
dr = 1:1.
65% isolated yield of 2.
It is important to note that the product of double alkenylation was not observed during these studies. Alkenylation of the fully substituted enoxysilane 1 must therefore proceed considerably faster under our conditions than alkenylation of product 2. Although speculatively, we attribute the observed selectivity to the difference in the conformational preferences of the bulky siloxy group.32 To demonstrate the application of such unusual selectivity, the synthesis of diketone 9 was undertaken (Scheme 2). Starting diketone 8 contains three reactive α-positions, C1, C8, and C10, and selective alkenylation at C8 represents a considerable challenge. To our delight, subjection of the bis-silyl enol ether derived from diketone 8 to our alkenylation conditions resulted in a selective reaction of the fully substituted siloxyalkene fragment, delivering monoalkenylated product 9 after a mild hydrolytic workup.
Scheme 2.
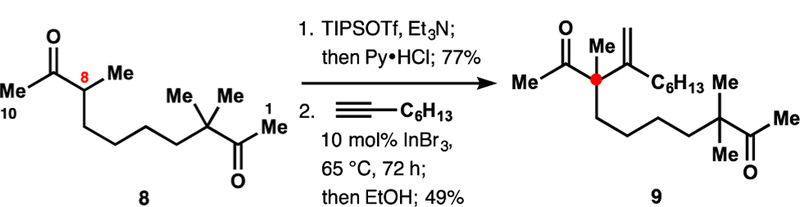
Synthesis of Diketone 9
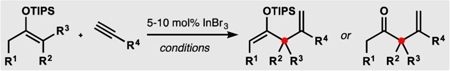
With a functional set of conditions in hand, we investigated the preliminary scope of this catalytic formal ene reaction. We found that alkyl acetylenes with aromatic, halide, and ether functionalities as well as conjugated alkynes successfully participate in the alkenylation process (dienes 10–14 in Table 2). The reaction also tolerated a Lewis basic ester group (diene 15), although more forcing conditions were required in this case.33 Trimethylsilylacetylene and internal alkynes did not participate in the alkenylation. The formal ene reaction was effective at forming quaternary centers containing four non-methyl substituents (dienes 16 and 17), and various substitution patterns of the cyclopentene ring were tolerated (dienes 18–21). Synthesis of dienes 19–21 demonstrates a convenient approach to diastereoselective vicinal difunctionalization of a simple alkenone34 and can be useful in the context of natural product synthesis.13 Cyclohexanone-derived silyl enol ethers also underwent successful alkenylation, and corresponding ketones 22 and 23 could be readily isolated after a mild hydrolytic workup.35 Furthermore, acyclic silyl enol ethers were suitable substrates for this formal ene reaction (e.g., see diene 24). Because the resulting 2-siloxy-1,4-dienes often proved hydrolytically unstable relative to their cyclic counterparts, the reaction mixtures were subjected to a mild hydrolytic workup, delivering the corresponding β,γ-unsaturated ketones (products 25–30). In almost all cases, the desired 1,1-disubstituted alkenes were produced in a highly regioselective manner, and only small quantities of corresponding 1,2-disubstituted isomers were observed.
Table 2.
Preliminary Substrate Scope of the Alkenylationa
 |
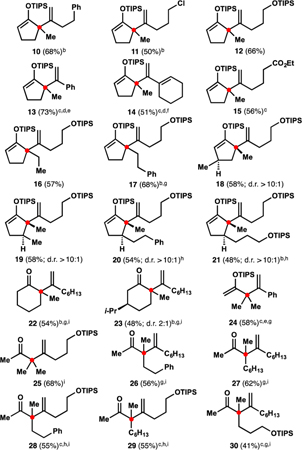 |
Typically, starting silyl enol ethers were mixtures of isomers; see SI for details. 1 equiv of silyl enol ether, 1.5 equiv of alkyne, 10 mol% of InBr3, (CH2Cl)2 (1 M in enoxysilane), 65 °C, ca. 24 h; rr ≥ 10:1 (except 15, rr 9:1; 24, rr 7:1). Siloxydienes (except 13, 14, and 24) contained ca. 5 mol% of inseparable allenes.
Heated to 50 °C.
Heated to 80 °C.
5 mol% of InBr3.
Neat. f5 M in silyl enol ether.
Heated for ca. 72 h.
Heated for ca. 48 h.
After hydrolysis.
We propose that the mechanism of this catalytic formal ene reaction involves initial nucleophilic attack of enoxysilane 1 on the alkyne–indium(III) bromide complex36 to form alkenylindium 31 (or the corresponding divinylindium derivative; Scheme 3).37 The formation and stereochemistry of intermediate 31 or related species are supported by observation of deuterated ketone 34 upon treatment of the reaction mixture with CD3OD.34 Similar alkenylindium derivatives have been previously isolated and characterized by Baba.24 Intermediate 31 can undergo protodemetalation with indium(III) bromide–ketone complex 32 to produce ketone 3 and enolate 33, respectively.26,38 Participation of ketone 3, which is formed early and whose concentration remains constant during the reaction,39 is supported by the observation that an additive of ketone 35 undergoes silylation in situ to form enoxysilane 36.34 Subsequent silylation of enolate 33 is expected to afford the desired siloxydiene 2.
Scheme 3.
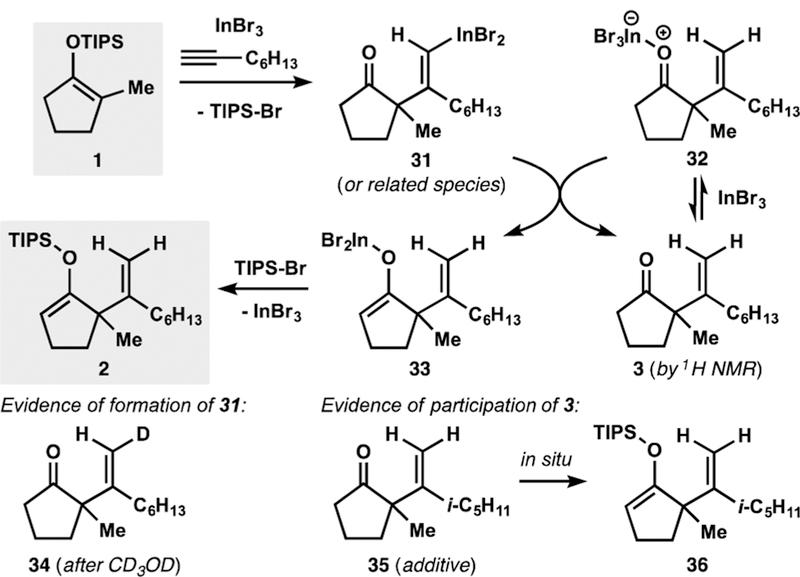
Proposed Mechanism of the Alkenylation
In summary, we disclose a new catalytic intermolecular ene reaction between ketone-derived silyl enol ethers and terminal alkynes. Among the features of this alkenylation are selective formation of highly sterically encumbered quaternary centers and direct access to 2-siloxy-1,4-dienes and corresponding β,γ-unsaturated ketones with synthetically challenging substitution patterns. The reaction has its limitations and future work will focus on expanding the substrate scope and further improving the efficiency of the process. However, this transformation has no alternatives among the current methods and can be expected to find broad application in natural product synthesis.
ACKNOWLEDGMENTS
Funding from the School of Physical Sciences at the University of California Irvine, Chao Family Comprehensive Cancer Center, and the National Science Foundation (DGE-1321846 to N.A.G.) is gratefully acknowledged. We thank Profs. Larry Overman, Chris Vanderwal, and Suzanne Blum for providing access to their instrumentation and helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b06847.
Detailed experimental procedures and characterization data for new compounds (PDF)
REFERENCES
- (1).(a) Kosugi M; Hagiwara I; Migita T Chem. Lett 1983, 12, 839–841. For application in synthesis see: [Google Scholar]; (b) Gracia J; Thomas EJJ Chem. Soc., Perkin Trans. 1 1998, 1, 2865–2871. [Google Scholar]; (c) Almendros P; Rae A; Thomas EJ Tetrahedron Lett 2000, 41, 9565–9568. [Google Scholar]
- (2).(a) Chieffi A; Kamikawa K; Åhman J; Fox JM; Buchwald SL Org. Lett 2001, 3, 1897–1900. [DOI] [PubMed] [Google Scholar]; (b) Hamada T; Buchwald SL Org. Lett 2002, 4, 999–1001. For application in synthesis see: [DOI] [PubMed] [Google Scholar]; (c) Riou M; Barriault LJ Org. Chem 2008, 73, 7436–7439. [DOI] [PubMed] [Google Scholar]
- (3).Huang J; Bunel E; Faul MM Org. Lett 2007, 9, 4343–4346. [DOI] [PubMed] [Google Scholar]
- (4).Lou S; Fu GC J. Am. Chem. Soc 2010, 132, 5010–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Cosner CC; Helquist P Org. Lett 2011, 13, 3564–3567. [DOI] [PubMed] [Google Scholar]; (b) Grigalunas M; Ankner T; Norrby P-O; Wiest O; Helquist P Org. Lett 2014, 16, 3970–3973. [DOI] [PubMed] [Google Scholar]; (c) Grigalunas M; Ankner T; Norrby P-O; Wiest O; Helquist PJ Am. Chem. Soc 2015, 137, 7019–7022. [DOI] [PubMed] [Google Scholar]; (d) Grigalunas M; Norrby P-O; Wiest O; Helquist P Angew. Chem., Int. Ed 2015, 54, 11822–11825. [DOI] [PubMed] [Google Scholar]
- (6).Su W; Raders S; Verkade JG; Liao X; Hartwig JF Angew. Chem., Int. Ed 2006, 45, 5852–5855. [DOI] [PubMed] [Google Scholar]
- (7).For a relevant Heck reaction of alkyl vinyl ethers see: Datta GK; Larhed M Org. Biomol. Chem 2008, 6, 674–676. [DOI] [PubMed] [Google Scholar]
- (8).For a recent review of alkenylation of enolates see: Ankner T; Cosner CC; Helquist P Chem. - Eur. J 2013, 19, 1858–1871. [DOI] [PubMed] [Google Scholar]
- (9).See refs 1a, 5c, and 6. For an elegant solution see ref 4.
- (10).For relevant examples of alkenylation see: (a) Ooi T; Goto R; Maruoka K J. Am. Chem. Soc 2003, 125, 10494–10495. [DOI] [PubMed] [Google Scholar]; (b) Nandi RK; Takeda N; Ueda M; Miyata O Tetrahedron Lett 2016, 57, 2269–2272. [Google Scholar]
- (11).See refs 1a and 1b for relevant discussions.
- (12).(a) Piers E; Marais PC J. Org. Chem 1990, 55, 3454–3455. [Google Scholar]; (b) Piers E; Renaud JJ Org. Chem 1993, 58, 11–13. [Google Scholar]; (c) Wang T; Cook JM Org. Lett 2000, 2, 2057–2059. [DOI] [PubMed] [Google Scholar]; (d) Sole D; Peidro E; Bonjoch J Org. Lett 2000, 2, 2225–2228. [DOI] [PubMed] [Google Scholar]; (e) Zhao S; Liao X; Cook JM Org. Lett 2002, 4, 687–690. [DOI] [PubMed] [Google Scholar]; (f) Yu J; Wang T; Liu X; Deschamps J; Flippen-Anderson J; Liao X; Cook JM J. Org. Chem 2003, 68, 7565–7581. [DOI] [PubMed] [Google Scholar]; (g) Sole D; Diaba F; Bonjoch J J. Org. Chem 2003, 68, 5746–5749. [DOI] [PubMed] [Google Scholar]; (h) Cao H; Yu J; Wearing XZ; Zhang C; Liu X; Deschamps J; Cook JM Tetrahedron Lett 2003, 44, 8013–8017. [Google Scholar]; (i) Zhou H; Liao X; Cook JM Org. Lett 2004, 6, 249–252. [DOI] [PubMed] [Google Scholar]; (j) Yu J; Wearing XZ; Cook JM J. Org. Chem 2005, 70, 3963–3979. [DOI] [PubMed] [Google Scholar]; (k) Sole D; Urbaneja X; Bonjoch J Org. Lett 2005, 7, 5461–5464. [DOI] [PubMed] [Google Scholar]; (l) Dounay AB; Humphreys PG; Overman LE; Wrobleski AD J. Am. Chem. Soc 2008, 130, 5368–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Shen L; Zhang M; Wu Y; Qin Y Angew. Chem., Int. Ed 2008, 47, 3618–3621. [DOI] [PubMed] [Google Scholar]; (n) Yao Y; Liang G Org. Lett 2012, 14, 5499–5501. [DOI] [PubMed] [Google Scholar]
- (13).George DT; Kuenstner EJ; Pronin SV J. Am. Chem. Soc 2015, 137, 15410–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).The utility of silyl enol ethers in modification of corresponding ketones has been appreciated for a long time: Brownbridge P Synthesis 1983, 1983, 1–28. [Google Scholar]
- (15).(a) Drouin J; Boaventura M-A; Conia J-M. J. Am. Chem. Soc 1985, 107, 1726–1729. Also see: [Google Scholar]; (b) Drouin J; Boaventura MA Tetrahedron Lett 1987, 28, 3923–3926. For application in synthesis see: [Google Scholar]; (c) Forsyth CJ; Clardy JJ Am. Chem. Soc 1990, 112, 3497–3505. [Google Scholar]; (d) Huang H; Forsyth CJ J. Org. Chem 1995, 60, 2773–2779. [Google Scholar]; (e) Huang H; Forsyth CJ J. Org. Chem 1995, 60, 5746–5747. [Google Scholar]
- (16).Imamura K; Yoshikawa E; Gevorgyan V; Yamamoto Y Tetrahedron Lett 1999, 40, 4081–4084. [Google Scholar]
- (17).(a) Maeyama K; Iwasawa N J. Am. Chem. Soc 1998, 120, 1928–1929. [Google Scholar]; (b) Iwasawa N; Maeyama K; Kusama H Org. Lett 2001, 3, 3871–3873. [DOI] [PubMed] [Google Scholar]; (c) Kusama H; Yamabe H; Iwasawa N Org. Lett 2002, 4, 2569–2571. [DOI] [PubMed] [Google Scholar]; (d) Iwasawa N; Miura T; Kiyota K; Kusama H; Lee K; Lee PH Org. Lett 2002, 4, 4463–4466. [DOI] [PubMed] [Google Scholar]; (e) Grandmarre A; Kusama H; Iwasawa N Chem. Lett 2007, 36, 66–67. [Google Scholar]
- (18).Nevado C; Cardenas DJ; Echavarren AM Chem. - Eur. J 2003, 9, 2627–2635. [DOI] [PubMed] [Google Scholar]
- (19).(a) Staben ST; Kennedy-Smith JJ; Huang D; Corkey BK; LaLonde RL; Toste FD Angew. Chem., Int. Ed 2006, 45, 5991 For selected applications in synthesis see: [DOI] [PubMed] [Google Scholar]; (b) Linghu X; Kennedy-Smith JJ; Toste FD Angew. Chem., Int. Ed 2007, 46, 7671–7673. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC; Tria GS; Edmonds DJ; Kar MJ Am. Chem. Soc 2009, 131, 15909–15917. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huwyler N; Carreira EM Angew. Chem., Int. Ed 2012, 51, 13066–13069. [DOI] [PubMed] [Google Scholar]; (e) Lu Z; Li Y; Deng J; Li A Nat. Chem 2013, 5, 679–684. [DOI] [PubMed] [Google Scholar]; (f) Moreno J; Picazo E; Morrill LA; Smith JM; Garg NK J. Am. Chem. Soc 2016, 138, 1162–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li Y; Zhu S; Li J; Li AJ Am. Chem. Soc 2016, 138, 3982–3985. [DOI] [PubMed] [Google Scholar]
- (20).(a) Barabe F; Betournay G; Bellavance G; Barriault L Org. Lett 2009, 11, 4236–4238. [DOI] [PubMed] [Google Scholar]; (b) Sow B; Bellavance G; Barabe F; Barriault L Beilstein J. Org. Chem 2011, 7, 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barabe F; Levesque P; Korobkov I; Barriault L Org. Lett 2011, 13, 5580–5583. [DOI] [PubMed] [Google Scholar]
- (21).(a) Ito H; Ohmiya H; Sawamura M Org. Lett 2010, 12, 4380–4383. [DOI] [PubMed] [Google Scholar]; (b) Iwai T; Okochi H; Ito H; Sawamura M Angew. Chem., Int. Ed 2013, 52, 4239–4242. [DOI] [PubMed] [Google Scholar]
- (22).Han Y; Zhu L; Gao Y; Lee C-S Org. Lett 2011, 13, 588–591. [DOI] [PubMed] [Google Scholar]
- (23).(a) Corkey BK; Toste FD J. Am. Chem. Soc 2007, 129, 2764–2765. [DOI] [PubMed] [Google Scholar]; (b) Brazeau J-F; Zhang S; Colomer I; Corkey BK; Toste FD J. Am. Chem. Soc 2012, 134, 2742–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nishimoto Y; Moritoh R; Yasuda M; Baba A Angew. Chem., Int. Ed 2009, 48, 4577–4580. [DOI] [PubMed] [Google Scholar]
- (25).(a) Yamaguchi M; Tsukagoshi T; Arisawa M J. Am. Chem. Soc 1999, 121, 4074–4075. [Google Scholar]; (b) Arisawa M; Miyagawa C; Yamaguchi M Synthesis 2002, 2002, 138–145. Also see: [Google Scholar]; (c) Jones IL; Moore FK; Chai CLL Org. Lett 2009, 11, 5526–5529. [DOI] [PubMed] [Google Scholar]
- (26).Nakamura M; Endo K; Nakamura EJ Am. Chem. Soc 2003, 125, 13002 See also: [DOI] [PubMed] [Google Scholar]; Itoh Y; Tsuji H; Yamagata K.-i.; Endo K; Tanaka I; Nakamura M; Nakamura E J. Am. Chem. Soc 2008, 130, 17161–17167 and references therein. [DOI] [PubMed] [Google Scholar]
- (27).For intramolecular examples see refs 17c, 17d, and 18.
- (28).Stork G; Hudrlik PF J. Am. Chem. Soc 1968, 90, 4462–4464. [Google Scholar]
- (29).Formation of trace quantities of strong Brønsted acids can be associated with processes catalyzed by metal salts: Wabnitz TC; Yu J-Q; Spencer JB Chem. - Eur. J 2004, 10, 484–493. [DOI] [PubMed] [Google Scholar]
- (30).Formation of 4 likely involves a regioisomeric carbometalation of alkyne followed by an intramolecular hydride migration. For relevant observations see: Harrak Y; Simonneau A; Malacria M; Gandon V; Fensterbank L Chem. Commun 2010, 46, 865–867. [DOI] [PubMed] [Google Scholar]
- (31).TIPS-enol ether precursors are readily available from corresponding ketones. See SI for preparation.
- (32).Such selectivity for fully substituted silyl enol ethers is not exclusive. See SI for a detailed discussion.
- (33).Application to aldehyde-, ketone-, nitrile-, and amide-containing alkynes was unsuccessful thus far.
- (34).See SI for details.
- (35).Attempted purification of corresponding siloxydienes was unsuccessful.
- (36).Surendra K; Corey EJ J. Am. Chem. Soc 2014, 136, 10918–10920. [DOI] [PubMed] [Google Scholar]
- (37).For a review of organoindium compounds see: Shen Z-L; Wang S-Y; Chok Y-K; Xu Y-H; Loh T-P Chem. Rev 2013, 113, 271–401. [DOI] [PubMed] [Google Scholar]
- (38).Indium(III) bromide–ketone complex 32 can be expected to be a strong Brønsted acid: Ren J; Cramer CJ; Squires RR J. Am. Chem. Soc 1999, 121, 2633–2634. [Google Scholar]
- (39).Protodemetalation of intermediate 10 or related species with alkyne–indium(III) bromide complex can be responsible for initial generation of ketone 3, which cannot be accounted for by the presence of residual TIPSOH or adventitious moisture.