

ABSTRACT
We have recently shown that chemotherapy with immunogenic cell death (ICD)-inducing agents can be advantageously combined with fasting regimens or caloric restriction mimetics (CRMs) to achieve superior tumor growth control via a T cell-dependent mechanism. Here, we show that the blockade of the CD11b-dependent extravasation of myeloid cells blocks such a combination effect as well. Based on the characterization of the myeloid and lymphoid immune infiltrates, including the expression pattern of immune checkpoint proteins (and noting a chemotherapy-induced overexpression of programmed death-ligand 1, PD-L1, on both cancer cells and leukocytes, as well as a reduced frequency of exhausted CD8+ T cells positive for programmed cell death 1 protein, PD-1), we then evaluated the possibility to combine ICD inducers, CRMs and targeting of the PD-1/PD-L1 interaction. While fasting or CRMs failed to improve tumor growth control by PD-1 blockade, ICD inducers alone achieved a partial sensitization to treatment with a PD-1-specific antibody. However, definitive cure of most of the tumor-bearing mice was only achieved by a tritherapy combining (i) ICD inducers exemplified by mitoxantrone and oxaliplatin, (ii) CRMs exemplified by hydroxycitrate and spermidine and substitutable for by fasting, and (iii) immune checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 interaction. Altogether, these results point to the possibility of synergistic interactions among distinct classes of anticancer agents.
KEYWORDS: Caloric restriction mimetics, immune checkpoint blockers, chemotherapy, combination therapies, tumor immune infiltrate
Introduction
In spite of ramping progress in cancer therapy, definitive cure is still a close-to-utopian goal. Even immunotherapies with immune checkpoint inhibitors (ICIs) that have become en vogue over that last decade only achieve cure in rather exceptional circumstances (apart from the treatment of melanoma), meaning that they usually delay tumor progression, and this in a limited fraction of patients (in the range of 20–30%) that carry cancers for which the ICI is clinically approved.1–9
Over the past decade, it has become increasingly accepted that long-term effects of conventional chemotherapies involve an immunological component.10–12 Indeed, chemotherapy with a specific subclass of cytotoxic agents that is referred to as ‘immunogenic cell death’ (ICD) inducers13 only delayed tumor growth when administered to mice bearing an intact immune system.14,15 ICD induced by such chemotherapeutics, exemplified by mitoxantrone (MTX) and oxaliplatin (OXA), is characterized by a series of premortem stress responses in cancer cells that allow them to alert innate immune effectors, in particular dendritic cells (DCs), to initiate an anticancer response by cross-presenting tumor antigens to cytotoxic T lymphocytes (CTLs).16–19 ICD inducers are widely used in cancer therapy and are still undergoing clinical evaluation.20,21
One of the premortem responses that is elicited by ICD-inducing chemotherapeutics is autophagy,22 which in turn facilitates the release of adenosine triphosphate (ATP) from dying cancer cells.23 Extracellular ATP acts on purinergic receptors to attract DC precursors into the tumor bed and to facilitate their local activation.16,24 Of note, it appears that immunostimulatory autophagy can be activated by dietary manipulations, in particular short-term starvation, or by a new class of pharmacological agents dubbed ‘caloric restriction mimetics’ (CRMs) that induce autophagy in a non-immunosuppressive fashion.25–28 In numerous preclinical models, fasting, caloric restriction and CRMs have proven healthy benefits, precisely by extending longevity and life expectancy in good health, by slowing down neurodegeneration, or decreasing the incidence of many pathologies, such as cardiovascular, metabolic, and inflammatory diseases, as well as cancer; this prophylactic efficacy being investigated in humans with some corroborating evidences recently published.29–38 In a therapeutic setting, the combination of CRMs with ICD inducers yields superior outcome compared to monotherapies with ICD-stimulatory pharmacological compounds or to the administration of CRMs alone (which usually do not affect tumor growth). Of note, the efficacy of the combination of CRMs with ICD inducers also relies on the immune system, meaning that depletion of CD8+ T cells suffices to abolish tumor growth reduction.27
Clinically approved ICIs either target cytotoxic T lymphocyte-associated protein 4 (CTLA-4) or the interaction between programmed cell death 1 (PD-1) and programmed cell death-ligand 1 (PD-L1).1–9,39,40 Several different monoclonal antibodies targeting PD-1/PD-L1 are now used as a sort of general therapy against multiple distinct cancer types, thus representing the only truly transversal antineoplastic strategy. Notwithstanding their broad application, the efficacy of immunotherapies targeting PD-1/PD-L1, alone or in combination with CTLA-4 is limited, requiring combination with available or yet-to-be-developed anticancer drugs.41,42
In the recent past, together with other groups, we have launched the hypothesis that ICD inducers might be used to sensitize cancers to ICI-based immunotherapy.43–45 Indeed, cancers that are pretreated with two ICD inducers (OXA and cyclophosphamide) are sensitized to subsequent ICIs.43,45 Here, we examined the hypothesis that CRMs might be advantageously combined with ICIs as well. While CRMs alone failed to sensitize to ICIs, combination treatments relying on the use of ICD inducers plus CRMs were particularly successful in rendering mouse cancers susceptible to complete remission mediated by ICIs. In other words, a triple combination involving ICD inducers, CRMs, and ICIs targeting the PD-1/PD-L1 interaction, allowed to cure established mouse cancers. We are now aiming at evaluating such tritherapy in cancer patients.
Results
CD11b blockade interferes with the anticancer effects of hydroxycitrate upon chemotherapy
The combination of the progesterone analog medroxyprogesterone (MPA) and repeated DNA damage by gavage with 2,4-dimethoxybenzaldehyde (DMBA) is highly efficient in inducing mammary carcinomas when administered to young female BALB/c mice (Figure 1(a,b)). In this model, the combination of MTX-based chemotherapy and the CRM hydroxycitrate (HC) demonstrates a potent efficacy in reducing tumor growth and prolonging mouse survival, much more so than MTX (Figure 1(c–h), Supplemental Table 1) or HC (data not shown) alone.27 These results were obtained in a ‘realistic’ setting in which treatments were started when the cancers could be detected by palpation and hence reached a surface of 25 mm2. Repeated injections of a monoclonal antibody (M1/70), that blocks CD11b-dependent extravasation of myeloid cells,16 significantly (**p= .0025) interfered with the tumor growth control by MTX + HC (Figure 1(c–g), Supplemental Table 1). Very similar results were obtained in a syngeneic model of transplantable MCA205 fibrosarcoma developing on immunocompetent C57Bl/6 mice (Figure 2(a)). Again, the combination treatment with MTX + HC was more successful in reducing tumor growth and in prolonging survival than MTX alone, and the efficacy of this treatment was decreased by CD11b blockade (Figure 2(b–j), Supplemental Table 2).
Figure 1.
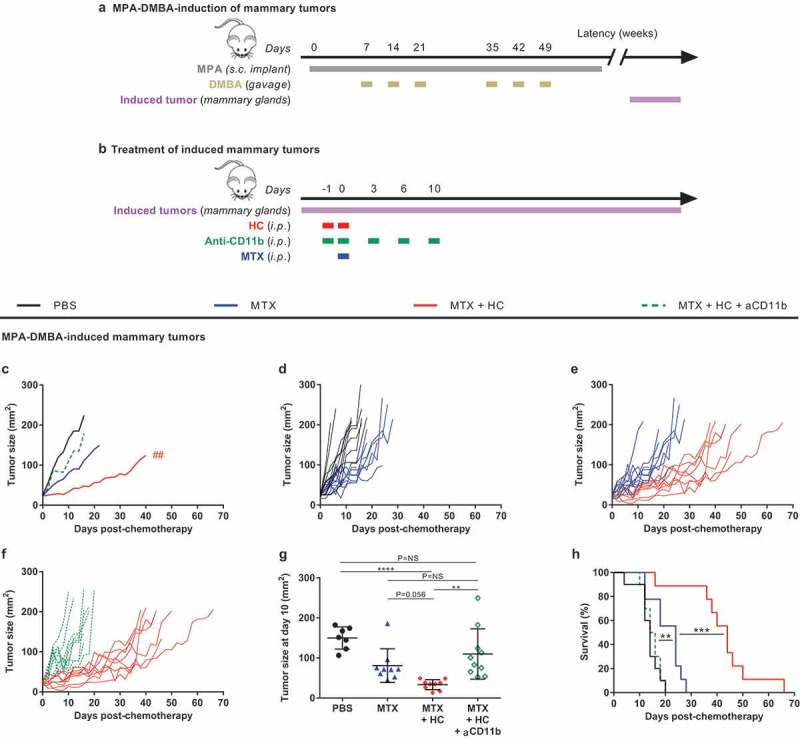
Myeloid cells contribute to the benefic action of the CRM HC to chemotherapy in a hormone- and carcinogen-induced mammary tumor model. (a) Experimental schedule of the MPA-DMBA-mediated induction of mammary tumors. Balb/c mice were implanted subcutaneously with a tablet continuously releasing the hormone MPA (day 0) followed by repeated oral gavages of the carcinogen DMBA (day 7, 14, 21, 35, 42 and 49). Breast cancer lesions become palpable a few weeks after the last gavage of DMBA. (b) Experimental schedule of the treatment of palpable mammary tumors. When the tumors reached ~25 mm2, mice were randomly assigned to the different treatment groups. Treatments consisted of the administration of (i) PBS (day 0; untreated control mice), (ii) chemotherapy alone (anthracycline MTX at day 0), (iii) chemotherapy + HC (delivered at day −1 and 0), (iv) chemotherapy + HC + anti-CD11b (CD11b-neutralizing antibodies injected at day −1, 0, 3, 6, 10). (c) Mean tumor growth curves of the treatment groups (n = 10/group). Curves were interrupted when more than 50% of the group had reached endpoint. (d–f) Individual tumor growth curves within each treatment group. (g) Dot plot illustrating the size of each individual tumor at day 10 post-chemotherapy. Mean ± SD is displayed. (h) Kaplan–Meier curves. Of note, some mice are shared with a previously reported data set (see Pietrocola F, Pol J et al. Cancer cell. 2016).27 ****p< .0001, ***p< .001, **p< .01 (comparisons with PBS or explicitly denoted by a segment); ##p< .01 (comparison between MTX + HC and MTX + HC + anti-CD11b); p= NS, not significant. For a detailed account of all comparisons, see Supplemental Table 1. CRM, caloric restriction mimetic; DMBA, 7,12-Dimethylbenz[a]anthracene; HC, hydroxycitrate; i.p., intraperitoneal; MPA, medroxyprogesterone acetate; MTX, mitoxantrone; PBS, phosphate-buffered saline; s.c., subcutaneous.
Figure 2.
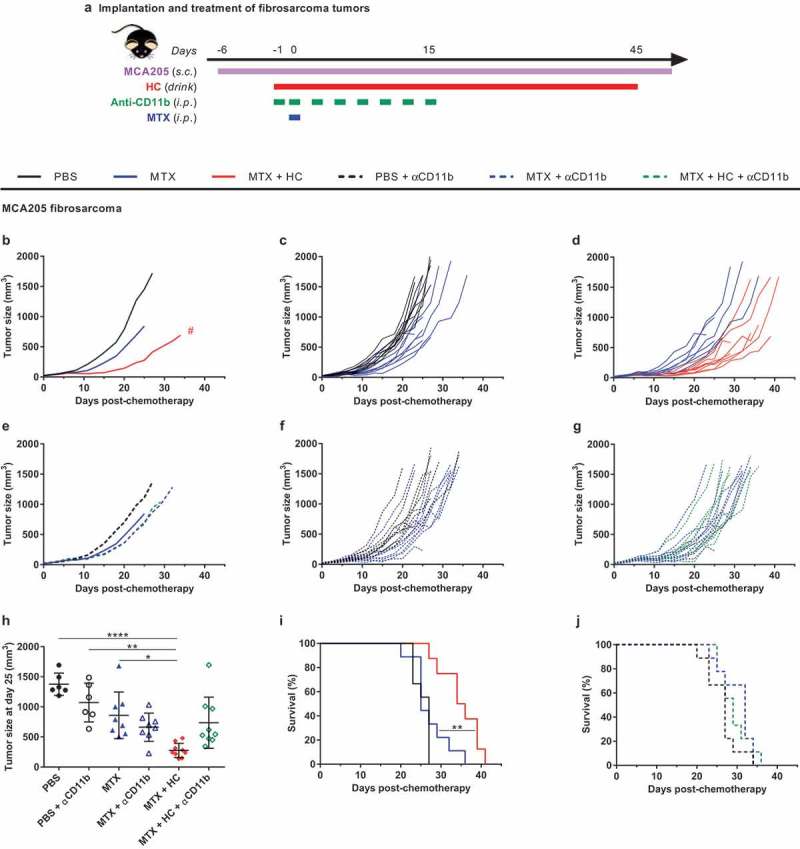
Myeloid cells are required for the benefic action of HC to chemotherapy in a fibrosarcoma tumor model. Experimental schedule of the implantation and treatment of syngeneic subcutaneous fibrosarcoma in C57Bl/6 mice (a) When tumors reached ~20 mm3, mice were randomly assigned to the different treatment groups. Treatments consisted of the administration of (i) PBS + isotype control antibody (untreated control mice), (ii) chemotherapy + isotype control antibody, (iii) chemotherapy + HC + isotype control antibody, (iv) PBS + anti-CD11b, (v) chemotherapy + anti-CD11b, (vi) chemotherapy + HC + anti-CD11b. The CRM HC was ingested through drinking water from day −1 to 45. Chemotherapy consisted of one injection of the anthracycline MTX at day 0 (or PBS in untreated controls). CD11b-neutralizing antibodies and isotype controls were injected at day −1, 0, 3, 6, 8, 10, 13 and 15. Mean (b) and individual (c,d) tumor growth curves of the treatment groups without CD11b neutralization. Mean (e) and individual (f,g) tumor growth curves of the treatment groups with CD11b neutralization. Of note, mean tumor growth curves in the panels B and E (n = 9/group) were interrupted when more than 50% of the group had reached endpoint. Dot plot (h) illustrating the size of each individual tumor at day 25 post-chemotherapy. Mean ± SD is displayed. Kaplan-Meier survival curves (i,j). ****p< .0001, **p< .01, *p< .05 (comparisons with PBS or explicitly denoted by a segment); #p< .05 (comparison between MTX + HC and MTX). For a detailed account of all comparisons, see Supplemental Table 2. CRM, caloric restriction mimetic; HC, hydroxycitrate; i.p., intraperitoneal; MTX, mitoxantrone; PBS, phosphate-buffered saline; s.c., subcutaneous.
Altogether, these results support the idea that myeloid cells (and presumably antigen-presenting cells) play a major role in the therapeutic efficacy of the combination of MTX + HC.
Effects of CRMs on the myeloid and lymphoid cancer immune infiltrate
Based on the aforementioned results, we decided to investigate the impact of fasting and of two different CRMs, namely HC and spermidine (Spd), on the composition of the immune infiltrate of cancers in the context of MTX-based chemotherapy. At day 3 post-chemotherapy (that was optionally preceded by a 2-day fasting regimen or by a 24-h treatment with either HC or Spd; the latter being maintained/repeated – Supplemental Figure 1(a)), no significant increments in the innate immune cell infiltrates (i.e. DC and natural killer cells) were detected in response to fasting, HC or Spd, perhaps because of the signs of immunodepletion mediated by MTX; a well-documented acute phenomenon46 (Supplemental Figure 1(b–e), Supplemental Table 8). However, RNA-Seq analyses of whole tumors yielded convincing evidence in favor of local immunomodulation by fasting or CRMs upon chemotherapy. Indeed, 480 genes involved in immunological activities were significantly affected between MTX chemotherapy alone versus MTX + fasting (n = 82) or MTX + Spd (n = 429) (Supplemental Figure 2(a)). Among them, 31 modulated genes were common to both combinatorial regimens and a REACTOME pathway analysis revealed a significant enrichment in 20 cellular pathways including the signaling of interleukins (i.e. Il1rap, Il6st, Il4ra, Il1rl1, Il13ra2) (Supplemental Figure 2(b)).
Thereafter, we concentrated our effort on immunophenotyping the leukocyte subsets infiltrating the tumor bed at day 11 post-chemotherapy (Figure 3(a)). At this later time point, MTX-induced adaptive antitumor immunity is activated and starts impairing tumor growth (Figure 2(b)). Chemotherapy-treated cancers contained a higher density of CD45+ leukocytes, more so when the animals were starved or received HC (Figure 3(b), Supplemental Table 3). Each of the co-treatments had a differential impact on the composition of the myeloid infiltrate. Thus, supplementation with HC caused an increased infiltration of the granulocyte neutrophils (phenotype: Ly6C+Ly6GhiCD11b+) (Figure 3(c), Supplemental Table 3) and of a particular monocyte-derived dendritic cell (moDC) subpopulation with activation/maturation markers (phenotype: Ly6ChiLy6G−CD11b+CD11c+CD80+MHC-IIhi), in comparison to MTX alone (Figure 3(d), Supplemental Table 3). Preconditioning by fasting (nutrient-free, NF) led to the expansion of a less activated moDC subpopulation (phenotype: Ly6G−Ly6ChiCD11b+CD11c+CD80+MHC-IIlo) (Figure 3(e), Supplemental Table 3). Supplementation with Spd further expanded a macrophage subpopulation with an M1 phenotype (Ly6G−F4/80+CD11c−CD11b+CD38+) (Figure 3(f), Supplemental Table 3) as compared to chemotherapy alone. The effects of starvation and CRMs were also determined at the level of the T lymphocyte infiltrate. Chemotherapy alone expanded the population of T cells, including CD8+ T lymphocytes, residing within the tumor bed (Figure 4(a,b), Supplemental Table 4), and increased the ratio of CD8+ over CD4+CD25+FoxP3+ regulatory T (Treg) cells (Supplemental Figure 3(a), Supplemental Table 9). Additionally, the activation status of the overall T cell compartment was analyzed. Upon MTX treatment, the production of type-1 cytokines, namely interferon-γ (IFNγ), tumor necrosis factor-α (TNFα) and interleukin-2 (IL-2), tended to increase following ex vivo stimulation with phorbol myristate acetate (PMA) + ionomycin (Supplemental Figure 3(b–d), Supplemental Table 9). Moreover, the CD8+ T cell subset demonstrated a wider spread and more intense expression of the early activation marker inducible T cell costimulator (ICOS), as well as a less frequent expression of the late activation/exhaustion marker PD-1 (Figure 4(c–f), Supplemental Table 4). The two activation molecules ICOS and PD-1 were detectable on half of the tumor-infiltrating CD8+ T lymphocytes. In comparison to chemotherapy alone, no significant impact of CRM supplementation or fasting preconditioning were observed on the CD8+ T cells/Tregs ratio (Supplemental Figure 3(a), Supplemental Table 9) or type-1 T helper/T cytotoxic (Th1/Tc1) response (Supplemental Figure 3(b–d), Supplemental Table 9). Still, a trend toward an increased production of TNFα was witnessed upon combination with HC (p= .061; Supplemental Figure 3(c), Supplemental Table 9). When combined with MTX, NF (but neither HC nor Spd) caused an increase in the density of total CD3+ and CD8+ T cell infiltrate (Figure 4(a,b), Supplemental Table 4); an observation that was recently reported in the literature with another anthracycline, namely doxorubicin, combined with a fasting-mimicking diet.47 At the same time, NF preconditioning supported chemotherapy-stimulated activation of CD8+ T lymphocytes (Figure 4(c), Supplemental Table 4) but promoted the engagement toward an exhausted phenotype at a level similar to untreated controls (Figure 4(e), Supplemental Table 4). As compared to MTX alone, HC supplementation showed clear evidence of an increased proportion of early activated lymphocytes (MTX versus MTX + HC: p= .078; a comparison that actually reached significance in our previous study when associated with the proliferation marker Ki6727) (Figure 4(c,d), Supplemental Table 4), yet without favoring exhaustion (Figure 4(e,f), Supplemental Table 4). Finally, Spd supplementation failed to further improve the overall T cell activation status as illustrated by the unchanged expression profile of ICOS and of PD-1 (Figure 4(c–f), Supplemental Table 4).
Figure 3.
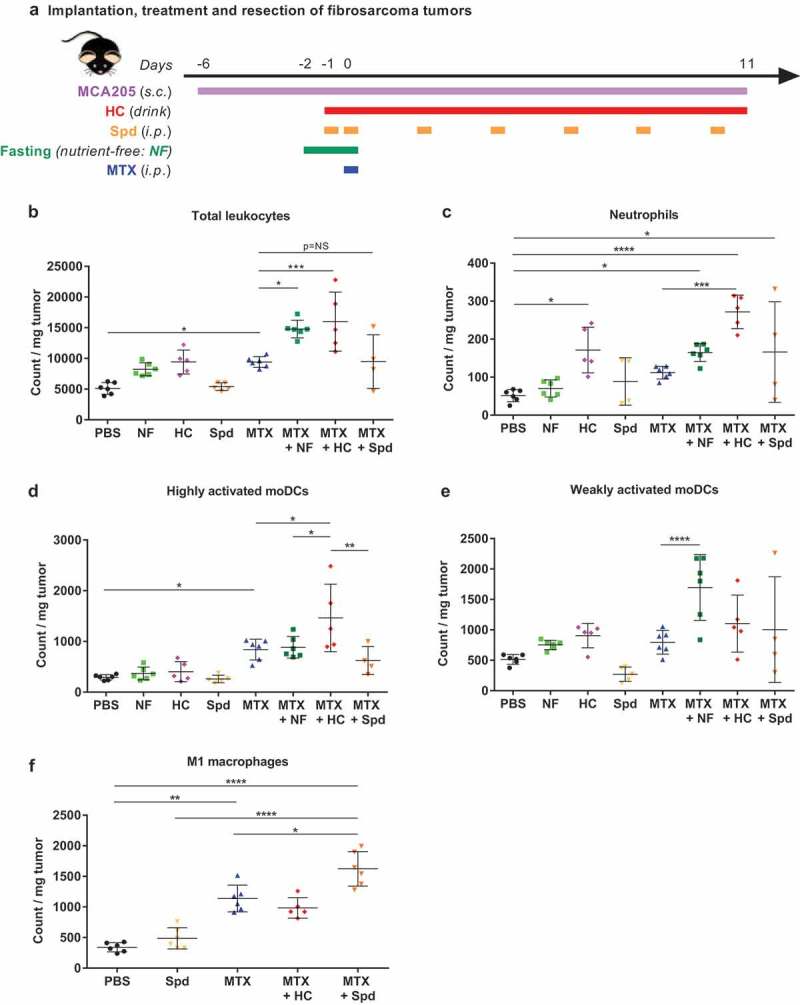
CRMs modulate tumor-infiltrating myeloid cell subsets. (a) Experimental schedule of the implantation and treatment of syngeneic subcutaneous fibrosarcoma in C57Bl/6 mice. When tumors reached ~20 mm3, mice were randomly assigned to the different treatment groups. Monotherapy regimens consisted of: (i) PBS (untreated control mice), (ii) MTX-based chemotherapy, (iii) fasting (labeled NF for “nutrient-free”), (iv) HC or (v) Spd. Bitherapies consisted of: (i) MTX + NF, (ii) MTX + HC or (iii) MTX + Spd. Chemotherapy consisted of one i.p. injection of the anthracycline MTX at day 0 (or PBS in untreated controls). Fasting lasted for 48 h starting at day −2. The CRM HC was continuously delivered through the drinking water starting at day −1. The CRM Spd was injected i.p. at day −1, 0, and then every 2–3 days. Eleven days post-chemotherapy, tumors were resected, processed and myeloid cell subsets were immunostained before flow cytometry-assisted analysis. (b) Total population of leukocytes (CD45+). (c) Neutrophils (CD45+CD11b+Ly-6C+Ly-6Ghi). (d) Highly activated moDCs (CD45+Ly-6ChiLy-6G−CD11b+CD11c+CD80+MHC-IIhi). (e) Weakly activated moDCs (CD45+Ly-6ChiLy-6G−CD11b+CD11c+CD80+MHC-IIlo). (f) Classically activated macrophages (M1; CD45+F4/80+CD11b+CD11c−CD38+). Dot plots illustrate the count of immune cells normalized per mg of tumor. Mean ± SD is displayed. ****p< .0001, ***p< .001, **p< .01, *p< .05; p= NS, not significant. For a detailed account of all comparisons, see Supplemental Table 3. CRM, caloric restriction mimetic; HC, hydroxycitrate; i.p., intraperitoneal; moDCs, monocyte-derived dendritic cells; MTX, mitoxantrone; NF, nutrient-free; PBS, phosphate-buffered saline; s.c., subcutaneous; Spd, spermidine.
Figure 4.
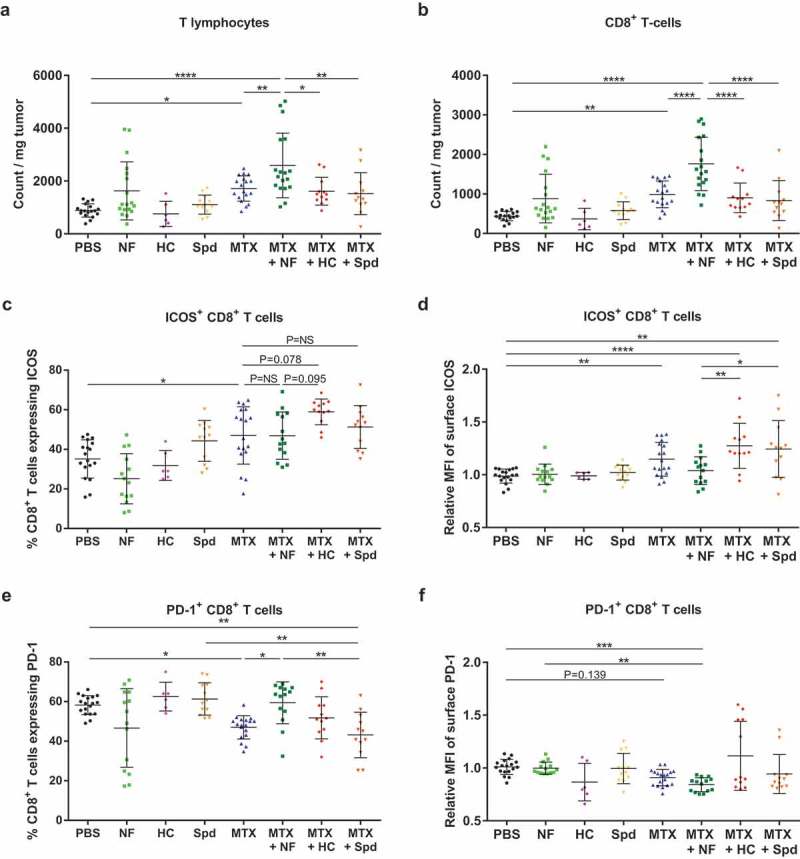
CRMs modulate tumor-infiltrating lymphoid cell subsets. Following the experimental schedule illustrated in Figure 3(a), T cell populations infiltrating the tumor microenvironment were analyzed by flow cytometry. (a) Total population of T lymphocytes (CD3+). (b) Total population of CD8+ T cells. (c) Percentage of CD8+ T cells expressing the early activation marker ICOS. (d) Level of expression of ICOS at the surface of CD8+ T cells (relative MFI). (e) Percentage of CD8+ T cells expressing the late activation/exhaustion molecule PD-1. (f) Level of expression of PD-1 at the surface of CD8+ T cells (relative MFI). Dot plots illustrate mean ± SD. ****p< .0001, ***p< .001; **p< .01, *p< .05; p= NS, not significant. For a detailed account of all comparisons, see Supplemental Table 4. HC, hydroxycitrate; ICOS, inducible T-cell costimulator; MFI, mean fluorescence intensity; MTX, mitoxantrone; NF, nutrient-free; PBS, phosphate-buffered saline; PD-1, programmed cell death protein 1; Spd, spermidine.
Altogether, it appears that fasting and CRMs further stimulated chemotherapy-induced activation/expansion of T cells and/or antigen-presenting cells, but achieved these effects through distinct mechanisms.
CRM-mediated sensitization to immune checkpoint blockade
We observed that treatment of MCA205 tumor-bearing mice with MTX induced the upregulation of PD-L1 both on non-leukocytes from the malignant tissue (CD45− cells, mostly cancer cells) (Figure 5(a,b), Supplemental Table 5) and in CD45+ leukocytes (Figure 5(c,d), Supplemental Table 5). This effect was not altered by co-treatment with starvation or the CRM HC (Figure 5(a–d)). Within tumor-infiltrating CD8+ T cells, no changes were observed in the distribution of surface expression of PD-1 (Figure 4(e–f)) and CTLA-4 (data not shown) in response to MTX alone versus its combination with CRMs. Nevertheless, fasting contributed to a wider spread of PD-1 expression (Figure 4(e)). MTX also induced an increase in PD-L2 expression in CD45− cells that was not affected by starvation nor by HC (Supplemental Figure 4, Supplemental Table 10).
Figure 5.
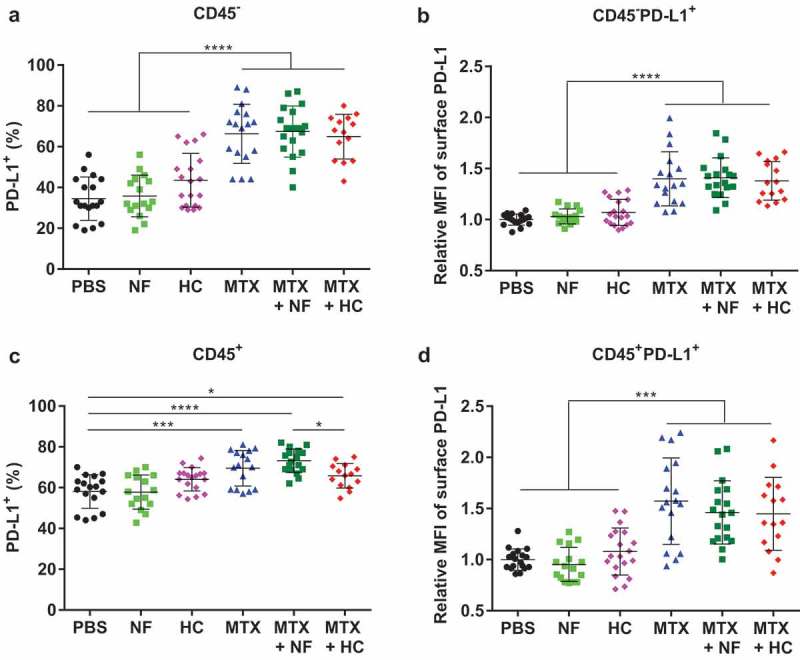
MTX-based chemotherapy impacts PD-L1 expression on both malignant cells and tumor-infiltrating immune cells, independently of CRMs. Following the experimental schedule illustrated in Figure 3(a), cell populations constituting the tumor microenvironment were analyzed by flow cytometry. Percentage of CD45− cells, mostly tumor cells, which express PD-L1 (a) and its surface expression level illustrated as relative MFI (b) Percentage of leukocytes (CD45+) cells expressing PD-L1 (c) and its surface expression level illustrated as relative MFI (d) Dot plots display mean ± SD. ****p< .0001, ***p< .001, *p< .05. For a detailed account of all comparisons, see Supplemental Table 5. HC, hydroxycitrate; MFI, mean fluorescence intensity; MTX, mitoxantrone; NF, nutrient-free; PBS, phosphate-buffered saline; PD-L1, programmed death-ligand 1.
Based on these results, we decided to investigate the possibility that MTX-based chemotherapy would sensitize the tumors to combinatorial immunotherapy targeting CTLA-4 and PD-1. To this aim, MCA205 fibrosarcoma-bearing mice received MTX-based chemotherapy alone or in association with fasting or CRMs (HC or Spd), followed by optional treatment with CTLA-4/PD-1-blocking antibodies starting at day 8 post-MTX (Figure 6(a)). Of note, in the absence of chemotherapy, MCA205 fibrosarcomas poorly responded to PD-1 blockade (2-day extension of the median survival in comparison to untreated controls), and this immunotherapy did not benefit from a pretreatment with starvation or the CRMs HC or Spd (Supplemental Figure 5, Supplemental Table 11). However, tumors pretreated with MTX responded to immunotherapy leading to complete tumor regression of a significant fraction of mice (2 out of 9) (Supplemental Figure 6, Supplemental Table 12). This fraction increased when MTX pretreatment was associated with starvation (4 out of 9 tumor-free mice), HC (8 out of 9 tumor-free mice) or Spd (7 out of 9 tumor-free mice) (Figure 6(b–d, f–h, j–l), Supplemental Table 6). Complete tumor regressions were associated with a remarkable extension of the survival but did not prevent most animals from succumbing to well-documented MTX-related long-term toxicity48 (Figure 6(e,i,m), Supplemental Table 6). Upon MTX plus CRM pretreatment, PD-1 blockade on its own was as efficient at curing mice as the dual therapy targeting both PD-1 and CTLA-4, while CTLA-4 blockade failed to match the performance (Supplemental Figure 7, Supplemental Table 13).
Figure 6.
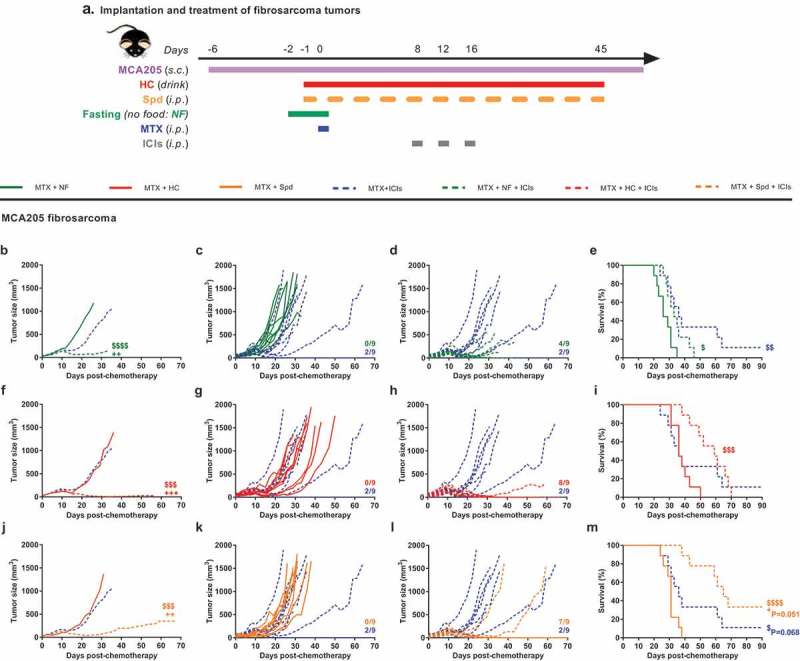
CRMs improve combinatorial treatment with MTX-based chemotherapy + ICIs. Experimental schedule of the implantation and treatment of syngeneic subcutaneous fibrosarcoma in C57Bl/6 mice (a) When tumors reached ~20 mm3, mice were randomly assigned to the different treatment groups. Monotherapy regimens consisted of: (i) PBS (untreated control mice) or (ii) MTX-based chemotherapy. Bitherapies consisted of: (iii) MTX + NF, (iv) MTX + HC, (v) MTX + Spd, and (vi) MTX + ICIs (cocktail of anti-PD-1 + anti-CTLA-4). Additionally, ICIs were evaluated in tritherapy regimens consisting of: (i) MTX + NF + ICIs, (ii) MTX + HC + ICIs, (iii) MTX + Spd + ICIs. Chemotherapy consisted of one i.p. injection of the anthracycline MTX at day 0 (or PBS in untreated controls). Fasting lasted for 48 h starting at day −2. The CRM HC was continuously delivered through the drinking water starting at day −1. The CRM Spd was injected i.p. at day −1, 0, and then every 2–3 days. ICIs were administered i.p. at day 8, 12, and 16. Mean (b, f, j) and individual (c, d, g, h, k, l) tumor growth curves of mice treated with bi- and tri-therapies. Of note, mean tumor growth curves (n = 9/group) were interrupted when more than 50% of the group had reached endpoint. On the panels displaying individual tumor growth curves, the number of animals that underwent complete tumor regression is indicated on the right end side. Kaplan–Meier curves (e, i, m). +++p < .001, ++p < .01 (comparisons to MTX + ICIs); $$$$p < .0001, $$$p < .001, $$p < .01, $p < .05 (comparisons to MTX + CRMs/NF). For a detailed account of all comparisons, see Supplemental Table 6. CRMs, caloric restriction mimetics; CTLA-4, cytotoxic T lymphocyte–associated protein 4; HC, hydroxycitrate; ICIs, immune checkpoint inhibitors; i.p., intraperitoneal; MTX, mitoxantrone; NF, nutrient-free; PBS, phosphate-buffered saline; PD-1, programmed cell death 1; s.c., subcutaneous; Spd, spermidine.
Rather similar results were obtained when MTX was replaced by another less toxic chemotherapeutic agent, oxaliplatin (OXA) (Figure 7(a)). Again, OXA alone sensitized to immunotherapy targeting solely PD-1 (i.e. without CTLA-4 blockade) and led to complete tumor regression in 8 out of 20 fibrosarcoma-bearing mice (Supplemental Figure 8, Supplemental Table 14). This cure rate of 40% with OXA + anti-PD-1 increased to 90% (9 out of 10 mice), 80% (16 out of 20 mice) and 70% (7 out of 10 mice) when fasting, HC and Spd, respectively, were added to the bitherapeutic regimen (Figure 7(b–m), Supplemental Table 7). Cancer-free mice failed to develop tumors when rechallenged with the cancer cell type from that they had been cured (MCA205) (Supplemental Figure 9(a–c)), yet allowed for the growth of an antigenically different malignancy (TC-1 lung cancer) (Supplemental Figure 9(d,e)). This observation supported the induction of a potent CTL response together with the establishment of long-lasting cancer-specific immune memory.
Figure 7.
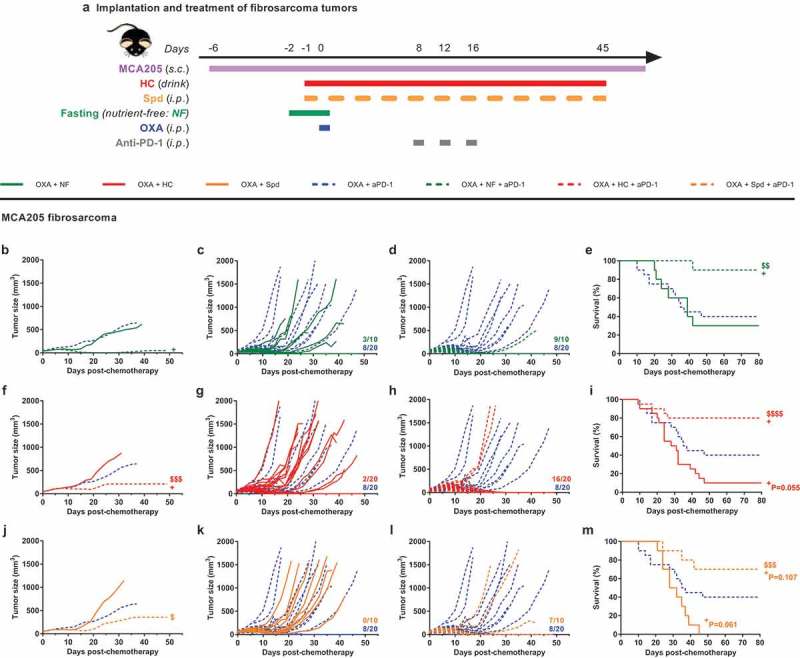
CRMs improve OXA + anti-PD-1 bitherapy. Experimental schedule of the implantation and treatment of syngeneic subcutaneous fibrosarcoma in C57Bl/6 mice (a). When tumors reached ~20 mm3, mice were randomly assigned to the different treatment groups. Monotherapy regimens consisted of: (i) PBS (untreated control mice) or (ii) OXA-based chemotherapy. Bitherapies consisted of: (i) OXA + NF, (ii) OXA + HC, (iii) OXA + Spd, and (iv) OXA + anti-PD-1. Anti-PD-1 was also evaluated in a tritherapy regimen consisting of: (i) OXA + NF + anti-PD-1, (ii) OXA + HC + anti-PD-1, (iii) OXA + Spd + anti-PD-1. Chemotherapy consisted of one i.p. injection of the platinum salt OXA at day 0 (or PBS in untreated controls). Fasting lasted for 48 h starting at day −2. The CRM HC was continuously delivered through the drinking water starting at day −1. The CRM Spd was injected i.p. at day −1, 0, and then every 2–3 days. Anti-PD-1 neutralizing antibodies were administered i.p. at day 8, 12, and 16. Mean (b, f, j) and individual (c, d, g, h, k, l) tumor growth curves of mice treated with bi- and tri-therapies. Of note, mean tumor growth curves (n = 10–20/group) were interrupted when more than 50% of the group had reached endpoint. On the panels displaying individual tumor growth curves, the number of animals that underwent complete tumor regression is indicated on the right end side. Kaplan–Meier curves (e, i, m). +p< .05 (comparisons to OXA + anti-PD-1); $$$p< .0001, $$$p< .001, $$p< .01, $p < .05 (comparisons to OXA + CRMs/NF). Data of the groups PBS, OXA + HC, OXA + anti-PD-1 and OXA + HC + anti-PD-1 consist of a pool of two independent experiments. For a detailed account of all comparisons, see Supplemental Table 7. CRM, caloric restriction mimetic; HC, hydroxycitrate; i.p., intraperitoneal; NF, nutrient-free; OXA, oxaliplatin; PBS, phosphate-buffered saline; PD-1, programmed cell death protein 1; s.c., subcutaneous; Spd, spermidine.
Altogether, these results demonstrate that immunogenic chemotherapeutics (such as MTX or OXA) sensitize to immunotherapy targeting the PD-1/PD-L1 interaction and that this sensitization effect can be amplified by starvation or CRMs.
Discussion
Recently, we have reported that CRMs enhance the antitumor activity of ICD-inducing chemotherapies such as the anthracycline MTX or the platinum salt OXA. The efficacy of the combination treatment relies on both tumor cell intrinsic and extrinsic mechanisms. Precisely, CRMs further stimulate the autophagy-dependent release of ATP from stressed cancer cells exposed to ICD inducers. This extracellular ATP acts as a chemoattractant that promotes immune cell recruitment into the tumor microenvironment. In particular, CD8+ T lymphocytes appears critical for the efficacy of the dual treatment by mediating immunosurveillance of malignant cells spared by chemotherapy.27
During ICD, in addition to the danger signals emitted by dying cancer cells, tumor antigens are captured, processed and cross-presented by antigen-presenting cells.16,49 These latter hence fill the gap between cancer cell ICD and the induction of the adaptive antitumor immunity. In the present article, we started out by characterizing the contribution of myeloid cells to the combinatorial effects of ICD inducers plus CRMs. We found out that the blockade of the extravasation of CD11b+ cells curtailed therapeutic efficacy. Despite signs of an early acute leukodepletion, anthracyclines ultimately enhanced the abundance of some myeloid cells and T lymphocytes in the tumor bed. These signs of local immunostimulation could be improved by fasting or by the CRMs HC and Spd, particularly at the myeloid cell (as well as transcriptomic) level and in a treatment-specific fashion. Indeed, an increase in activated moDCs that were either MHC-IIlo or MHC-IIhi was witnessed with starvation and HC, respectively, as well as an increase in M1 macrophages in response to Spd. Thus, supplementation of ICD-inducing chemotherapy with CRMs or fasting stimulated tumor infiltration of some myeloid cell subsets able to perform antigen cross-presentation. Interestingly, while all CRMs and fasting homogeneously induce autophagy in tumor cells, their end results on the myeloid compartment differed. Thus, each CRM and fasting seem to have additional modulatory effects on tumor cells and/or stromal entities that affect, likely not exclusively, the profile of chemokines and other attractants. In this sense, we did preview a differential immune signature in our RNA-Seq data set at early time point post-treatment.
Considering the critical role of CD8+ T lymphocytes and the improved efficacy of the bitherapies over chemotherapy alone, we anticipated a stronger Tc1 response (i.e. CD8+ T cells producing IFNγ, TNFα and IL-2) and a higher CD8+ T cells/Tregs ratio (whose value is commonly used as a prognostic indicator) with CRMs/fasting. Following unspecific PMA+ionomycin (re-)stimulation of the immune cells, we observed a trend toward an increased Tc1/Th1 population upon dual treatments in comparison to untreated controls, but no difference as compared to chemotherapy alone. Moreover, we had previously reported that HC treatment, in combination with MTX or on its own, reduced by 30% to 50% the population of immunosuppressive Tregs infiltrating MCA205 fibrosarcoma or KRAS-induced lung tumors in mice, respectively.27 Yet, similarly to Spd and fasting, HC remained unable to further increase the CD8+ T cells/Tregs ratio as compared to monotherapy with an ICD inducer. One explanation to our failure to visualize the superior effector response of bitherapies could result from a technical limitation. The latter consists of our inability to phenotype tumor-specific T cell subsets (due to the lack of known fibrosarcoma-associated antigens in the present model) rather than the whole T cell compartment. Meanwhile, it is plausible that CD8+ T cell subsets mediating tumor growth control are of limited size, rendering their detection challenging. Additionally, further investigations will determine if other types of effectors, such as NK cells, are contributing to the therapeutic benefit of CRMs to chemotherapy. At the moment, detection of surface ICOS remains fitted to monitor the expansion of activated CD8+ T lymphocytes upon chemotherapy and its further increase in combination with HC.
In parallel, we studied the exhaustion phenotype of tumor-infiltrating CD8+ T lymphocytes. Treatment with the anthracycline MTX reduced the proportion of cells harboring PD-1. Still, this immunoinhibitory molecule remained detectable on half of the CD8+ subset. Moreover, the expression of PD-L1 was increased by chemotherapy, both on cancer cells and on leukocytes. Consequently, based on the characterization of the immune infiltrate, it was possible to predict that chemotherapy mediated by ICD inducers would sensitize to subsequent PD-1 blockade. Preconditioning by fasting further improved the efficacy of such combination of chemotherapy plus anti-PD1. One explanation could reside in the increased pool of exhausted PD-1+ CD8+ T cells amenable to reactivation by checkpoint blockade. Nonetheless, supplementation with CRMs did not interfere with the modulatory action of MTX on the distribution and level of expression of PD-1 and PD-L1 in the tumor bed. Thus, based on these results obtained 11 days post-chemotherapy, there is no obvious explanation for the fact that ICD-inducing drugs become more potent in their PD-1-sensitizing effect when they are combined with CRMs. Some elements might explain such observation. First, we showed that CRMs have subtle and treatment-specific effects on myeloid cells (and likely other cell types) which might reflect a more favorable tumor microenvironment for antitumor immunity. Also, CRMs may positively modulate the PD-1/PD-L1 axis, not only in the myeloid compartment, but also potentially in the lymphoid alcove, without impacting the expression level of these surface molecules. Moreover, some tumors pretreated with the combination of ICD inducers and CRMs tended to be smaller when immunotherapy was started, perhaps rendering them more amenable to subsequent immune-dependent control.
If the triple combination of chemotherapy, anti-PD1 immunotherapy and CRMs (or fasting) demonstrated a remarkable antineoplastic efficacy, it also raised an immune memory that protected cured animals from tumor relapse. The induction of an immune memory is one property of ICD inducers.50,51 Nevertheless, if we showed that CRMs can potentiate the effector immune response, a recent report provided the first evidence in favor of their additional propensity to stimulate the development and persistence of the immune memory compartment. Precisely, in a model of influenza infection, Puleston et al. demonstrated that Spd was able to stimulate intrinsically, in an autophagy-dependent manner, the development of memory T cells following vaccination. This property actually potentiated vaccine efficacy in elderly animals which commonly show defects in mounting proper immune memory.52 Consequently, prolonged delivery of CRMs could not only benefit to ICD-inducing cancer treatments but also to other types of immunotherapies which rely on primary and/or secondary expansion of tumor-specific T lymphocytes (e.g. adoptive T cell therapies, cancer vaccines).53–59
Notwithstanding the aforementioned limitations, our results delineate a strategy for tumor treatment that involves a therapy consisting in the administration of three drug classes: (i) ICD inducers exemplified by MTX and OXA, (ii) CRMs exemplified by HC and Spd and substitutable for fasting (if the nutritional status of the patient allows it) and (iii) ICIs targeting the PD-1/PD-L1 interaction. As shown here, such a triple combination has the potential to cure the majority of mice bearing established fibrosarcomas. It will be important to investigate other ICD-inducing chemotherapies, CRMs and ICIs, different treatment schedules (sequential versus simultaneous), as well as antigenically different cancer types, to generalize and optimize these preclinical findings. Considering the safety profile of CRMs, and their access over the counter like HC, translation into the clinic is manageable and affordable. Clinical evaluations of CRM supplementation to cancer patients treated with ICD-inducers plus ICIs are encouraged.
Materials and methods
Mouse strains and accommodation
6- to 8-week-old female Balb/c and C57Bl/6 mice were purchased from Envigo France. Animals were maintained in a temperature-controlled and specific pathogen-free environment, respecting cycles of 12 h of light and 12 h of darkness, and fed ad libitum (unless specified otherwise) with the SAFE A04 diet (SAFE™) together with free access to water. Experiments were performed in compliance with the EU Directive 63/2010 and the protocols 4462-2016031108383932v3 and 4552-2016031417225217v3 that were approved by the Ethics Committee of the Gustave Roussy Campus Cancer (D9407611) and the protocols 03981.02, 4661-2016031018452249v3, 2088-2016041518388910v4 and 21307–2019070217181551 approved by the Ethics Committee of the Cordeliers Research Center (A750612; B750612).
Cell culture
The murine methylcholanthrene-induced fibrosarcoma MCA205 and lung carcinoma TC-1 cell lines were grown in Dulbecco’s Modified Eagle Medium (DMEM) and Roswell Park Memorial Institute medium (RPMI), respectively, supplemented with 10% (v/v) of fetal bovine serum and 100 IU/ml of penicillin G and 100 mg/ml streptomycin. Cells were maintained under standard culture conditions (37°C, 5% CO2, >90% humidity).
In vivo – murine cancer models
Hormone- and carcinogen-induced mammary tumor model. Mammary tumors were induced in young (7-week-old) female BALB/c mice by implantation of medroxyprogesterone acetate (MPA)-releasing pellets followed by gavage with the DNA damaging agent 7,12-dimethylbenz[a]anthracene (DMBA) for the following 6 weeks. The overall scheme of tumor induction is illustrated in Figure 1(a). Implanted fibrosarcoma tumor model. 0,3 x 106 MCA205 cells were resuspended in 100 µL of PBS and subcutaneously (s.c.) injected in the right flank of mice under anesthesia with 2.5% isoflurane. Tumor growth was monitored via repeated measurements of the tumor size using a digital caliper. The area of mammary tumors was calculated as follows: tumor size (mm2) = length x width. The volume of fibrosarcoma tumors was calculated using the following formula: tumor size (mm3) = (length x width x height)/8 x 4/3 x π. Mean tumor growth curves were calculated by carrying over the last tumor size values of the mice that reached endpoint and interrupted when more than 50% of the group had reached endpoint. Mouse survival was carefully monitored. Tumor size exceeding 250 mm2 or 2000 mm3, tumor ulceration, weight loss superior to 20% as compared to the beginning of the treatment and poor body condition were considered as endpoints.
In vivo – combination therapies
Once DMBA:MPA-induced mammary or fibrosarcoma tumors reached ~25 mm2 or ~20 mm3, respectively, mice were randomized across the different groups. Treatment schedules are detailed in the Figures 1(b), 2(a), 3(a), 6(a) and 7(a) and supplemental Figures 1(a), 5(a) and 7(a). Potassium hydroxycitrate tribasic monohydrate (HC) was injected intraperitoneally (i.p.) at 100 mg/kg in Earle’s Balanced Salt Solution (EBSS) or given orally via the drinking water at 5 mg/ml. Spermidine (Spd) was administered i.p. at 50 mg/kg in EBSS. Fasting consisted of a 48 h-period with complete food deprivation while maintaining ad libitum access to water. Mitoxantrone dihydrochloride (MTX) and oxaliplatin (OXA) were dissolved in saline water and injected i.p. at 5.17 and 10 mg/kg, respectively (day 0). PBS was injected i.p. in untreated control animals the same day as chemotherapy. All the aforementioned compounds were purchased from Sigma Aldrich (St. Louis, MO, USA). Anti-CD11b (clone M1/70), anti-PD-1 (clone 29F.1A12), and anti-CTLA-4 (clone 9D9) antibodies were injected i.p. at 200, 200 and 100 µg per mouse, respectively. Isotype controls (clone LTF-2 or clone 2A3) were delivered i.p. at the corresponding dose. All neutralizing antibodies were purchased from BioXCell.
Ex vivo – tumor processing
Tumors were harvested, weighed and transferred on ice in gentleMACS C tubes (Miltenyi Biotec™) containing 1 ml of Roswell Park Memorial Institute medium (RPMI). Tumors were dissociated mechanically with scissors, then enzymatically using Miltenyi Biotec™ mouse tumor dissociation kit and gentleMACS Octo Dissociator following the manufacturer’s instructions. Tumor homogenates were filtered through 70 µM MACS® SmartStrainers (Miltenyi Biotec™) and washed twice with PBS. Finally, bulk tumor cells were homogenized in PBS at a concentration corresponding to 250 mg of the initial tumor weight per mL.
Ex vivo – phenotyping of the tumor immune infiltrate
Bulk tumor cell homogenates, each corresponding to 50 mg of the initial tumor sample, were stained with LIVE/DEAD™ Fixable Yellow dye (Thermo Fisher Scientific™). Fc receptors were blocked with anti-mouse CD16/CD32 (clone 2.4G2, Mouse BD Fc Block™, BD Pharmingen™). To assess the production of type-1 cytokines (i.e. IFNγ, TNFα, IL-2) by effector T lymphocytes, cells were (re-)stimulated for 5 h in serum-free CTL-Test™ PLUS Medium (ImmunoSpot®) containing 20 ng/ml PMA (Calbiochem), 1 µg/ml ionomycin (Sigma), and brefeldin A (BD GolgiPlugTM, BD Biosciences, dilution 1:100). Surface staining of murine immune cell populations infiltrating the tumor was performed with the following fluorochrome-conjugated antibodies: 1) “Myeloid cell” panel: anti-CD45 APC-Fire750 (clone 30F-11, BioLegend™), anti-Ly-6G PE (clone 1A8, BD Pharmingen™), anti-Ly-6C FITC (clone AL-21, BD Pharmingen™), anti-CD11b V450 (clone M1/70, BD Pharmingen™), anti-CD11c PE-Vio770 (REA754, Miltenyi Biotec™), anti-CD80 PerCP-Cy5.5 (16-10A1, BD Pharmingen™), and anti-I-A/E (MHC-II) APC (clone M5/114.15.2, BioLegend™); 2) “T-cell activation/exhaustion” panel: anti-CD3 APC V450 (clone 17A2, Thermo Fisher Scientific™), anti-CD8 PE (clone 53–6.7, BD Pharmingen™), anti-CD4 PerCP-Cy5.5 (clone RM4-5, Thermo Fisher Scientific™), anti-CD25 PE-Cy7 (clone PC61.5, Thermo Fisher Scientific™), anti-ICOS BV421 (clone 7E.17G9, BD Pharmingen™), and anti-PD-1 APC-Fire750 (clone 29F.1A12, BioLegend™); 3) “T-cell restimulation” panel: anti-CD3 BV421 (clone 145-2C11, BD Pharmingen™), anti-CD8 FITC (clone 53–6.7, BD Pharmingen™), anti-CD4 PerCP-Cy5.5 (clone RM4-5, Thermo Fisher Scientific™); 4) PD-L1/2-expressing cell panel: anti-CD45 AlexaFluor647 (clone 30F-11, BioLegend™), anti-PD-L1 BV421 (clone MIH5, BD Pharmingen™), and anti-PD-L2 PE-Dazzle594 (clone TY25, BioLegend™); 5) NK(T) cell panel: anti-CD3 FITC (clone 17A2, Thermo Fisher Scientific™), and anti-NK1.1 PerCP-Cy5.5 (clone PK136, BD Pharmingen™). Then, cells were fixed and permeabilized in BD Cytofix/Cytoperm™ buffer (BD Pharmingen™), with the exception of the samples undergoing T cell immunophenotyping that were incubated instead in eBioscience™ Foxp3/Transcription Factor Staining Buffer (Thermo Fisher Scientific™). To complete the “T-cell activation/exhaustion” panel, an intranuclear staining was performed with anti-FoxP3 FITC (clone FJK-16s, Thermo Fisher Scientific™). To complete the “T-cell restimulation” panel, an intracellular cytokine staining was performed with anti-IFNγ APC (clone XMG1.2, BioLegend™), anti-TNFα APC-Cy7 (clone MP6-XT22, BioLegend™), and anti-IL-2 PE-Dazzle594 (clone JES6-5H4, BioLegend™). Finally, stained samples were run through a BD LSR II flow cytometer. Data were acquired using BD FACSDiva™ software (BD biosciences) and analyzed using FlowJo software (TreeStar, Inc.). Absolute counts of leukocytes and tumor cells were normalized considering the following parameters: weight of the harvested tumor and total volume of the dissociated tumor cell suspension (cell concentration typically set to 250 mg/ml in PBS), proportion of the whole cell suspension stained (typically 200 μl containing 50 mg of bulk tumor cell suspension) and proportion of the stained cell suspension ran through the flow cytometer (typically 300 out of 400 μl of the stained cell suspension).
Ex vivo – RNA sequencing (RNA-seq)
Tumor was harvested 3 days post-chemotherapy and stored at 4°C in RNAlater (QIAGEN) until RNA extraction. Tissues were incubated in RLT lysis buffer (QIAGEN) before undergoing two cycles of 20 s at 5,500 rpm in a Precellys 24 tissue homogeneator (Bertin Technologies, Montigny-le-Bretonneux, France). Tissue extracts were then centrifuged at 12,000 g (4°C) and supernatants were collected. Total RNA from tumor homogenates was purified using RNeasy Mini Kit (QIAGEN). The libraries were prepared following the Tru-seq mRNA protocol from Illumina, starting from 1 µg of high-quality total RNA. Paired end (2 × 75) sequencing was performed on an Illumina Nextseq 500 platform. Fluorometric Qubit RNA HS assay (Life Technologies, Grand Island, New York, USA) was applied to measure RNA concentration. RNA quality (RNA integrity number 8.2) was assessed on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). To build libraries, 1 µg of high quality total RNA sample (RIN >8) was processed using Truseq stranded mRNA kit (Illumina) according to the manufacturer’s instructions. Briefly, after purification of poly-A containing mRNA molecules, nucleic acids were fragmented and reverse-transcribed using random primers. Replacement of dTTP by dUTP during the second strand synthesis permitted to achieve strand specificity. Addition of a single A base to the cDNA was followed by ligation of adapters. Libraries were quantified by qPCR using the KAPA Library Quantification Kit for Illumina Libraries (KapaBiosystems, Wilmington, MA) and library profiles were assessed using the DNA High Sensitivity LabChip kit on an Agilent Bioanalyzer. Libraries were sequenced on an Illumina Nextseq 500 instrument using 75 base-length read V2 chemistry in a paired-end mode. After sequencing, a primary analysis based on AOZAN software (ENS, Paris) was applied to demultiplex and control the quality of the raw data (based of FastQC modules/version 0.11.5). Obtained fastq files were then aligned using Star algorithm (version 2.5.2b) and quality control of the alignment realized with Picard tools (version 2.8.1). Reads were then counted using Feature count (version Rsubread 1.24.1) and the statistical analyses of the read counts were performed with the DESeq2 package version 1.14.1 to determine the proportion of differentially expressed genes between two samples.
In silico – RNA-seq data analysis
RNA-Seq data analysis was performed by GenoSplice technology (www.genosplice.com). Sequencing, data quality, reads repartition (e.g., for potential ribosomal contamination), and insert size estimation were performed using FastQC, Picard-Tools, Samtools and rseqc. Reads were mapped using STARv2.4.060 on the mm10 Mouse genome assembly. Gene expression regulation study was performed as already described.61 Briefly, for each gene present in the Mouse FAST DB v2016_1 annotations, reads aligning on constitutive regions (that are not prone to alternative splicing) were counted. Based on these read counts, normalization and differential gene expression were performed using DESeq262 on R (v.3.2.5). Only genes expressed in at least one of the six compared experimental conditions were further analyzed. Genes were considered as expressed if their fragments per kilobase million (FPKM) value was greater than 97.5% of the background FPKM value based on intergenic regions. Results were considered statistically significant for uncorrected p-values ≤0.05 and fold-changes ≥1.5. Hierarchical clustering using Euclidean distance and heatmaps have been performed using Morpheus (https://software.broadinstitute.org/morpheus). Pathways enriched in the genes regulated in both comparisons, MTX + NF versus MTX and MTX + Spd versus MTX, were searched in KEGG, REACTOME and GO databases, using DAVID V6.8.63,64
Statistical analyses
Linear mixed-effects models were applied for longitudinal comparison of tumor growth curves using TumGrowth web tool (https://kroemerlab.shinyapps.io/TumGrowth/).65 GraphPad Prism software was used for data graphing and other statistical analyses. Multiple comparison of tumor sizes or tumor-infiltrating immune cell subsets was conducted using Kruskal–Wallis Test with Dunn’s post hoc when most data sets were not normally distributed, and a Holm-Sidak’s test when normally distributed. For testing data normality, we applied the Shapiro–Wilk test and considered that distribution was not normal when p-value < 0.05. For comparing mouse survivals, a Log-rank (Mantel-Cox) test was performed.
Funding Statement
SL is supported by a scholarship of the Fondation pour la Recherche Médicale (FRM FDT201805005722) for her last year of PhD program. JGP is supported by a fellowship of the Seerave Foundation. GK’s team is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Acknowledgments
We thank the staffs of the histology, cell imaging and flow cytometry platform (CHIC) and of the animal facility (CEF) of the Cordeliers Research Center (Paris, France) as well as of the Genom’IC facility of the Cochin Institute (Paris, France) for the RNA-Seq analysis.
Conflicts of interest
JGP, GK, and SL are inventors on a patent application on the « use of caloric restriction mimetics for potentiating chemoimmunotherapy » filed by Institut National de la Santé et de la Recherche Médicale (INSERM), Assistance Publique-Hôpitaux De Paris (AP-HP), Université Sorbonne Paris Cité (Université Paris Descartes – Paris 5 and Université Paris Diderot – Paris 7), Sorbonne Université (Université Pierre et Marie Curie – Paris 6), Université Paris-Sud – Paris 11, and Gustave Roussy. GK and LZ received a research grant by Elior..
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Wallis CJD, Butaney M, Satkunasivam R, Freedland SJ, Patel SP, Hamid O, Pal SK, Klaassen Z.. Association of patient sex with efficacy of immune checkpoint inhibitors and overall survival in advanced cancers: a systematic review and meta-analysis. JAMA Oncol. 2019. doi: 10.1001/jamaoncol.2018.5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP.. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1–15. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 3.Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, Minenza E, Linardou H, Burgers S, Salman P, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–2104. doi: 10.1056/NEJMoa1801946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell. 2018;33:853–861.e4. doi: 10.1016/j.ccell.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A, Carlino MS, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378:1789–1801. doi: 10.1056/NEJMoa1802357. [DOI] [PubMed] [Google Scholar]
- 6.Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, Zahurak M, Yang SC, Jones DR, Broderick S, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;379:e14. doi: 10.1056/NEJMc1808251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budczies J, Mechtersheimer G, Denkert C, Klauschen F, Mughal SS, Chudasama P, Bockmayr M, Jöhrens K, Endris V, Lier A, et al. PD-L1 (CD274) copy number gain, expression, and immune cell infiltration as candidate predictors for response to immune checkpoint inhibitors in soft-tissue sarcoma. Oncoimmunology. 2017;6:e1279777. doi: 10.1080/2162402X.2017.1279777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 10.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28:690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G. The anticancer immune response: indispensable for therapeutic success? J Clin Invest. 2008;118:1991–2001. doi: 10.1172/JCI35180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 13.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 14.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005;202:1691–1701. doi: 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. 2016;16:759–773. doi: 10.1038/nrc.2016.91. [DOI] [PubMed] [Google Scholar]
- 16.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Kroemer G, Senovilla L, Galluzzi L, Andre F, Zitvogel L. Natural and therapy-induced immunosurveillance in breast cancer. Nat Med. 2015;21:1128–1138. doi: 10.1038/nm.3944. [DOI] [PubMed] [Google Scholar]
- 18.Bezu L, Sauvat A, Humeau J, Leduc M, Kepp O, Kroemer G. eIF2alpha phosphorylation: a hallmark of immunogenic cell death. Oncoimmunology. 2018;7:e1431089. doi: 10.1080/2162402X.2018.1490854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoll G, Ma Y, Yang H, Kepp O, Zitvogel L, Kroemer G. Pro-necrotic molecules impact local immunosurveillance in human breast cancer. Oncoimmunology. 2017;6:e1299302. doi: 10.1080/2162402X.2017.1299302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology. 2017;6:e1386829. doi: 10.1080/2162402X.2017.1386829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pol J, Vacchelli E, Aranda F, Castoldi F, Eggermont A, Cremer I, Sautes-Fridman C, Fucikova J, Galon J, Spisek R, et al. Trial watch: immunogenic cell death inducers for anticancer chemotherapy. Oncoimmunology. 2015;4:e1008866. doi: 10.1080/2162402X.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castoldi F, Vacchelli E, Zitvogel L, Maiuri MC, Pietrocola F, Kroemer G. Systemic autophagy in the therapeutic response to anthracycline-based chemotherapy. Oncoimmunology. 2019;8:e1498285. doi: 10.1080/2162402X.2018.1498285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 24.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 25.Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Madeo F, Carmona-Gutierrez D, Hofer SJ, Kroemer G. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 2019;29:592–610. doi: 10.1016/j.cmet.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 27.Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, Levesque S, Castoldi F, Jacquelot N, Yamazaki T, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–160. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pietrocola F, Pol J, Vacchelli E, Baracco EE, Levesque S, Castoldi F, Maiuri MC, Madeo F, Kroemer G. Autophagy induction for the treatment of cancer. Autophagy. 2016;12:1962–1964. doi: 10.1080/15548627.2016.1214778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 31.Pietrocola F, Lachkar S, Enot DP, Niso-Santano M, Bravo-San Pedro JM, Sica V, Izzo V, Maiuri MC, Madeo F, Mariño G, et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015;22:509–516. doi: 10.1038/cdd.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–1438. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balasubramanian P, Howell PR, Anderson RM. Aging and caloric restriction research: a biological perspective with translational potential. EBioMedicine. 2017;21:37–44. doi: 10.1016/j.ebiom.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiechl S, Pechlaner R, Willeit P, Notdurfter M, Paulweber B, Willeit K, Werner P, Ruckenstuhl C, Iglseder B, Weger S, et al. Higher spermidine intake is linked to lower mortality: a prospective population-based study. Am J Clin Nutr. 2018;108:371–380. doi: 10.1093/ajcn/nqy102. [DOI] [PubMed] [Google Scholar]
- 35.Redman LM, Smith SR, Burton JH, Martin CK, Il’yasova D, Ravussin E. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 2018;27:805–15 e4. doi: 10.1016/j.cmet.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pifferi F, Terrien J, Marchal J, Dal-Pan A, Djelti F, Hardy I, Limbani V, Steinberg J, Zengini E, Warsame K, et al. Caloric restriction increases lifespan but affects brain integrity in grey mouse lemur primates. Commun Biol. 2018;1:30. doi: 10.1038/s42003-018-0052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lope V, Martin M, Castello A, Ruiz A, AM C, JM B-C, Antolín S, Ramos-Vázquez M, García-Sáenz JÁ, Muñoz M, et al. Overeating, caloric restriction and breast cancer risk by pathologic subtype: the EPIGEICAM study. Sci Rep. 2019;9:3904. doi: 10.1038/s41598-019-39346-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levesque S, Pol JG, Ferrere G, Galluzzi L, Zitvogel L, Kroemer G. Trial watch: dietary interventions for cancer therapy. Oncoimmunology. 2019;8:1591878. doi: 10.1080/2162402X.2019.1591878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cabo M, Offringa R, Zitvogel L, Kroemer G, Muntasell A, Galluzzi L. Trial watch: immunostimulatory monoclonal antibodies for oncological indications. Oncoimmunology. 2017;6:e1371896. doi: 10.1080/2162402X.2017.1371896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanpouille-Box C, Lhuillier C, Bezu L, Aranda F, Yamazaki T, Kepp O, Fucikova J, Spisek R, Demaria S, Formenti SC, et al. Trial watch: immune checkpoint blockers for cancer therapy. Oncoimmunology. 2017;6:e1373237. doi: 10.1080/2162402X.2017.1373237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. 2018;48:417–433. doi: 10.1016/j.immuni.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, Dong H. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. 2018;9:1739. doi: 10.3389/fimmu.2018.01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44:343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing anti-tumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol. 2019;30:219–235. doi: 10.1093/annonc/mdy551. [DOI] [PubMed] [Google Scholar]
- 45.Zhou J, Yang T, Liu L, Lu B. Chemotherapy oxaliplatin sensitizes prostate cancer to immune checkpoint blockade therapies via stimulating tumor immunogenicity. Mol Med Rep. 2017;16:2868–2874. doi: 10.3892/mmr.2017.6908. [DOI] [PubMed] [Google Scholar]
- 46.Fox EJ. Mechanism of action of mitoxantrone. Neurology. 2004;63:S15–8. doi: 10.1212/wnl.63.12_suppl_6.s15. [DOI] [PubMed] [Google Scholar]
- 47.Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng C-W, Cacciottolo M, Martin-Montalvo A, de Cabo R, Wei M, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell. 2016;30:136–146. doi: 10.1016/j.ccell.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alderton PM, Gross J, Green MD. Comparative study of doxorubicin, mitoxantrone, and epirubicin in combination with ICRF-187 (ADR-529) in a chronic cardiotoxicity animal model. Cancer Res. 1992;52:194–201. [PubMed] [Google Scholar]
- 49.Bloy N, Garcia P, Laumont CM, Pitt JM, Sistigu A, Stoll G, Yamazaki T, Bonneil E, Buqué A, Humeau J, et al. Immunogenic stress and death of cancer cells: Contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev. 2017;280:165–174. doi: 10.1111/imr.12582. [DOI] [PubMed] [Google Scholar]
- 50.Humeau J, Lévesque S, Kroemer G, Pol JG. Gold standard assessment of immunogenic cell death in oncological mouse models. Methods Mol Biol. 2019;1884:297–315. doi: 10.1007/978-1-4939-8885-3_21. [DOI] [PubMed] [Google Scholar]
- 51.Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G.. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife. 2014;3. doi: 10.7554/eLife.03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L. Trial watch: adoptively transferred cells for anticancer immunotherapy. Oncoimmunology. 2017;6:e1363139. doi: 10.1080/2162402X.2017.1363139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sprooten J, Ceusters J, Coosemans A, Agostinis P, De Vleeschouwer S, Zitvogel L, Kroemer G, Galluzzi L, Garg AD. Trial watch: dendritic cell vaccination for cancer immunotherapy. Oncoimmunology. 2019;1–21. doi: 10.1080/2162402X.2019.1638212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pol JG, Levesque S, Workenhe ST, Gujar S, Le Boeuf F Clements DR, et al. Trial watch: oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology. 2018;7:e1503032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pol JG, Acuna SA, Yadollahi B, Tang N, Stephenson KB, Atherton MJ, Hanwell D, El-Warrak A, Goldstein A, Moloo B, et al. Preclinical evaluation of a MAGE-A3 vaccination utilizing the oncolytic maraba virus currently in first-in-human trials. Oncoimmunology. 2019;8:e1512329. doi: 10.1080/2162402X.2018.1512329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pol JG, Atherton MJ, Bridle BW, Stephenson KB, Le Boeuf F, Hummel JL, Martin CG, Pomoransky J, Breitbach CJ, Diallo J-S, et al. Development and applications of oncolytic maraba virus vaccines. Oncolytic Virother. 2018;7:117–128. doi: 10.2147/OV.S154494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gujar S, , Pol JG, Kroemer G. Heating It Up: Oncolytic Viruses Make Tumors 'Hot' and Suitable for Checkpoint Blockade Immunotherapies. Oncoimmunology 2018; 7:e1442169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pierini S, Perales-Linares R, Uribe-Herranz M, Pol JG, Zitvogel L, Kroemer G, Facciabene A, Galluzzi L. Trial watch: DNA-based vaccines for oncological indications. Oncoimmunology. 2017;6:e1398878. doi: 10.1080/2162402X.2017.1398878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Noli L, Capalbo A, Ogilvie C, Khalaf Y, Ilic D. Discordant growth of monozygotic twins starts at the blastocyst stage: a case study. Stem Cell Rep. 2015;5:946–953. doi: 10.1016/j.stemcr.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 64.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Enot DP, Vacchelli E, Jacquelot N Zitvogel L, Kroemer G. Tumgrowth: An Open-access Web Tool for The Statistical Analysis of Tumor Growth Curves. Oncoimmunology. 2018:7:e1462431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.