

ABSTRACT
The benefits of anti-cancer agents extend beyond direct tumor killing. One aspect of cell death is the potential to release antigens that initiate adaptive immune responses. Here, a diffusion enhanced formulation, INT230-6, containing potent anti-cancer cytotoxic agents, was administered intratumorally into large (approx. 300mm3) subcutaneous murine Colon26 tumors. Treatment resulted in regression from baseline in 100% of the tumors and complete response in up to 90%. CD8+ or CD8+/CD4+ T cell double-depletion at treatment onset prevented complete responses, indicating a critical role of T cells in promoting complete tumor regression. Mice with complete response were protected from subcutaneous and intravenous re-challenge of Colon26 cells in a CD4+/CD8+ dependent manner. Thus, immunological T cell memory was induced by INT230-6. Colon26 tumors express the endogenous retroviral protein gp70 containing the CD8+ T-cell AH-1 epitope. AH-1-specific CD8+ T cells were detected in peripheral blood of tumor-bearing mice and their frequency increased 14 days after treatment onset. AH-1-specific CD8+ T cells were also significantly enriched in tumors of untreated mice. These cells had an activated phenotype and highly expressed Programmed cell-death protein-1 (PD-1) but did not lead to tumor regression. CD8+ T cell tumor infiltrate also increased 11 days after treatment. INT230-6 synergized with checkpoint blockade, inducing a complete remission of the primary tumors and shrinking of untreated contralateral tumors, which demonstrates not only a local but also systemic immunological effect of the combined therapy. Similar T-cell dependent inhibition of tumor growth was also found in an orthotopic 4T1 breast cancer model.
KEYWORDS: Endogenous vaccine, tumor model, anticancer agent, T cells, intratumoral delivery, PD-1, chemotherapy, combination therapy, immunogenic cell death
Introduction
Cancer is the second leading cause of death worldwide, and it was predicted that 4,700 new cancer patients will be diagnosed daily in the United States in 2018.1 Cancer treatments range from local therapies such as surgery and radiation to systemically administered therapeutics including cytotoxic agents, hormones, targeted agents and immunotherapy with combinations of different treatment regimens becoming more frequent.2,3 In recent years, immunotherapy, such as checkpoint inhibitors anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and anti-Programmed cell death protein 1 (PD-1), have had meaningful impact in multiple tumor types, including advanced melanoma, lung, gastric, hepatocellular, cervical head and neck and renal cancer, by eliciting durable responses.4–7 However, many patients do not respond to current immunotherapies, and this is thought to be due to their lack of immune infiltration especially CD8+ T cells and interferon signature, in the tumor.7–10
Many efforts have been made to characterize immune responses in order to enhance the immunogenicity of tumors. It was noticed that not only immunotherapy but also more traditional therapies such as cytotoxic agents can induce immunomodulatory responses at least in mice.3,11 Cisplatin, for example, is a platinum-based compound that hydrolyses within the cell to form a platinum-DNA adduct, which was shown to increase Major Histocompatibility Complex (MHC) class I on Colon 26 tumor cells and antigen presenting cells (APC) in a mesothelioma model.12 Another example is vinblastine, a drug that destabilizes microtubules to arrest cell division, which also stimulates maturation of dendritic cells (DCs) by upregulation of CD40, CD80, CD86 and MHC class II.13,14 Even though these immunomodulatory stress responses are important, cell death itself has by far the biggest impact. Death by cytotoxic agents releases a vast amount of tumor antigens and damage-associated molecular pattern (DAMP) signals which bind to and cause a large influx of leukocytes to prime the immune system.15,16 Chemotherapy-driven immunogenic cell death (ICD) has been further characterized for agents, such as oxaliplatin, that release intracellular components such as calreticulin, adenosine triphosphate (ATP)17 and high-mobility group box 1 (HMGB1)18 which lead to receptor binding on APCs and uptake of cell debris, T cell activation, effector function induction and memory formation11,16 Cisplatin has not been found to cause calreticulin transport to the surface when given alone, and is therefore not classified as an ICD inducer16,19–21 although it does induce HMGB1 and ATP release, and cisplatin combined with other inducers of an ER stress responses such as radiation, tunicamycin or thapsigargin can result in calreticulin exposure.16,20,21 However, in a murine model of plasmacytoma, intravenous cisplatin resulted in tumor clearance and mice were protected upon re-challenge. This protection was not observed in Nude mice or upon T cell depletion prior to cisplatin treatment22 This suggests that mechanisms of immune activation after cell death may be diverse and may need to be further explored. Moreover, studies into immune responses to cell death by systemically delivered cytotoxic agents in patients have been limited due to toxicity-limiting doses.
In recent years, it was shown that therapies previously given systemically may also be efficacious when delivered locally. Intratumoral (IT) IL-2, for example, was shown to be effective in the early stages of melanoma patients.23,24 Our laboratory has published multiple studies delivering effective cancer vaccines subcutaneously next to the tumor site rather than intramuscularly.25–27 Local administration has the advantage of delivering a higher dose to the tumor site with the potential for less systemic toxicity. Local cancer vaccine treatment may also help to overcome a lack of immune response in cold tumors. Immunological death of tumor cells may cause an immune response that had not occurred spontaneously, converting the tumor into an endogenous cancer vaccine. This is the approach that we have explored here. We hypothesized that local administration of cytotoxic agents may maximize the release of a large variety of antigens from dying cells to induce strong adaptive immunity while limiting systemic dissemination of the agents to normal tissues.
In this study, INT230-6, a formulation that consists of fixed concentrations of widely used agents, cisplatin and vinblastine, and the cell penetration enhancer IT-006, is utilized as a model therapeutic (Bender et al. 2018 submitted). Bender et al. demonstrated that intratumorally delivered INT230-6 diffused throughout the tumor tissue and was taken up quickly by cells within the tumor, resulting in regression with complete response of Colon26 (C26) tumors and limited toxicity. We show here that INT230-6 turns tumors into in situ vaccines by relying on CD4+ and CD8+ T cells for its efficacy in the C26 colon and orthotopic 4T1 breast cancers, inducing long-term immunological memory, reducing burden of distant micrometastases and synergizing with checkpoint inhibitors to induce systemic immunity and regression of distant tumors.
Results
Intratumoral INT230-6 induces immunological responses
In accordance with Bender et al. 2018, INT230-6 delayed the growth of C26 tumors compared to untreated control mice and 100% regression from the baseline tumor volume and 50% complete response was achieved in this experiment but up to 90% in other experiments (Figure 1a). INT230-6 had a similar degree of efficacy in female and male WT BALB/c mice (data not shown). To assess whether an adaptive immune response was mediating the antitumoral effect of INT230-6, CD4+ and/or CD8+ T cells were depleted at the onset of INT230-6 treatment. In Figure 1b, INT230-6 treatment with control IgG antibody significantly improved survival over vehicle controls; all mice regressed from baseline and 80% had complete responses. Depletion of CD4+ T cells did not significantly alter the effect and resulted in a 70% complete response and similar overall survival. Depletion of CD8+ T cells significantly shortened survival; although most tumors showed tumor shrinkage from baseline for the first 7 days, no complete response was obtained. Similarly, depletion of CD4+ and CD8+ T cells significantly reduced survival compared to INT230-6 with IgG control antibodies (and approached the no treatment control). This suggests that the initial reduction in tumor mass was due to the direct cytotoxic effect of the chemotherapy but that long-term eradication of tumor, and hence complete responses and improvement in survival, were dependent on effector CD8+ T cells. When we repeated this experiment in RAG1-deficient (RAG1−/-) mice that lack T and B cells, we confirmed that INT230-6 had almost no effect on survival in the absence of adaptive immunity (Figure 1c). Interestingly, when looking at the individual growth curves in RAG1−/- mice, even though we saw some limited regression and growth delay in the initial phase, we did not observe regression from baseline, suggesting a role for adaptive immunity even in the initial regression. Finally, we tracked tumor antigen-specific CD8+ T cells in peripheral blood (Figure 1d). The immunodominant epitope of C26 is the AH-1 peptide presented by H-2Ld. By weekly monitoring of AH-1-specific CD8+ T cells, we confirmed that during the regression phase, approximately 14 days after treatment onset, a significantly increased proportion of AH-1 tetramer-reactive CD8+ T cells could be detected in the periphery. These data together with the depletion studies demonstrate that INT230-6 treatment induces tumor-specific T cell responses and that the ability of the INT230-6 treatment regimen to induce long-term rejection of tumors is dependent on these cells.
Figure 1.
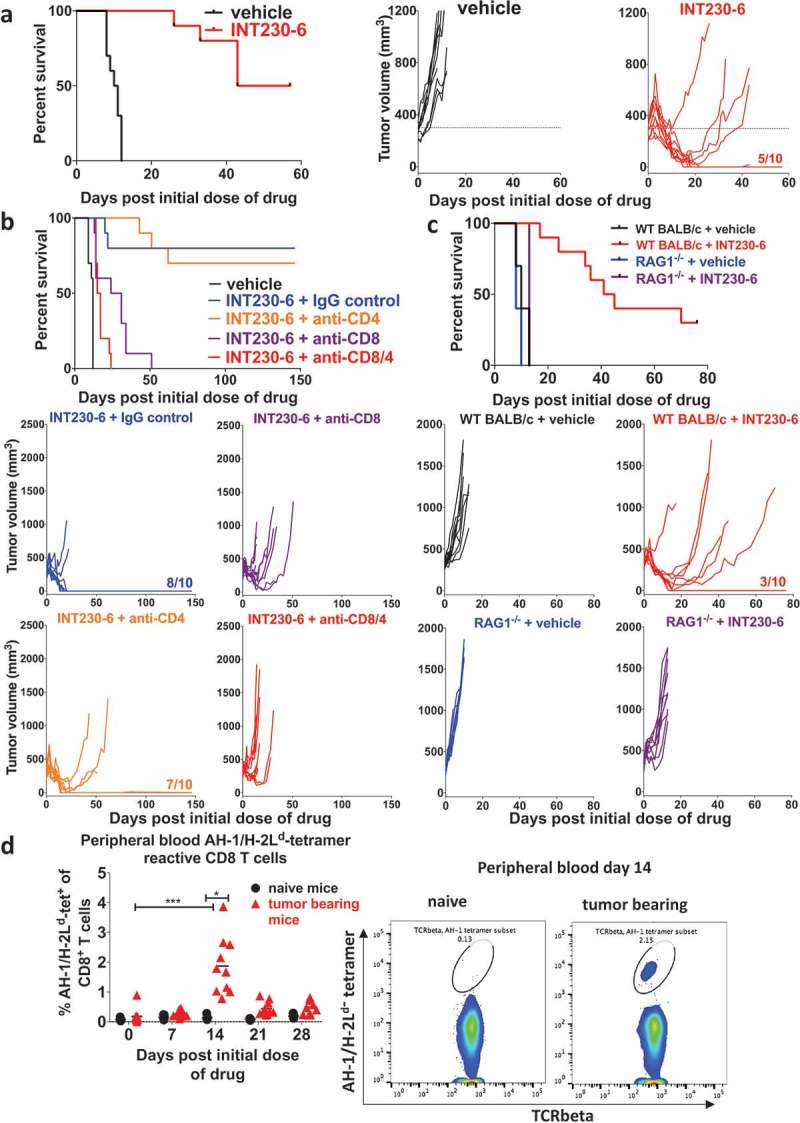
INT230-6 efficacy is dependent on CD8+ T cell.
Naïve mice were SC challenged with 1 × 106 C26 cells into the right flank. Vehicle or INT230-6 (0.5 mg/ml cisplatin, 0.1 mg/ml vinblastine, 10 mg/ml IT-006 cell penetration enhancer) were intratumorally (IT) administered into 300 mm3 (approx. 8.5 mm in diameter, 100 µl/400 mm3 volume C26 tumor) SC tumors (n = 10/group) for 5 sequential days (day 0 to 4) and tumor growth was monitored. a) Kaplan-Meier plot (left graph) and individual responses (right graphs) are displayed of a representative experiment of at least five repeats with similar or better results; vehicle (black) and INT230-6 (red) treatment are shown. The fraction 5/10 indicates the number of complete responders. The Log-rank test indicated a significant difference between the groups (p < .0001). b) Kaplan-Meier plot (top) and individual responses (below) of a representative experiment (of 3 with similar results) are shown illustrating vehicle (black) and INT230-6 with either IgG control antibody (300 µg, blue), anti-CD4 (100 µg, orange), anti-CD8 (200 µg, purple) or anti-CD4 and anti-CD8 (red) depletion antibodies. Antibodies were administered intraperitoneally (IP) on day 0, 1, 5, 8 and 15 after the initial INT230-6 dose (n = 10/group). Fractions (e.g. 8/10) indicate the number of complete responders. By log-rank test, differences were significant (p < .0001 unless stated otherwise) between vehicle and all other groups as well as INT230-6 + IgG control vs INT230-6 + anti-CD8 (p < .0006) or INT230-6 + anti-CD8/4, INT230-6 + anti-CD4 vs INT230-6 + anti-CD8 or INT230-6 + anti-CD8/4. c) Vehicle or INT230-6 (50 μl/400 mm3 C26 tumor) were administrated for 3 sequential days (day 0 to 2) and tumor growth was monitored. Kaplan-Meier plots (top) and individual responses (below) of WT BALB/c and RAG1−/- mice are shown. This experiment was performed twice with similar results. The fraction (3/10) indicates the number of complete responders. Log-rank test was significantly different between WT BALB/c + INT230-6 vs all other groups (p < .0001), WT BALB/c + vehicle vs RAG1−/- + vehicle (p < .05), WT BALB/c + vehicle vs RAG1−/- + INT230-6 (p < .01) as well as RAG1−/- + vehicle vs RAG1−/- + INT230-6 (p < .0001). d) INT230-6 (100 μl/400 mm3 C26 tumor) was administrated IT for 5 sequential days (n = 10). Untreated tumor-bearing mice as well as naïve mice (n = 5) were bled before treatment (day 0). Treated mice and naïve mice were bled on day 7, 14, 21 and 28 after the initial dose of drug. Peripheral blood was stained for tumor antigen-specific (AH-1/H-2Ld-tetramer-positive) CD8+ T cells and analyzed by flow cytometry. A representative experiment (of 2 with similar results) is shown. Percentage of AH-1/H-2Ld-tetramer-reactive CD8+ T cells amongst CD8+ T cells are shown as individual values with mean. Percentage of AH-1/H-2Ld-tetramer-reactive CD8+ T cells was significantly increased (p < .05) by Kruskal–Wallis post hoc Dunn’s test on day 14 after treatment onset. Representative flow cytometry plots of AH-1/H-2Ld-tetramer reactive populations on day 14 are shown (right panels).
Intratumoral INT230-6-mediated immunological memory requires both CD4+ and CD8+ T cells
Since INT230-6 induced complete remission of the tumors with no further relapse, we tested whether this treatment generated long-term immunological memory. To test this hypothesis, mice with complete responses were re-challenged with C26 cells SC or IV and compared to naïve mice (Figure 2a–b). After SC re-challenge (Figure 2a), naïve mice died from their tumor burden whereas of re-challenged mice, 86% were resistant to developing a tumor, demonstrating that INT230-6-treated mice had developed immunological memory. IV re-challenge of mice with complete response also resulted in partial protection (50%) demonstrating systemic memory was induced (Figure 2b). Additionally, we asked whether immunological memory was T-cell dependent. Mice underwent CD4+ and/or CD8+ T-cell depletion before the SC re-challenge (Figure 2a). Depletion of CD4+ or CD8+ cells resulted in partial loss of protection; however, when both T cell subsets were depleted, a total loss of protection was observed, as demonstrated by shortened survival comparable to that of naïve mice. Interestingly, when re-challenging mice with complete response who had CD4 depletion prior to INT230-6 treatment, immunologic memory was lost in 60% (data not shown). This highlights the role of CD4+ T cell help during the initial immune priming to achieve functional immunologic memory.28 This result indicates that CD4+ and CD8+ T cell-dependent immunological memory is generated by INT230-6 and both are necessary for the long-term protection.
Figure 2.
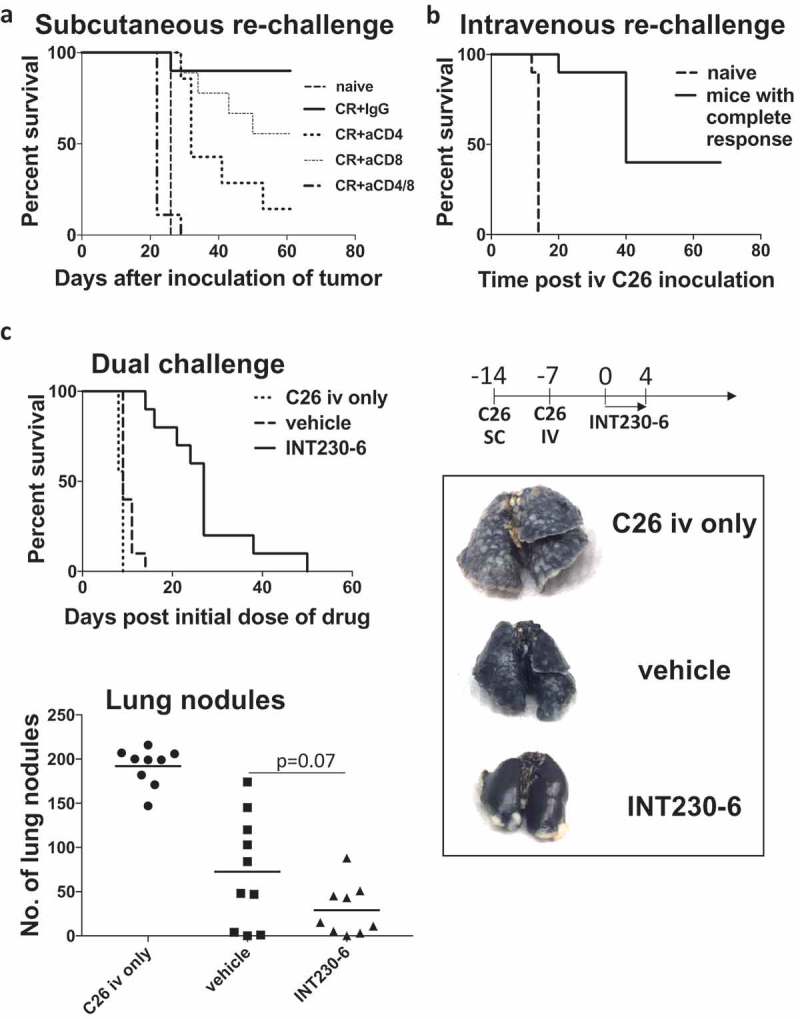
Mice with complete response (CR) were protected from tumor re-establishment in a T cell-dependent manner.
a) Mice with complete response (CR) and naïve ( ) mice were challenged with 1 × 106 C26 tumor cells by SC injection into the right flank (n = 10/group). Mice with CR underwent prior depletion of CD4 (
) mice were challenged with 1 × 106 C26 tumor cells by SC injection into the right flank (n = 10/group). Mice with CR underwent prior depletion of CD4 ( ), CD8 (
), CD8 ( ), CD4/8 (
), CD4/8 ( ) or control IgG injections (
) or control IgG injections ( ), (150 µg anti-CD8/4, 300 µg IgG, day −1, 3, 5, 12 and 19) by IP administration. Kaplan-Meier plot is shown of a representative experiment (of 2 with similar results) and a Log-rank test indicated significant differences (p < .0001: naïve vs CR+IgG, naïve vs CR+aCD4, naïve vs CR+aCD8, CR+IgG vs CR+aCD4/8, CR+CD8 vs CR+CD4/8; p < .001: CR+CD4 vs CR+CD4/8; p < .01: naïve vs CR+aCD4, CR+IgG vs CR+aCD4). b) C26 tumor cells (1x106) were inoculated IV into mice with CRs (solid line) and naïve mice (dashed line, n = 10/group) to produce lung metastases. Kaplan-Meier plot is shown of a representative experiment (of 3 with similar results) and a log-rank test showed a significant difference (p < .0001) between groups. c) C26 tumor cells (1x106) were inoculated SC into the right flank and 2.5 × 105 C26 tumor cells were inoculated IV 7 days later. An additional 10 mice received IV tumor cells only (dotted line). Vehicle (dashed line) or INT230-6 (solid line) were administered IT into 275mm3 (50µl/400mm3 C26 tumor) SC tumors (n = 10/group) for 5 sequential days (day 0 to 4) starting on day 14 and tumor growth was monitored. Kaplan-Meier plot of a representative experiment (of 2 with similar results) is shown (top left), and a log-rank test indicated significant differences (p < .01: C26 iv only vs vehicle; p < .0001: C26 iv only vs INT230-6, vehicle vs INT230-6). Quantification of lung nodules at the time of death (n = 9–10/group) is presented (bottom left). Median test was applied for vehicle vs INT230-6 treatment groups. Representative lungs that had been injected with 15% black India ink are shown on the right. Healthy lung tissue is black and smooth whereas tumor nodules appear white and nodular.
), (150 µg anti-CD8/4, 300 µg IgG, day −1, 3, 5, 12 and 19) by IP administration. Kaplan-Meier plot is shown of a representative experiment (of 2 with similar results) and a Log-rank test indicated significant differences (p < .0001: naïve vs CR+IgG, naïve vs CR+aCD4, naïve vs CR+aCD8, CR+IgG vs CR+aCD4/8, CR+CD8 vs CR+CD4/8; p < .001: CR+CD4 vs CR+CD4/8; p < .01: naïve vs CR+aCD4, CR+IgG vs CR+aCD4). b) C26 tumor cells (1x106) were inoculated IV into mice with CRs (solid line) and naïve mice (dashed line, n = 10/group) to produce lung metastases. Kaplan-Meier plot is shown of a representative experiment (of 3 with similar results) and a log-rank test showed a significant difference (p < .0001) between groups. c) C26 tumor cells (1x106) were inoculated SC into the right flank and 2.5 × 105 C26 tumor cells were inoculated IV 7 days later. An additional 10 mice received IV tumor cells only (dotted line). Vehicle (dashed line) or INT230-6 (solid line) were administered IT into 275mm3 (50µl/400mm3 C26 tumor) SC tumors (n = 10/group) for 5 sequential days (day 0 to 4) starting on day 14 and tumor growth was monitored. Kaplan-Meier plot of a representative experiment (of 2 with similar results) is shown (top left), and a log-rank test indicated significant differences (p < .01: C26 iv only vs vehicle; p < .0001: C26 iv only vs INT230-6, vehicle vs INT230-6). Quantification of lung nodules at the time of death (n = 9–10/group) is presented (bottom left). Median test was applied for vehicle vs INT230-6 treatment groups. Representative lungs that had been injected with 15% black India ink are shown on the right. Healthy lung tissue is black and smooth whereas tumor nodules appear white and nodular.
INT230-6 treatment reduces metastatic burden at a distant site
To further characterize whether T cell immunity could induce systemic protection against micrometastasis, we challenged mice with C26 cells not only SC but also IV seven days before treatment onset (Figure 2c). Mice that received C26 tumor cells only IV died from lung tumor burden, whereas mice with vehicle-treated SC tumors in addition to IV challenge had reduced metastatic burden in the lung but died from SC tumors, suggesting that the presence of tumor at the SC site perhaps induced an immune response which reduced the tumor burden at the secondary tumor site (lung). Mice that had tumor at both sites and received INT230-6 treatment into the SC tumors had a significant survival advantage. The majority died from the primary (SC) tumor burden, not the lung tumors. INT230-6 treated animals had a trend (p = 0.07) towards greater reduction in the numbers of lung nodules than mice whose SC tumors had been treated with vehicle only, suggesting that T cells induced by the local treatment of the SC tumor were able to reduce metastatic lung tumor burden.
PD-1hiCD44hicd69+ AH-1-specific CD8+ T cells were raised against untreated C26 tumors but were unable to clear untreated tumors
Because INT230-6 treatment induces CD8+ T cell effector and memory responses, we wanted to further phenotype CD8+ T cells within the C26 tumor model. When staining for AH-1/Ld-tetramer-specific CD8+ T cells in untreated C26 tumor-bearing mice, we identified a significant proportion of these tumor antigen-specific CD8+ T cells in the tumor microenvironment, which were present at much lower frequencies in the periphery (Figure 3a). Compared to CD8+ T cells in the periphery, total tumor CD8+ T cells and especially antigen-specific CD8+ T cells not only had an activated phenotype (CD44hiCD69hiCD62Llow), but the majority also expressed high levels of PD-1 as seen in humans also29 suggesting that C26 tumors induced AH-1-specific CD8+ T-cell activation and proliferation, and these activated T cells homed to the tumor site.
Figure 3.
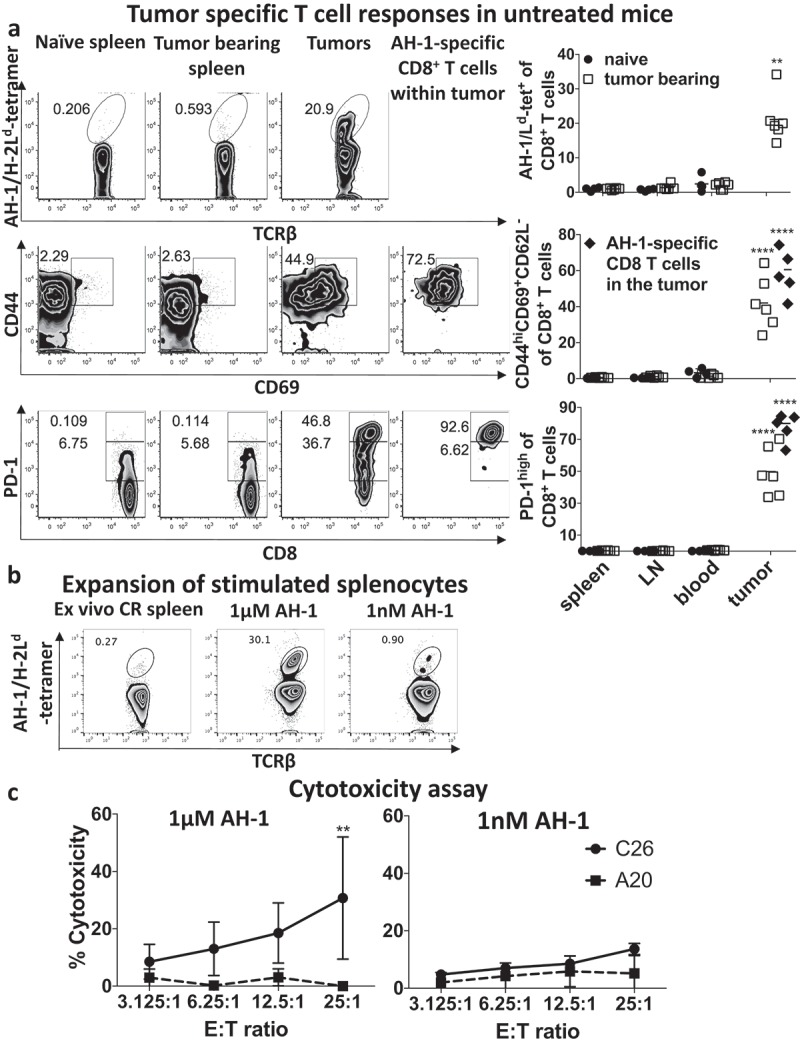
PD-1hi, CD44hiCD69+ AH-1-specific CD8+ T cells were induced by growth of untreated C26 tumors.
a) Mice with bulky (1000mm3) C26 tumors (untreated) as well as naïve controls (n = 4–6/group) were bled and euthanized to harvest and prepare single cell suspensions of tumors, spleen and draining lymph nodes (LN). Lymphocytes were stained for AH-1/Ld-tetramer reactive populations (top panel), activation markers (CD44hiCD69+CD62−, middle panel) and PD-1 expression (lower panel) on CD8+ T cells and analyzed by flow cytometry. Individual responses and mean values are shown of pooled data from two experiments. Kruskal–Wallis with Tukey post hoc test was performed and indicated that AH-1 specific CD8+ T cells are significantly enriched (p < 0.01) in the tumor compared to the periphery (blood, spleen or lymph nodes) of tumor-bearing or naive mice. Total tumor CD8+ T cells and AH-1 specific CD8+ T cells were significantly more activated (p < 0.0001) and expressed higher levels of PD-1 (p < 0.0001) than in the periphery of tumor-bearing or naïve mice. b) Splenocytes, harvested from mice with CR, were stimulated for one week with high (1 μM, left graph) or low (1 nM, right graph) AH-1 peptide concentration with syngeneic BALB/c splenocytes as antigen-presenting cells. Lymphocytes were stained for AH-1/Ld-tetramer reactive CD8+ T populations. c) Lactate dehydrogenase (LDH) cytotoxicity assay was performed with expanded splenocytes. For this, C26 cells were applied as AH-1 expressing target (T) cells (solid line). A20 cells were utilized as an AH-1 negative target cell line (dashed line). These cells were incubated with different ratios of CD8+ effector (E) T cells for 4 h and LDH assay was performed to calculate cytotoxicity. Mean ± standard deviation is shown of three pooled experiments. Two-way ANOVA with Sidak’s multiple comparison test was performed and showed significant difference (**p < 0.01) at the 25:1 ratio. Representative flow cytometry plots (bottom) illustrate the expansion of AH-1 specific CD8+ T cells.
Tumor-specific CD8+ T cells in mice with complete response are functional
To determine the functionality of AH-1-specific CD8+ T cells, we performed a cytotoxic assay. In order to obtain sufficient tumor antigen-specific CD8+ T cells, we expanded them from splenocytes from mice with complete responses (Figure 3b). Splenocytes were stimulated for one week with high (1 μM) or low (1 nM) AH-1 peptide concentration to expand CD8+ T cells of low and high functional avidities, respectively, before determining lytic activity (Figure 3c). Stimulation of CD8+ T cells with high AH-1 peptide concentration presumably expands T cells with low as well as high functional avidity (Figure 3b). Expansion (about 100-fold) was observed by flow cytometry and expanded CD8+ T cells could lyse C26 cells but not the control AH-1-negative syngeneic tumor cell line, A20 (Figure 3c). Stimulation of CD8+ T cells with low AH-1 peptide concentration, in order to expand high avidity T cells, did not result in significant expansion and the stimulated pool of cells were consequently not able to significantly lyse C26 tumor cells. Hence, in vitro, a pool of splenocytes enriched in AH-1-specific CD8+ T cells were capable of lysing tumor cells, suggesting that suppression of AH-1-specific CD8+ T cells may hamper their ability to control tumor in the tumor microenvironment in vivo. However, most CD8+ T cells responding to the AH-1 epitope may have low functional avidity, as we could not induce expansion with low concentrations of peptide, consistent with known tolerization of high avidity T cells in the thymus.
CD8 infiltration increases 11 days after treatment onset
To confirm the local immune response of INT230-6 treatment, histology was performed on tumors before treatment and over 14 days after treatment onset. H&E staining detected leukocyte infiltration into the tumor (Figure 4 top row). Immunohistochemistry revealed that CD8+ T cell infiltration was very low at baseline (Figure 4, 2nd, 3rd row, and graph). Consistent with the known timeline for a T cell response to develop, and also consistent with the appearance of tumor-specific T cells at day 14 but not day 7 in Figure 1d, we found that the density of CD8+ T cells did not change significantly before day 8 after treatment onset, but increased thereafter with the most apparent effects on day 11 and 14 (combined day 11 + 14 significantly higher than the earlier time points p = 0.04).
Figure 4.
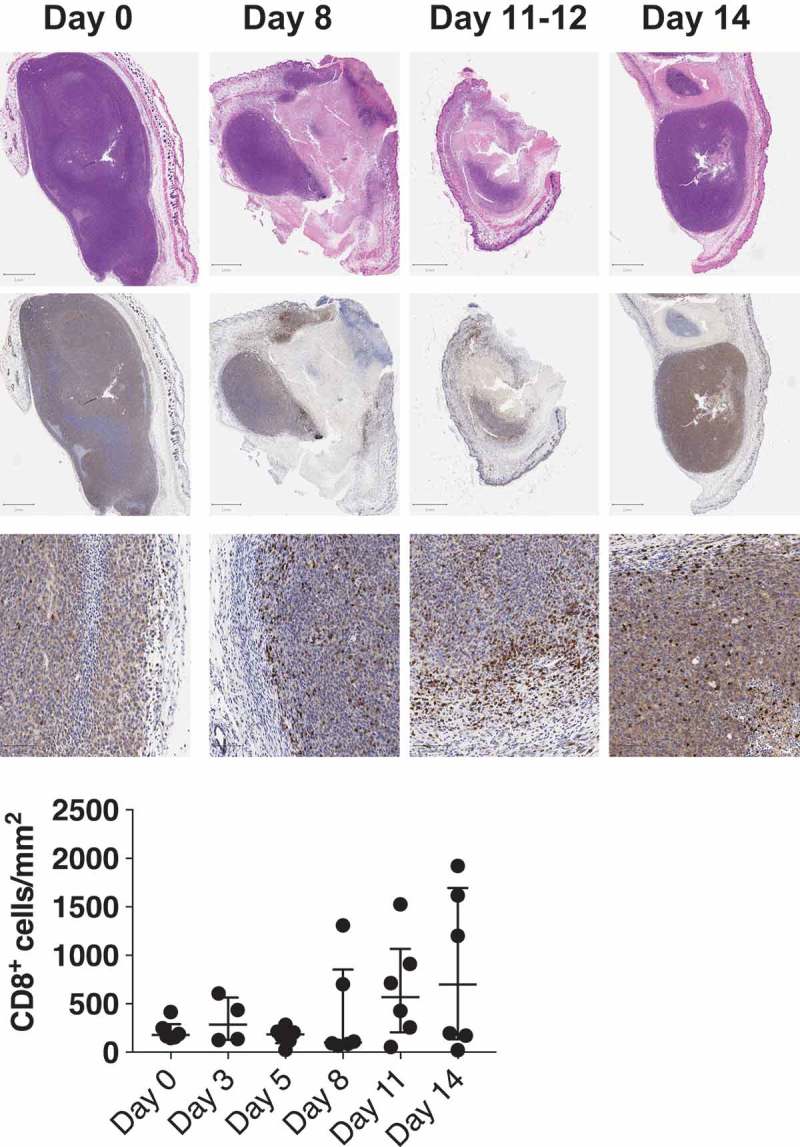
CD8+ T cells infiltrate increases 11 days after treatment onset.
Top panel shows representative images of histology of C26 tumors before treatment (day 0) and after treatment (day 8, 11–12 and 14). H&E staining is depicted in the top row, CD8+ T cells are presented in the middle and bottom row at different magnifications (see scale bar). The bottom graph is a quantification of CD8+ T cells per mm2 of tumor tissue (n = 6 per time point). Each point represents a different mouse tumor, as mice has to be euthanized to harvest tumors at each time point. The 12 specimens at days 11 + 14 differ from those at the earlier time points (p = 0.04) by a Kruskal–Wallis test.
INT230-6 treatment of primary tumors results in regression of tumors at distant sites when combined with anti-CTLA-4
Systemic T cell responses should control tumors in other sites. To determine if systemic responses mounted upon INT230-6 treatment can be enhanced with checkpoint inhibitors, we performed a dual contralateral tumor experiment in which we applied the combinatorial treatment of IT INT230-6 with either IP anti-PD-1 (Figure 5a–d) or IP anti-CTLA-4 (Figure 5e–h). As an additional control to vehicle-treated primary tumors some mice received tumors only at the contralateral site and were left untreated (Figure 5d,h). In Figure 5a with anti-PD-1, the contralateral tumors were kept small by inoculating only three days before treatment onset. INT230-6 had significant efficacy on the primary tumor compared to vehicle only (Figure 5c). This activity in the injected tumor was enhanced by addition of anti-PD-1 (doubling the rate of complete response despite no complete responses seen with the checkpoint inhibitor given alone). At the contralateral tumor site, the INT230-6-treated group had significantly different tumor sizes only from the group bearing a tumor at that contralateral site alone, not from the vehicle control (Figure 5d, h), but nevertheless resulted in significantly improved survival (Figure 5b, f). Anti-PD-1 monotherapy had limited efficacy on either tumor site, not significantly different in tumor size from vehicle control. However, tumor sizes were found to be smaller than in mice with contralateral tumor alone (in the absence of a primary tumor) at the untreated contralateral site (Figure 5d). The combination of INT230-6 and anti-PD-1 did significantly improve the response of primary tumors over INT230-6 treatment alone on day 16 and 18 (Figure 5c) and on secondary tumors on day 18 (Figure 5d), but this did not translate into a statistically significant survival advantage (Figure 5b).
Figure 5.
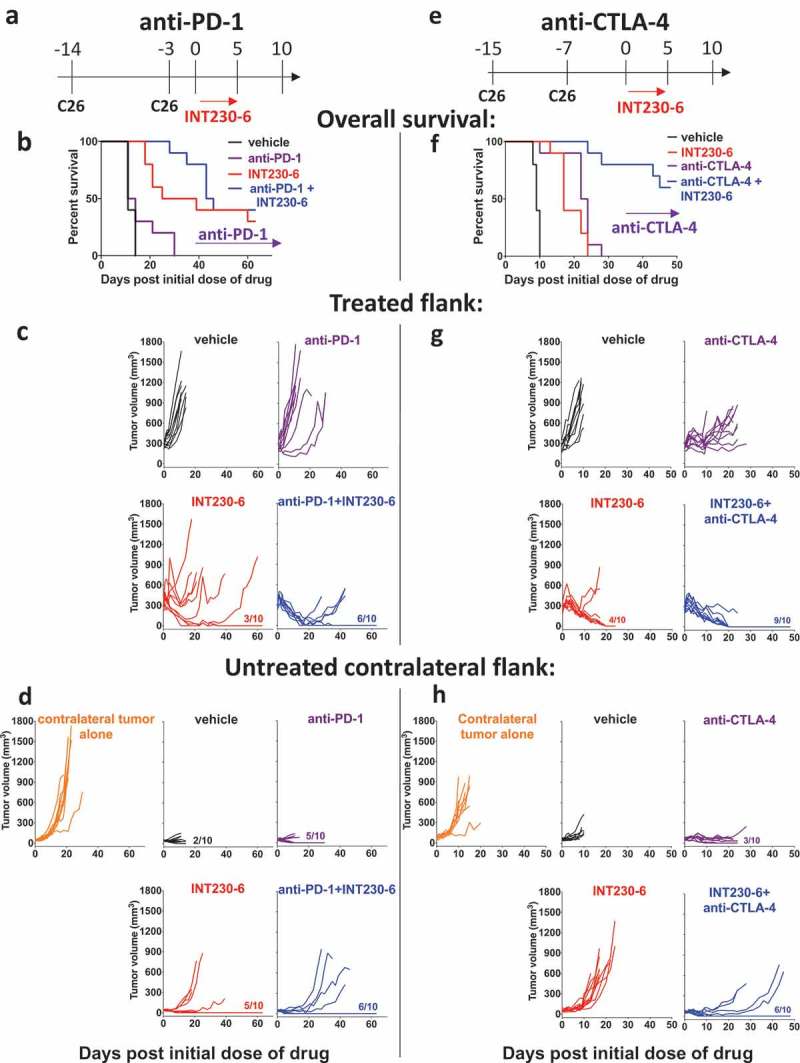
Contralateral tumor response can be induced by INT230-6 in combination with anti-CTLA-4 but not with anti-PD-1.
a) Illustration of INT230-6 and anti-PD-1 treatment regimen (top). C26 (1x106) were inoculated into the right flank (day −14). Contralateral tumors were inoculated 11 days after primary tumors (day −3). Primary tumors were treated with INT230-6 (50 μl/400 mm3 tumor, 5 sequential days) starting on day 0. Average primary tumor volume on day 0 was 290mm3 and average contralateral tumor volume was 42mm3. Anti-PD-1 treatment (100 μg) was given on day 0, 3, 7 and 10. b) Kaplan-Meier plot (below illustration) and individual responses of c) treated (ipsilateral) (middle) and d) contralateral flank (bottom) are shown of vehicle (black), anti-PD-1 (purple), INT230-6 (red) and anti-PD-1+ INT230-6 (blue) treatment as well as contralateral tumor only control (orange, n = 10/group). Fractions (e.g. 3/10) indicate the number of mice that completely lost tumor that the indicated flank. Log-rank test was significantly different between vehicle and INT230-6 (p < 0.0001), vehicle and anti-PD-1+ INT230-6 (p < 0.0001), anti-PD-1 and INT230-6 (p < 0.01) and anti-PD-1 and anti-PD-1 + INT230-6 (p < 0.0001). Two-way ANOVA with Sidak’s multiple comparison test of growth curves showed that untreated contralateral tumors only were significantly different (p < 0.0001) from all groups on secondary site. Furthermore, vehicle was significantly different (p < 0.0001) from INT230-6 and INT230-6+ anti-PD-1 on day 8 and 11 on the primary site. Vehicle was significantly different (p < 0.05) from INT230-6+ anti-PD-1 on day 11 on the secondary site. INT230-6 was significantly different anti-PD-1 or INT230-6+ anti-PD-1 on day 16 (p < 0.01) and day 18 (p < 0.001) on the primary tumor site. INT230-6 was significantly different (p < 0.01) from INT230-6 + anti-PD-1 on day 18 on the contralateral site. Anti-PD-1 was significantly different (p < 0.0001) from INT230-6+ anti-PD-1 on day 8 and 11 at the primary site only. e) Illustration of INT230-6 and anti-CTLA-4 treatment regimen (top). C26 (1x106) tumor cells were inoculated into the right flank (day −15). Contralateral tumors were inoculated 7 days after primary tumors (day −8). Primary tumors were treated with INT230-6 (50 μl/400 mm3 tumor, 5 sequential days) starting on day 0. Average primary tumor volume on day 0 was 250mm3 and average contralateral tumor volume was 60mm3. Anti-CTLA-4 treatment (100 μg) was given on day 0, 3 and 6. f) Kaplan-Meier plot (below illustration) and individual responses of g) treated (ipsilateral) (middle) and h) contralateral flank (bottom) are shown of vehicle (black), anti-CTLA-4 (purple), INT230-6 (red) and anti-CTLA-4+ INT230-6 (blue) treatment as well as contralateral tumor only control (orange, n = 10/group). Fractions (e.g. 4/10) indicate the number of mice that completely lost tumor in the indicated flank. Log-rank test was significantly different between vehicle and all other groups (p < 0.0001), INT230-6 and anti-CTLA-4+ INT230-6 (p < 0.0001) and anti-CTLA-4 and anti-CTLA-4+ INT230-6 (p < 0.0001). All experiments were performed twice. Two-way ANOVA with Sidak’s multiple comparison test of growth curves showed that untreated contra-later tumors were significantly different (p < 0.0001) from all other groups on the contralateral site. Vehicle was significantly different (p < 0.0001) from INT230-6, anti-CTLA-4 and anti-CTLA-4+ INT230-6 on day 6 and 8 at the primary (ipsilateral) site. Vehicle was significantly different from anti-CTLA-4 (day 8: p < 0.0001) and anti-CTLA-4+ INT230-6 (day 6: p < 0.001, day 8: p < 0.0001) at the contralateral site. INT230-6 was significantly different from anti-CTLA-4 at the primary site (day 8: p < 0.05, day 10: 0.001) and secondary site (day 8: p < 0.01, day 9: p < 0.001, day 10: p < 0.0001). INT230-6 was significantly different from anti-CTLA-4 + INT230-6 at the contralateral site only (day 6: p < 0.05, day 8–10: p < 0.0001). Anti-CTLA-4 was significantly different from anti-CTLA-4 + INT230-6 at the primary site (day 8: p < 0.01, day 9: p < 0.05, day 10: p < 0.0001) and at the secondary site (day 6: p < 0.05).
In Figure 5e studying anti-CTLA-4, contralateral tumors were 30% larger than in Figure 5a as C26 cells were inoculated seven days before treatment onset. Mice that received vehicle treatment had a shorter survival than in Figure 5b due to the larger tumor burden at the contralateral site. Again, INT230-6 treatment alone resulted in a significant response at the primary tumor site. Anti-CTLA-4 monotherapy significantly reduced tumor sizes in the primary site (Figure 5g), although none of the mice had a complete response. Anti-CTLA-4 also had a significant impact on contralateral tumors compared to vehicle and untreated contralateral tumors alone by inducing the complete response of contralateral tumors in 3 out of 10 mice (Figure 5h). Anti-CTLA-4 synergized with INT230-6, resulting in significantly improved survival over INT230-6 or anti-CTLA-4 monotherapy (Figure 5f). The combination significantly increased the number of complete responses on both primary (9/10) and contralateral (6/10) tumors compared with either treatment alone. This suggests that CTLA-4 may be a more critical checkpoint in T cell responses to these distant untreated tumors, at least under these conditions, and rejuvenation of intra-tumor antigen-specific CD8+ T cells (as expected to be mediated by anti-PD-1) may be less critical than enhanced activation of T cells and promotion of T cell entry into the tumor microenvironment by anti-CTLA-4. Thus, although both checkpoint inhibitors showed some activity in combination with INT230-6, the effect was more pronounced with anti-CTLA-4, especially on the contralateral tumors.
INT230-6 facilitates T cell mediated tumor regression in 4T1 orthotopic breast carcinoma model
To test the efficacy of INT230-6 intratumoral injection on inducing T cell mediated long-term tumor regression in a tumor growing orthotopically, we utilized the 4T1 breast carcinoma cell line. This cell line gives rise to mammary tumors when injected orthotopically into the mammary fat pad of BALB/c mice. 0.5 × 106 cells were injected into the mammary fat pad adjacent to the 4th nipple of BALB/c mice. Once the tumors reached an average size of ~200 mm3, we performed intratumoral injection with either vehicle or INT230-6 for five consecutive days. We found that tumors treated with INT230-6 were significantly smaller and grew at a much slower rate (Figure 6a) compared to the vehicle-treated ones (Figure 6b). Furthermore, we found that the survival of mice carrying 4T1 tumors treated with INT230-6 was significantly longer than those treated with vehicle (Figure 6c). Although unlike the C26 tumor model, we did not observe a CR in 4T1 orthotopic mode (probably because mortality was driven by early lung metastases in this model), the size of INT230-6 treated tumor remained significantly smaller, even two weeks after the last dose of INT230-6. Therefore, we hypothesized that this prolonged protection may be due to activation of the adaptive immune system.
Figure 6.
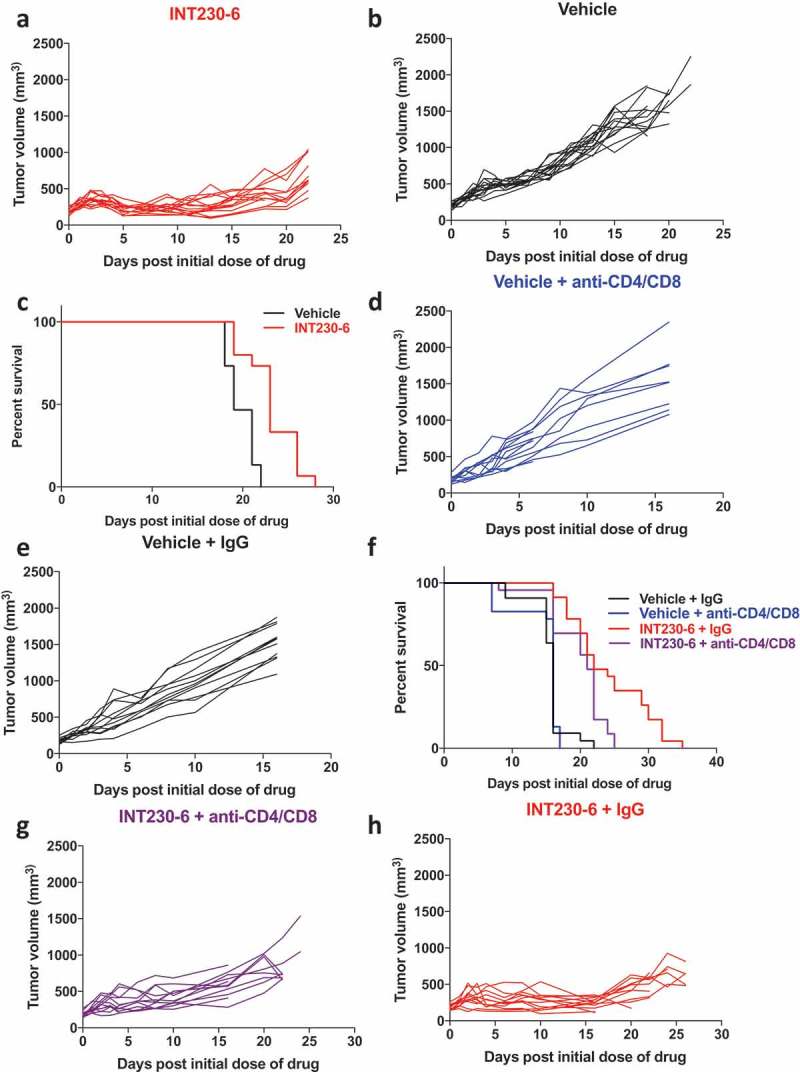
Intratumoral administration of INT230-6 delays tumor growth rate and improves overall survival, in a T cell-dependent manner, in 4T1 orthotopic breast carcinoma tumor models: a & b) 4T1 tumor growth curve of individual mice when treated with either INT230-6 (a) or vehicle (b). The INT230-6 treated 4T1 mammary tumors grew at a significantly slower rate than the vehicle-treated tumors (n = 15, **p < 0.003). c) Kaplan-Meier survival curve shows a significant difference between the vehicle-treated 4T1 mammary tumor-bearing mice as compared to INT230-6 treated ones (n = 15, *p < .03). d & e) Vehicle treated 4T1 tumor grow at a similar rate with (d) or without (e) CD4/CD8 depletion. f) Kaplan-Meier survival curve depicting the differences in survival probabilities of mice carrying 4T1 mammary tumor upon treatment with either INT230-6 or vehicle, with or without depletion of CD4 and CD8 cells. g-h) The CD4/CD8 depleted INT230-6 group (g) shows a significantly faster tumor growth rate compared to INT230-6 & IgG treated (h) ones(***p < 0.0003). There was a significant difference in survival probability of INT230-6 + IgG (red curve) treated mice (n = 22) compared to INT230-6 + anti-CD4/CD8 treated mice (purple curve) (n = 22, **p < 0.008). No significant difference, either in survival or tumor growth rate, was observed in vehicle-treated groups, with (e) or without (f) CD4/CD8 depletion. There was a significant difference in overall survival of vehicle versus INT230-6 treated tumor-bearing mice when both were T-cell depleted (panel f, blue vs purple curve **p < 0.001).
To evaluate the role of the adaptive immune system in mediating tumor regression after INT230-6 treatment in the 4T1 orthotopic model, we depleted both CD4+ and CD8+ T cells at the time of treatment onset. To do so, we injected anti-CD4 (100 µg/mouse) and anti-CD8 (200 µg/mouse) antibodies intraperitoneally to mice carrying 4T1 mammary tumors treated with either INT230-6 or vehicle. Rat IgG (300 µg/mouse) was used as a control. We did not find any significant effect on the growth kinetics (Figure 6d–e), as well as overall survival (Figure 6f-black and blue curve), between CD4/CD8 depleted and IgG-treated animals in the vehicle-treated groups. However, we observed a significant reduction in the survival probability of CD4/CD8 depleted mice carrying 4T1 mammary tumors treated with INT230-6 in comparison with those treated with INT230-6 and IgG (Figure 6f-purple and red curve). Additionally, the growth rate of CD4/CD8 depleted INT230-6 treated 4T1 mammary tumors was also significantly faster than that of the INT230-6 + control IgG-treated tumors (Figure 6g–h). This suggests a probable role of the T cell adaptive immune system in mediating the anti-tumoral effect of INT230-6 treatment. Additionally, we observed a significant difference in overall survival (**p < 0.001) of mice carrying 4T1 tumors when treated with either vehicle versus INT230-6 when CD4/CD8 depleted (Figure 6f- blue vs purple curves). Nevertheless, the INT230-6 treated and CD4/CD8 depleted tumors grew significantly more slowly than the vehicle-treated and CD4/CD8 depleted group (**p < 0.006) (Figure 6d vs h). This suggests that INT230-6 itself also can affect tumor growth through the chemotherapeutic effect partially independently of T cell immunity. However, on depleting T cells, the effect is partially lost (Figure 6g vs h), confirming a role of CD4/CD8 T cells in mediating the anti-tumor effect in INT230-6 treated 4T1 orthotopic breast tumor model.
Discussion
In this study, we demonstrate that the IT delivered cytotoxic formulation, INT230-6, converts tumors into endogenous vaccines. Specifically, we show not only that the efficacy of INT230-6 depends on CD8+ T cells but also that it induces systemic long-term T cell memory and protection against rechallenge. Tumor antigen-specific CD8+ T cells can also be detected at increased levels in the circulation during tumor regression, strengthening the evidence that tumor-specific immunity is induced. Limited efficacy of effector T cells induced by INT230-6 alone to shrink contralateral tumors can be substantially improved by synergy with anti-CTLA-4.
In situ vaccination and checkpoint inhibitors, such as anti-CTLA-4, have the advantage that antigen-specific T cell responses are tailored in vivo and therefore a wide range of antigens can be targeted simultaneously by the treatment-induced immune response. In contrast to recently developed antigen-specific adoptive cell therapy treatments30,31 in situ vaccination also has the advantage of reducing the risk of resistance and immunoediting compared to defined antigen-targeted therapies. In the clinic, current checkpoint blockade immunotherapies are given systemically, which in the majority of cases restricts efficacy to tumors already infiltrated with T lymphocytes. Here, we deliver a formulation directly into C26 colon or orthotopic 4T1 mammary tumors which results in complete response, responses at distant sites, and immunologic T-cell dependent memory, as well as prolonged survival in the C26 model, and in the case of 4T1, also delayed tumor growth and prolonged survival dependent on T cells (in the 4T1 model, prolonged survival (CR) is limited by the early metastasis of tumors from the primary site to the lungs). The mechanism is likely due to massive cell death, as its component cisplatin cross-links DNA and vinblastine inhibits microtubule formation. We discovered that this cell death is highly immunogenic and dependent on induction of adaptive immunity, as treatment of tumor-bearing RAG1−/- mice, which do not have lymphocytes, results in a temporary growth delay (due to local cytotoxicity) but no regression from baseline or complete response. Additionally, Bender et al. (2018) detected influx of dendritic cells in treated tumors by 8 days, which may contribute to induction of T cells.32 Although cisplatin by itself is not thought to induce immunogenic cell death because of a failure to induce calreticulin exposure (although it does induce HMGB1 and ATP release)16,19–21 combinations of cisplatin with other ER stress inducers, such as thapsigargin or tunicamycin or radiotherapy, can induce immunogenic cell death16,20,21 so it is possible that the combination with vinblastine and the stress of the intratumoral delivery contributed to this process. The main effector cell is the CD8+ T cell, as depletion of CD8+ cells at treatment onset results in abrogation of complete response, and tumor antigen-specific CD8+ T cells can be detected in peripheral blood during the regression phase of tumors. Long-term memory is also T cell-dependent, as is tumor growth slowing and improved survival in the 4T1 orthotopic model. Cancer vaccines are created to prime and expand tumor antigen-specific T cells,3 which makes the intratumoral formulations that induce cell death, such as INT230-6, ideal candidates to promote broad antigen presentation, which will result in an in situ vaccine-like effect.
Intratumoral delivery of anti-cancer agents is not a new concept, and even though benefits have been recorded, these have not been translated into standard care, mainly because of three assumptions or beliefs: 1) accessible primary tumors can be mostly resected anyway, 2) the needle track may spread tumor cells to produce metastases and 3) local treatment may not have an effect on metastases,33 suggesting that a role for systemic immunity in the efficacy of cytotoxic agents has not been widely recognized. In addition, most anti-cancer agents are not formulated specifically for intratumoral delivery as INT230-6 is. A recent study by Gao et al. 2017, for example, investigates the efficacy of an intratumoral methotrexate-loaded implant in a sarcoma model which caused delay in tumor growth, but the immune response was not investigated.34 On the other hand, a clinical study by Yu et al. 2015 compared intratumoral chemotherapy to ultra-minimum incision personalized intratumoral chemoimmunotherapy which consisted of an oxidant, a cytotoxic drug (cytosine arabinoside) and hapten.35 The authors treated 97 patients with advanced lung cancer and found significantly improved overall survival with their chemoimmunotherapy. It was also shown that patients on chemoimmunotherapy had more leukocyte infiltrate in their tumors.35 Even though the authors highlighted the ‘chemoimmunotherapy’, both regimens tested contained a cytotoxic agent that induced an immune response through cell death to different degrees, similar to this study, although a clear role for CD8+ T cells was not observed. The concept of using checkpoint inhibitors with intratumoral therapy is also supported by the observation that in a glioblastoma model, intratumoral chemotherapy improved the effect of anti-PD-1 whereas systemic chemotherapy diminished the benefit of anti-PD-1.36
A definite role of CD8+ T cells in response to intratumoral cytotoxic agents has not been described in the literature to our knowledge. However, Casares et al. have shown that injection of doxorubicin-treated CT26 tumor cells induces tumor rejection in BALB/c mice but not in nude mice or mice depleted of CD8+ cells prior to treated CT26 cell injection. This loss of protection suggested that immunogenic cell death was induced11 The authors then treated CT26 tumors intratumorally with doxorubicin in BALB/c and nude mice and showed that only immunocompetent mice were able to clear 40% of the tumors, suggesting the dependence of tumor clearance on immune responses. Our study has now definitely proven this concept and gone further in tracking of tumor-specific CD8+ T cells in the circulation in response to treatment, and in demonstrating CD8+ T-cell-dependent immunological memory, effects on distant untreated tumors, and synergy with checkpoint blockade. This has direct relevance to the clinic. A study by Scurr et al. demonstrated that low dose oral cyclophosphamide treatment in advanced stage colorectal cancer patients led to a marked expansion of tumor antigen, 5T4-specific CD8+ T cells expressing granzyme B in peripheral blood, which correlated with prolonged progression-free survival.37
Even though we have focused on treatment of a colorectal origin and orthotopic breast origin cancer, different cytotoxic agents have efficacy in different tumor types and may cause different types of ICD. Drugs composed of multiple agents with advanced cell uptake may, therefore, be able to maximize ICD pathways and consequently increase the probability of turning tumor tissue into a cancer vaccine. INT230-6’s in situ vaccine potential was tested only in the C26 and orthtopic 4T1 models here, but these models constitute a valuable proof of principle.
When looking more closely at the C26 tumor microenvironment, we were surprised to see that tumor antigen-specific CD8+ T cells can readily be detected in tumor tissue from treatment-naïve animals but were less prevalent in the circulation, suggesting activation and expansion of tumor-specific CD8+ T cells by C26 and homing to the tumor tissue. These cells expressed high levels of PD-1 and had an activated phenotype that was defined by CD69+CD44hiCD62L−. A recent study by Enamorado et al. 2017 showed that in order to induce efficacious cancer vaccines tissue resident memory CD8+ T cells need to infiltrate the tumor.38 These cells are defined by expression of CD69 and CD103. As we have not been able to look at the CD8+ T cells recovered from infiltrates of treated tumors (because they are rejected) or at CD103+ cells, we are not able to further subdivide the tumor-infiltrating CD8+ T cells. Nevertheless, we have clearly shown that upon treatment with INT230-6 CD8+ T cells home to the tumor tissue (Figure 4). The presence of lymphocyte infiltration into untreated C26 tumors suggests that tumors were ‘hot’ rather than ‘cold’. CT26, a sister cell line, has been characterized as an immunogenic tumor model.39 On the one hand, this limited our insight into de novo responses of less infiltrated tumors, but on the other hand, this allowed us to track and test the tumor antigen-specific CD8+ T cell responses. For example, we were able to examine functionality of tumor antigen-specific CD8+ T cells from mice with complete response. We found that tumor antigen-specific CD8+ T cells were functionally active and possibly of low functional avidity. This agrees with the fact that high avidity T cells against self are deleted in the thymus. As the tumor antigen epitope in this study, AH-1, is derived from the endogenous retroviral protein, gp70, that is not expressed in healthy murine tissue due to DNA methylation,40 it may not have been expressed in the thymus. Furthermore, low affinity or low avidity may also partially explain why tumor antigen-specific CD8+ T cells were not able to eradicate the untreated tumor despite being present in the tumor.
To clear primary C26 tumors, Bender et al. (2018 submitted) combined INT230-6 with anti-PD-1 or anti-CTLA-4. They determined that INT230-6 and anti-PD-1 synergized most efficiently. In this study, we studied combinational treatment on untreated contralateral tumors. Due to the high expression of PD-1 on tumor antigen-specific CD8+ T cells, we also expected a synergistic effect with anti-PD-1. However, using a dual tumor model we surprisingly found that synergy was achieved more effectively with anti-CTLA-4, rather than anti-PD-1. As anti-CTLA-4 promotes entry of new T cells into the tumor in addition to CD4+ and CD8+ T cell activation, whereas anti-PD-1 targets mainly rejuvenation of exhausted CD8+ T cells,41–44 it may be possible that anti-CTLA-4 synergizes by promoting entry of the endogenous vaccine-induced T cells into the small untreated contralateral tumors, which may not have recruited or induced their own T cells. Another explanation could be that there is limited expression of PD-L1 in the untreated contralateral tumors, making blockade of the PD-1/PD-L1 axis less critical. This can be further explored in the future.
The combinational studies with anti-PD-1 and anti-CTLA-4 were not run in parallel. In fact, the contra-lateral tumors that were studied in combination with anti-PD-1 were inoculated only three days before treatment onset. Interestingly, treatment of tumor with vehicle, which causes damage to the tumor structure, increased the immune response enough to reduce tumor burden, resulting in 2/10 mice losing their contralateral tumors. When contralateral tumors were given seven days before vehicle treatment so that tumors were more established (30% larger), all contralateral tumors persisted. This is consistent with the model of tumor immunosurveillance – the immune system is able to detect and destroy tumor cells, but after longer exposure will be overridden by escape mechanisms such as immunosuppression.45 Interestingly, this was recently also confirmed in patients on anti-PD-1 treatment. Response to the checkpoint inhibitor was correlated with tumor burden at treatment onset. As anti-PD-1 targets T cells, this can be interpreted in the wider sense that there is a threshold for T cells to induce regression.46 The fact that anti-PD-1 had lower efficacy in smaller contralateral tumors than anti-CTLA-4 did even in larger contralateral tumors suggests the greater efficacy at the distant site of anti-CTLA-4 even though experiments were not performed in parallel.
To highlight the important findings from this paper, it is best to compare two papers, studies by Sagiv-Barfi et al.47 and by Ariyan et al.48 The Sagiv-Barfi study induces in situ vaccination by agonistic anti-OX40 and CpG intratumoral delivery47 CpG, acting through Toll-like receptor 9, induces OX40 expression, allowing agonistic anti-OX40 to synergistically stimulate anti-tumor immunity. This treatment regimen induced complete regression of not only treated primary tumors but also contralateral tumors. This efficacy was not achieved by monotherapies, showing that stimulation of pattern recognition receptors (as induced by DAMPS) and activation of T cells need to be targeted in order to induce the in situ vaccine conversion, similar to cytotoxic agents’ synergy with a checkpoint inhibitor. Ariyan et al.48 combined local chemotherapy, melphalan, with systemic anti-CTLA-4 in pre-clinical models and clinical melanoma. The authors found that the combination increased response to local and systemic tumors with 62% having a complete response and remaining tumor free after one year. They determined that even though tumors and peripheral blood of patients had low T cell numbers at treatment onset, an increase could be measured for CD4+ and CD8+ T cells in both compartments suggesting a systemic immune response, which is in agreement with our study. However, the role of T cells in the Ariyan et al. study was inferred from correlations with response,48 whereas in the present study, we proved by T cell depletion that the induction of CRs of the primary tumor as well as long-term memory of 3 months or longer were both dependent on both CD4+ and CD8+ T cells.
To conclude, INT230-6 is composed of two cytotoxic agents with a diffusion enhancer agent. This study shows that this intratumoral treatment can cause immunogenic cell death that results in an endogenous personalized vaccine. This effect is likely due to release of DAMPS that can recruit presenting cells into the tumor, which in turn can process tumor antigens and induce an expanded population of antigen-specific CD8+ T cells and long-term T cell memory. These effects are augmented with the co-administration of checkpoint inhibitors. Our results suggest that novel intratumoral approaches can be designed to provide a new strategy for effective immunotherapy of cancer. INT230-6 is currently being evaluated in a Phase I/II clinical trial for solid tumors.
Materials and methods
Mice
Female and male BALB/c mice were obtained from Animal Production Colonies, Frederick Cancer Research Facility, National Cancer Institute (NCI, Frederick, MD). Female BALB/c RAG1-deficient (The Jackson Laboratory, Bar Harbor, ME) were bred under specific pathogen-free and Helicobacter-free conditions at the NCI. NCI’s Animal Care and Use Committee permitted and guided all experiments that were performed with mice. Mice were at least seven weeks of age.
Reagents
INT230-6 contains 0.5 mg/ml cisplatin (Tocris, Minneapolis, MN), 0.1 mg/ml vinblastine (Tocris) and 10 mg/ml SHAO cell penetration enhancer (Intensity Therapeutics, Westport CT) (Bender et al. 2018, submitted). The enhancer is 8-((2-hydroxybenzoyl)amino)octanoate or SHAO, with the structure:
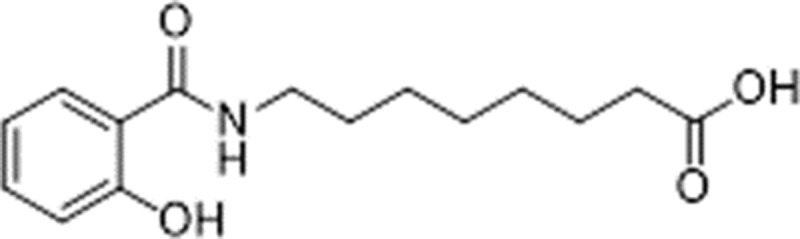
It has no pharmacological activity on its own. It is dissolved using NaOH and then Tween 80 (final concentration 0.2%) is added and the solution neutralized. The final formulation of INT230-6 contained 10 mg/ml of SHAO, 0.5 mg/ml of cisplatin, and 0.1 mg/ml of vinblastine. The vehicle control is just an aqueous solution of Tween 80 0.2% but neither of the cytotoxic agents or the enhancer. It was either prepared under sterile conditions in house or provided by Intensity Therapeutics in sterile vials. The selection of this compound was empirical. Intensity Therapeutics originally empirically screened six different cytotoxic agents for killing of tumor cells in vitro, and selected the two most cytotoxic, cisplatin and vinblastine, to combine in the INT230-6. The ratios were then optimized for maximum killing in vitro as well.
Cell lines
The Colon26 (C26) BALB/c murine carcinoma cell line (obtained from Charles River Laboratories, Raleigh) was cultured in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 100 units/ml Penicillin and 100 μg/ml Streptomycin, 2 mM L-glutamine and 10 mM HEPES.
4T1 mammary carcinoma cells were purchased from ATCC. The cells were cultured and expanded in RPMI-1640 medium supplemented with 10% Fetal Bovine Serum, 100 units/ml Penicillin and 100ug/ml Streptomycin. Low passage (0.5x106) cells (P4-5) were inoculated into BALB/c mice via mammary fat pad injection.
The A20 BALB/c murine B lymphoma cell line was cultured in T cell medium consisting of RPMI-1640, supplemented with 10% FCS, 100 units/ml penicillin and 100 μg/ml streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate, and 1x nonessential amino acids (Gibco Laboratories, Gaithersburg, MD) and 5 × 10−5 M 2-mercaptoethanol.
All cells were maintained at 37°C and 5% CO2.
Tumor models
A single cell suspension of C26 cells in Hank’s Balanced Salt Solution was prepared and inoculated subcutaneously (SC) into the right flank (1x106 cells/mouse). Tumor sizes were monitored by caliper gauge two to three times a week and converted to tumor volume (V) calculated from length (L) and width (W) as following: V = (W2 × L)/2 (Faustino-Rocha 13 Lab Animal). After tumors reached an average size of 300 mm3, mice were randomized into appropriate treatment groups. Intratumoral (IT) treatment of INT230-6 or vehicle was performed at 50 µl or 100 µl/400 mm3 for 3 to 5 sequential days.
Re-challenges were performed by inoculation of C26 (1x106 cells/mouse) cells SC into the left flank or intravenously (IV) before monitoring tumor growth either by caliper gauge or labored breathing. Lungs from IV-challenged mice were harvested, stained and fixed, and the number of nodules was quantified to determine tumor burden.49
Dual tumor challenges were performed by first inoculating C26 cells (1x106) SC into the right flank. The second challenge was either IV (2.5x105 C26 cells) 7 days after the first challenge or SC into the left flank (1x106 C26 cells) 7 to 11 days after the initial inoculation. Tumor growth was monitored at both tumor sites as described above. INT230-6 was given 14–15 days after primary tumor inoculation (50 µl/400 mm3 for 5 sequential days). Combinational therapy was performed by intraperitoneal (IP) injection of 0.1 mg anti-PD-1 (clone RMP1-14, Bio X Cell, West Lebanon, NH) on day 0, 3, 7 and 10 after INT230-6 treatment onset or 0.1 mg anti-CTLA-4 (clone 9H10, Bio X Cell) on day 0, 3 and 6 post INT230-6 treatment onset. Rat immunoglobulin G (IgG, 0.1 mg, Sigma-Aldrich, St. Louis, MO) was administered as a control for anti-PD-1 treatment and Syrian hamster IgG (Bio X Cell) as control for anti-CTLA-4.
For the 4T1 orthotopic breast cancer model, BALB/c female mice were shaved around the 4th nipple and right flank area. The mammary fat pad was located adjacent to the 4th nipple. A single cell suspension of 0.5 × 106 cells/100 µl was injected into the mammary fat pad. Tumors were allowed to grow up to an average size of ~200 mm3 before the beginning of treatment with either INT230-6 or vehicle.
In vivo T lymphocyte depletion
T cells were depleted in vivo by IP injection of 0.1 mg anti-CD4 (clone GK1.5, Harlan Laboratories, Frederick, MD) and/or 0.2 mg anti-CD8 (clone 2.43, Harlan Laboratories) during INT230-6 treatment experiments on days 0, 1, 5, 8 and 15 after the initial INT230-6 dose. Depletion in re-challenge studies was performed with 0.15 mg anti-CD8 and/or anti-CD4 on days −1, 3, 5, 12 and 19. Rat IgG (0.3 mg) was administered as a control for antibody treatments.
Immunohistochemistry staining of CD8
C26 tumors were collected at treatment onset and on day 3, 5, 8, 11 and 14 after onset. Tumors were fixed in neutral-buffered formalin, embedded in paraffin wax and cut into 5 µm thick sections. Sections were stained with hematoxylin and eosin (H&E). CD8 (clone YTS169.4, Abcam) staining was performed on destained H&E slides. Secondary anti-rat antibody was utilized before the DAB substrate was applied. Mounted slides were digitalized with Aperio ScanScope (Leica). Cell quantification was performed according to McCarty Jr et al.50 Cell quantification is reported as number of positive cells per mm2.
Splenocytes, peripheral blood, tumor draining lymph node and tumor infiltrating leukocytes
Retro-orbital bleeds were performed at treatment onset as well as on days 7, 14, 21 and 28 after onset when monitoring lymphocytes during the treatment course. Blood was collected in 0.1 M EDTA, lysed with Ammonium-Chloride-Potassium Lysing Buffer (Lonza, Allendale, NJ) and washed with RPMI-1640.
When euthanizing tumor-bearing mice, retro-orbital bleeds were performed before harvesting and generating single cell suspensions from spleens and tumor-draining lymph nodes (axillary and inguinal). Finally, tumor infiltrating leukocytes (TILs) were prepared by detaching tumors from the skin and homogenizing them on a gentleMACS Dissociators in gentleMACS™ C Tubes containing a digestion buffer (RPMI-1640 with 50 µg/ml Liberase TM (Roche Molecular Systems Inc., Branchburg, NJ), and 0.2 mg/ml DNase I, Sigma-Aldrich). Digestion followed by incubation at 37°C in a rotating water bath for 40 min at 100 rpm for agitation. Cells were then passed through a 100 µm nylon membrane, washed and fractionated by Histopaque 1083 to obtain leukocytes.
Flow cytometry
To monitor peripheral AH-1-specific CD8+ T cells in blood, cells were incubated with anti-CD16/CD32 (clone 93, Biolegend, San Diego, CA) and with AH-1/H-2Ld -tetramers (NIH Tetramer Facility) before staining with FITC-anti-TCR β chain (clone H57-597, Biolegend) and PE-anti-CD8a (clone 53–6.7, BD Biosciences, San Jose, CA).
Single cell suspensions from organs harvested from naïve and tumor-bearing mice were stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific, Waltham, MA) followed by incubation with anti-CD16/CD32 and AH-1/H-2Ld -tetramers. Finally, cells were stained with Alexa Fluor 700 anti-CD45 (clone 30-F11, Biolegend), APC-Cy7 anti-TCR β chain (clone H57-597, Biolegend), BV650 anti-CD8a (clone 53–6.7 Biolegend), FITC anti-CD69 (clone H1.2F3, Biolegend), PerCP-Cy5.5 anti-CD44 (clone 1M7, Biolegend), BV421 anti-CD62L (clone Mel-14, Biolegend), and anti-PD-1 (clone RMP1-30, Biolegend) for the evaluation of CD8+ T cell characteristics. Gates were set with fluorescent-1 (FMO) controls to account for spillover from other channels, but the antibodies used had been validated also previously with isotype controls.
All cells were visualized on an LSRII or FACSymphony using DIVA software (BD Biosciences). Data were analyzed by FlowJo (FlowJo, LLC, Ashland, Oregon).
Cytotoxic T lymphocyte killing assay
Splenocytes, from mice with complete response, were prepared and plated in 24-well culture plates (Corning, Corning, NY) at 4 × 106 cells/well and incubated for 7 days with 1 μM or 1 nM AH-1 (SPSYVYHQF) peptide (United BioSystems, Herndon, VA). Cultures were supplemented with 10% T-STIM Culture Supplement Rat, without CON A (Corning) on day 2 and 4. The CD8+ T cell cytotoxicity was measured with the Pierce™ LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Briefly, all cells were resuspended and incubated in T cell medium containing 5% FCS. Target cells (1x104, C26 and A20 cells) were cultured with different ratios of stimulated splenocytes containing effector cells (25:1 to 3.125:1 effector:target ratio) in a 96-well flat-bottom plate (Corning) for 4 h in triplicate. Supernatant was then transferred into new plates and incubated for 30 min with the reaction mix before stopping the reaction and measuring absorbance at 490 and 680 nm. Cytotoxicity (%) was calculated the following: (Sample LDH activity – spontaneous LDH activity)/(maximum LDH activity – spontaneous LDH activity) x 100.
Statistics
The data were analyzed using the Log-rank test, Mann–Whitney test, Kruskal–Wallis with Tukey post hoc test or two-way ANOVA with Sidak’s test using GraphPad Prism (version 7, GraphPad Software, Inc., La Jolla, CA). Significance was determined at p < 0.05, and all experiments were performed at least twice to guarantee reproducibility.
Funding Statement
This work was funded by the intramural research program of the Center for Cancer Research, National Cancer Institute, project [Z01-C-004020], and by a Collaborative Research and Development Agreement with Intensity Therapeutics. We also thank the Gui Foundation for partial funding support;National Cancer Institute.
Acknowledgments
We would like to thank the Vaccine Branch Flow Core, the NCI animal facilities and Dr. David Venzon for statistical analysis of the data, and Dr. Julian Burks for help with the orthotopic tumor model studies.
Disclosure statement
The NCI authors of no conflict of interest to disclose. LB and IW are employees of Intensity Therapeutics which has developed and owns the INT230-6 technology.
References
- 1.Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Palumbo MO, Kavan P, Miller WH Jr., Panasci L, Assouline S, Johnson N, Cohen V, Patenaude F, Pollak M, Jagoe RT, et al. Systemic cancer therapy: achievements and challenges that lie ahead. Front Pharmacol. 2013;4:57. doi: 10.3389/fphar.2013.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015;3:436–443. doi: 10.1158/2326-6066.CIR-15-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy. 2016;8:821–837. doi: 10.2217/imt-2016-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28:383–391. doi: 10.1093/intimm/dxw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 9.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bilusic M, Gulley JL. Local immunotherapy: a way to convert tumors from “Cold” to “Hot”. J Natl Cancer Inst. 2017;109:djx132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohtsukasa S, Okabe S, Yamashita H, Iwai T, Sugihara K. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J Cancer Res Clin Oncol. 2003;129:719–726. doi: 10.1007/s00432-003-0492-0. [DOI] [PubMed] [Google Scholar]
- 13.Kaur R, Kaur G, Gill RK, Soni R, Bariwal J. Recent developments in tubulin polymerization inhibitors: an overview. Eur J Med Chem. 2014;87:89–124. doi: 10.1016/j.ejmech.2014.09.051. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka H, Matsushima H, Nishibu A, Clausen BE, Takashima A. Dual therapeutic efficacy of vinblastine as a unique chemotherapeutic agent capable of inducing dendritic cell maturation. Cancer Res. 2009;69:6987–6994. doi: 10.1158/0008-5472.CAN-09-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 16.Galluzzi L, Zitvogel L, Kroemer G. Immunological mechanisms underneath the efficacy of cancer therapy. Cancer Immunol Res. 2016;4:895–902. [DOI] [PubMed] [Google Scholar]
- 17.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 18.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 19.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi: 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- 20.Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, Michaud M, Menger L, Gdoura A, Tajeddine N, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene. 2011;30:1147–1158. doi: 10.1038/onc.2010.500. [DOI] [PubMed] [Google Scholar]
- 21.Hato SV, Khong A, de Vries IJ, Lesterhuis WJ. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res. 2014;20:2831–2837. doi: 10.1158/1078-0432.CCR-13-3141. [DOI] [PubMed] [Google Scholar]
- 22.Nio Y, Hirahara N, Minari Y, Iguchi C, Yamasawa K, Toga T, Tamura K. Induction of tumor-specific antitumor immunity after chemotherapy with cisplatin in mice bearing MOPC-104E plasmacytoma by modulation of MHC expression on tumor surface. Anticancer Res. 2000;20:3293–3299. [PubMed] [Google Scholar]
- 23.Weide B, Eigentler TK, Pflugfelder A, Zelba H, Martens A, Pawelec G, Giovannoni L, Ruffini PA, Elia G, Neri D, et al. Intralesional treatment of stage III metastatic melanoma patients with L19-IL2 results in sustained clinical and systemic immunologic responses. Cancer Immunol Res. 2014;2:668–678. doi: 10.1158/2326-6066.CIR-13-0206. [DOI] [PubMed] [Google Scholar]
- 24.Murthy V, Minehart J, Sterman DH. Local immunotherapy of cancer: innovative approaches to harnessing tumor-specific immune responses. J Natl Cancer Inst. 2017;109:djx097. [DOI] [PubMed] [Google Scholar]
- 25.Terabe M, Robertson FC, Clark K, De Ravin E, Bloom A, Venzon D, Kato S, Mirza A, Berzofsky JA. Blockade of only TGF-β 1 and 2 is sufficient to enhance the efficacy of vaccine and PD-1 checkpoint blockade immunotherapy. Oncoimmunology. 2017;6:e1308616. doi: 10.1080/2162402X.2017.1308616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terabe M, Ambrosino E, Takaku S, O’Konek JJ, Venzon D, Lonning S, McPherson JM, Berzofsky JA. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res. 2009;15:6560–6569. doi: 10.1158/1078-0432.CCR-09-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakashima H, Terabe M, Berzofsky JA, Husain SR, Puri RK. A novel combination immunotherapy for cancer by IL-13Ralpha2-targeted DNA vaccine and immunotoxin in murine tumor models. J Immunol. 2011;187:4935–4946. doi: 10.4049/jimmunol.1102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 29.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 32.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–23 e4. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldberg EP, Hadba AR, Almond BA, Marotta JS. Intratumoral cancer chemotherapy and immunotherapy: opportunities for nonsystemic preoperative drug delivery. J Pharm Pharmacol. 2002;54:159–180. [DOI] [PubMed] [Google Scholar]
- 34.Gao L, Xia L, Zhang R, Duan D, Liu X, Xu J, Luo L. Enhanced antitumor efficacy of poly(D,L-lactide-co-glycolide)-based methotrexate-loaded implants on sarcoma 180 tumor-bearing mice. Drug Des Devel Ther. 2017;11:3065–3075. doi: 10.2147/DDDT.S143942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu B, Lu Y, Gao F, Jing P, Wei H, Zhang P, Liu G, Ru N, Cui G, Xu X, et al. Hapten-enhanced therapeutic effect in advanced stages of lung cancer by ultra-minimum incision personalized intratumoral chemoimmunotherapy therapy. Lung Cancer (Auckl). 2015;6:1–11. doi: 10.2147/LCTT.S70679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathios D, Kim JE, Mangraviti A, Phallen J, Park CK, Jackson CM, Helman LJ, Kastan MB, Knapp DW, Levin WJ, et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. 2016;8:370ra180. doi: 10.1126/scitranslmed.aaf0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, Bridgeman H, Adams R, Brewster A, Jones R, et al. Low-dose cyclophosphamide induces antitumor T-cell responses, which associate with survival in metastatic colorectal cancer. Clin Cancer Res. 2017;23:6771–6780. doi: 10.1158/1078-0432.CCR-17-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, Mejías-Pérez E, Esteban M, Melero I, Hidalgo A, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. 2017;8:16073. doi: 10.1038/ncomms16073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, Ohlfest JR, Hu P, Epstein AL. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurst TP, Magiorkinis G. Activation of the innate immune response by endogenous retroviruses. J Gen Virol. 2015;96:1207–1218. doi: 10.1099/jgv.0.000017. [DOI] [PubMed] [Google Scholar]
- 41.Tan LY, Martini C, Fridlender ZG, Bonder CS, Brown MP, Ebert LM. Control of immune cell entry through the tumour vasculature: a missing link in optimising melanoma immunotherapy? Clin Transl Immunol. 2017;6:e134. doi: 10.1038/cti.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and Anti-PD-1 checkpoint blockade. Cell. 2017;170:1120–33 e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swann JB, Smyth MJ. Immune surveillance of tumors. Jci. 2007;117:1137–1146. doi: 10.1172/JCI31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, Levy R. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med. 2018;10:eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ariyan CE, Brady MS, Siegelbaum RH, Hu J, Bello DM, Rand J, Fisher C, Lefkowitz RA, Panageas KS, Pulitzer M, et al. Robust antitumor responses result from local chemotherapy and CTLA-4 blockade. Cancer Immunol Res. 2018;6:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park JM, Terabe M, van Den Broeke LT, Donaldson DD, Berzofsky JA. Unmasking immunosurveillance against a syngeneic colon cancer by elimination of CD4+ NKT regulatory cells and IL-13. Int J Cancer. 2005;114:80–87. doi: 10.1002/ijc.20669. [DOI] [PubMed] [Google Scholar]
- 50.McCarty KS Jr., Miller LS, Cox EB, Konrath J, McCarty KS. Sr. Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch Pathol Lab Med. 1985;109:716–721. [PubMed] [Google Scholar]