

ABSTRACT
CD19-specific chimeric antigen receptor (CAR)+ T cells have demonstrated clinical efficacy and long-lasting remissions, concomitant with tolerable normal B-cell aplasia. However, many tumor-associated antigens (TAAs) are expressed on normal tissues, the destruction of which would lead to intolerable toxicity. Thus, there is a need to engineer CAR+ T cells with improved safety profiles to restrict toxicity against TAA-expressing normal tissues. Bioengineering approaches include: (i) targeting CAR+ T cells to the tumor site, (ii) limiting CAR+ T-cell persistence, and (iii) restricting CAR activation. We review and evaluate strategies to engineer CAR+ T cells to reduce the potential of on-target, off-tissue toxicity.
KEYWORDS: Bioengineering, chimeric antigen receptor, on-target toxicity, T cell
Abbreviations
- ADCC, antibody-dependent cell cytotoxicity; CAIX, carbonic anhydrase IX; CAR
chimeric antigen receptor
- CCR
chimeric co-stimulation receptor
- CEA
carcinoembryonic antigen
- EGFR
epidermal growth factor receptor
- FITC, fluorescein isothiocyanate; huEGFRt, truncated human epidermal growth factor receptor; HSV-TK
herpes simplex virus-derived thymidine kinase
- iCAR
inhibitory CAR
- iCasp9
inducible caspase 9
- ODDD
oxygen-dependent degradation domain
- PD-1
programmed death receptor 1
- PNE
peptide neo-epitope
- PSMA
prostate-specific membrane antigen
- SAE
serious adverse event
- scFv
single-chain variable sequence
- TAA
tumor-associated antigen
Introduction
Genetic modification of T cells to express chimeric antigen receptors (CARs) endows specificity to tumor-associated antigens (TAAs) via a single-chain variable sequence (scFv) derived from a monoclonal antibody (mAb) coupled to T-cell activation domains. Reports of phase I/II clinical trials of CD19-specific CAR+ T cells for B-cell malignancies reveal antitumor efficacy, long-term B-cell aplasia, and acute toxicity related to elevated serum cytokines.1-4 Human trials have identified three mechanisms of toxicity from CAR-engineered T cells: (i), cytokine release syndrome, (ii) anaphylactic response to foreign moieties, and (iii) on-target, off-tissue toxicity (Fig. 1). This review is focused on engineering approaches to limit on-target, off-tissue toxicity.
Figure 1.
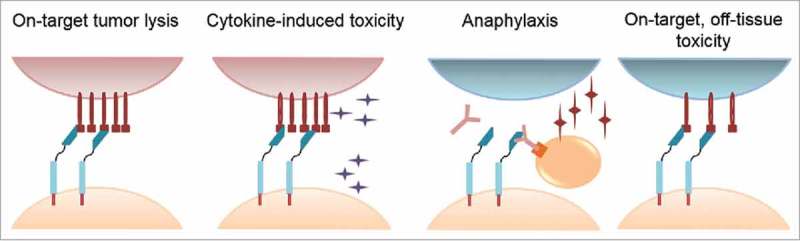
Potential mechanisms of toxicity mediated by infused chimeric antigen receptor (CAR)+ T cells. Cytokine-induced toxicity describes damage mediated by T-cell activation and release of inflammatory cytokines, as exhibited in clinical trials with CD19-specific CAR+ T cells. Anaphylactic response (anaphylaxis) describes development of an IgE-mediated immune response to foreign CAR moieties that degranulates mast cells. On-target, off-tissue toxicity describes CAR+ T-cell recognition of tumor-associated antigens (TAAs) expressed on normal tissue(s).
CD19 expression is restricted to B-lineage cells; therefore, targeting CD19 on B-lineage malignancies also eliminates normal B cells, which is considered tolerable for recipients with advanced leukemias and lymphomas.5-8 However, CAR+ T cells targeting TAAs with widespread normal tissue expression can result in serious adverse events (SAEs), which has restricted their application. Targeting renal cell carcinoma with a CAR redirected to carbonic anhydrase IX (CAIX) showed activity against normal tissues expressing CAIX, resulting in damage to bile ducts and reversible cholangitis.9 Infusion of HER2-redirected CAR T cells to treat colorectal cancer resulted in respiratory distress and death in one patient after massive pulmonary T-cell infiltration, attributed to normal expression of HER2 on pulmonary tissue.10
Paradoxically, CARs targeting HER2 and EGFR in recent trials for patients with osteosarcoma or EGFR-overexpressing tumors, respectively, have demonstrated good tolerability.11, 12 Differences in HER2-specific CAR designs and dosing may be responsible for the difference in toxicity profiles, including differences in (i) scFv, (ii) endodomain, (iii) dose-escalation trial design, and the absence of both (iv) lymphodepletion and (v) concomitant IL-2 administration. These data provide encouragement that approaches exist for harnessing CAR biology to safely target antigens with normal tissue expression.
Locoregional delivery of CAR+ T cells may restrict systemic circulation and circumvent toxicity to normal tissue beyond the biodistribution of the administered product. In a phase I study, carcinoembryonic antigen (CEA)-specific CAR+ T cells were infused via the hepatic artery to limit extrahepatic toxicity.13 Stable disease was achieved in one of the six patients, and no SAEs were reported. Minor adverse events, including fever and colitis, were attributed to IL-2, although a contribution of CAR T cells could not be excluded. A similar loco-regional delivery strategy applied IL13Rα2-specific CAR+ T cells for glioblastoma by direct delivery into the postsurgical resection cavity, and was associated with temporary, manageable inflammation.14 No other adverse events were reported. In a murine model of mesothelioma, intrapleural delivery of mesothelin-specific CARs demonstrated enhanced antitumor efficacy, long-term remission, and functional persistence. However, they also effectively eliminated extrathoracic tumor, indicating that locoregional delivery alone is insufficient to prevent normal tissue toxicity.15
Aside from route of delivery, CAR+ T cells can be bioengineered to reduce the potential for normal tissue toxicity. These approaches can be categorized as three main strategies: (i) targeting systemically-administered CAR+ T cells to the tumor site (Fig. 2), (ii) limiting CAR+ T-cell persistence (Fig. 3), and (iii) restricting CAR-mediated T-cell activation (Fig. 4).
Figure 2.
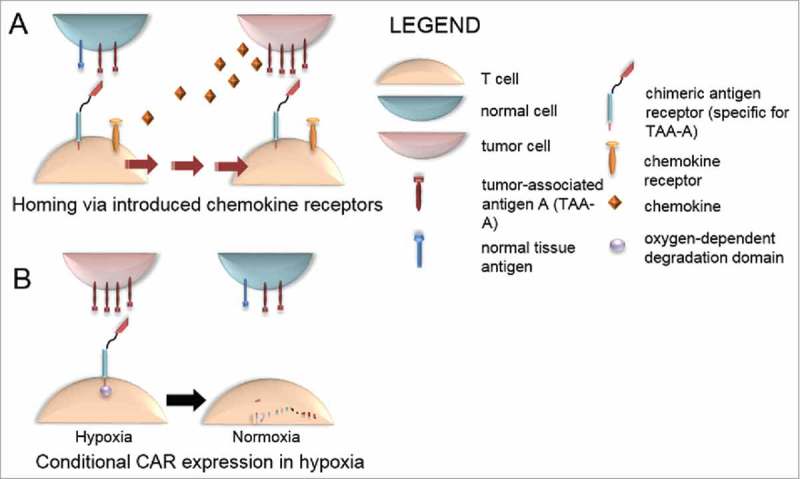
Strategies to limit on-target, off-tissue toxicity by targeting CAR+ T cells to the tumor site. (A) Homing via introduced chemokine receptors. Genetically introduced chemokine receptors enable CAR-modified T cells to home to chemokine-secreting malignancies, enriching biodistribution in the tumor microenvironment. (B) Conditional expression of CAR in hypoxia. CAR fused to an oxygen-dependent degradation domain results in degradation of CAR in normoxic, normal tissue, and selective expression of CAR in hypoxia, a common environmental condition in many malignancies.
Figure 3.
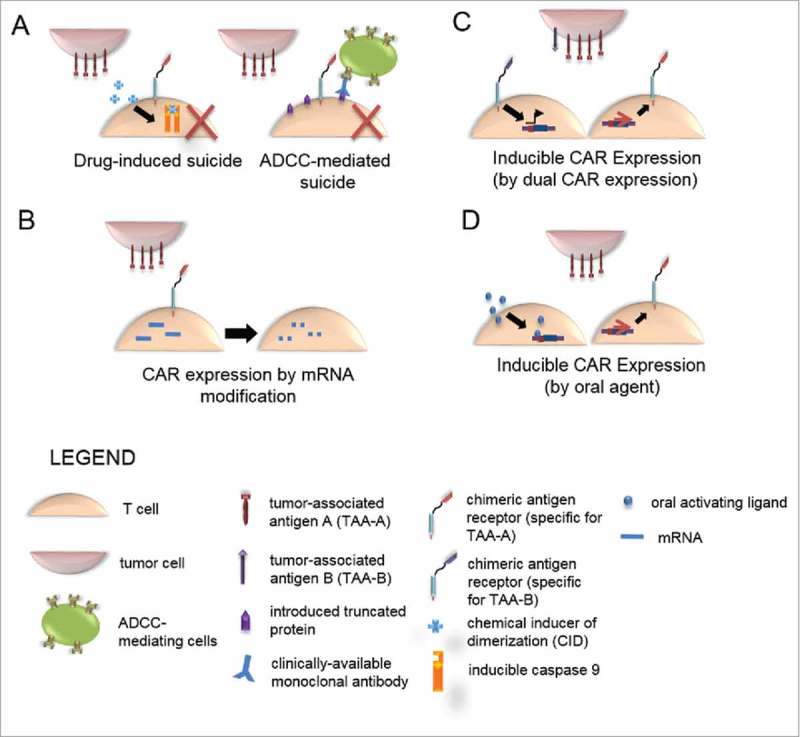
Strategies to limit on-target, off-tissue toxicity by limiting CAR T-cell persistence. (A) Suicide genes. Conditional, selective ablation of CAR-modified T cells can be achieved through enforced expression of suicide genes. Drug-induced suicide relies on administration of a drug, here a chemical inducer of dimerization, which bioactivates in the T cell by inducing dimerization of inducible caspase 9 to result in initiation of a suicide program. Antibody dependent cell cytotoxicity (ADCC)-mediated suicide occurs via introduction of a biologically inert, truncated protein on the surface of CAR-modified T cells. Administration of a clinically available monoclonal antibody activates PBMC to mediate ADCC. (B) CAR expression by mRNA modification results in transient, self-limiting expression of CAR to reduce long-term antigen recognition and temporally limit toxicity. (C) Inducible CAR expression can be achieved by dual expression of two chimeric molecules in which antigen A activates a syn-NOTCH receptor, which in turn activates NOTCH responsive elements in the CAR promoter to selectively express a CAR specific for antigen B in the presence of antigen A. (D) Inducible CAR expression may potentially be achieved by expression under a drug-inducible promoter, such as the Rheo-Switch Therapeutic System (RTS)® to allow for selective CAR expression upon administration of an oral activating ligand.
Figure 4.
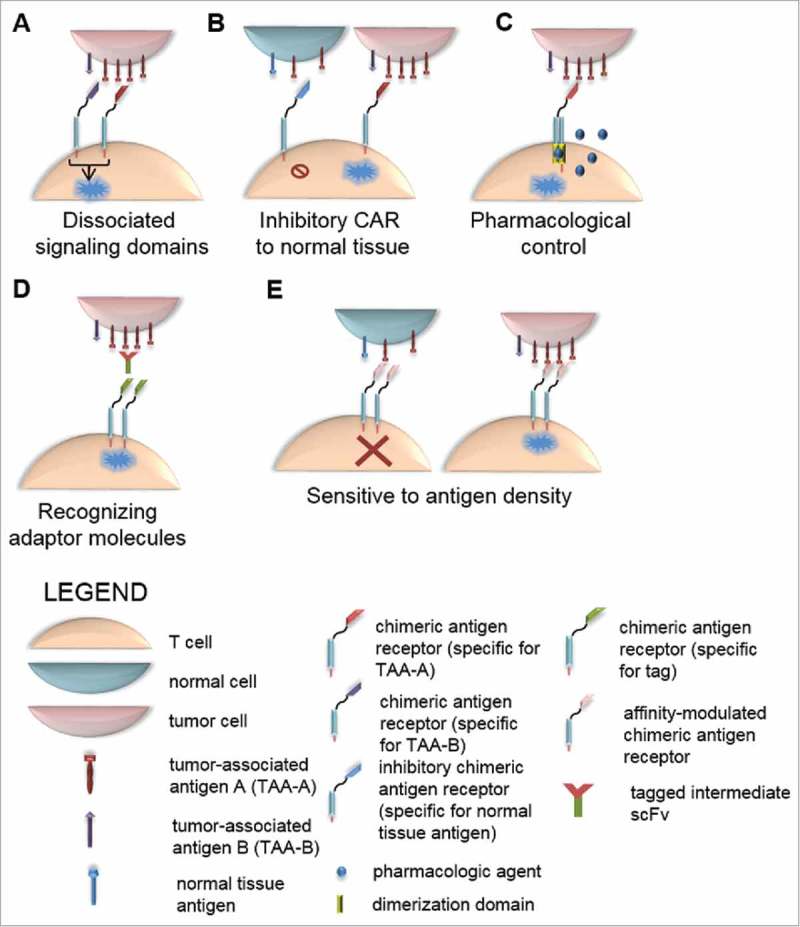
Strategies to limit on-target, off-tissue toxicity by restricting CAR T cell activation. (A) Dissociated signaling domains. Decoupling expression of CD3ζ and CD28 co-stimulation by expressing these intracellular endodomains in two separate chimeric proteins requires expression of two antigens to result in fully competent CAR activation. Selection of two antigens mutually co-expressed only on tumor cells limits recognition of normal tissue expressing only one antigen. (B) Inhibitory CAR to normal tissue. Expression of a CAR with an inhibitory intracellular signaling domain derived from PD-1 that is antigen-restricted to normal tissue can result in reversible inhibition of T-cell activation mediated by a second CAR specific for tumor antigen. (C) Pharmacological control of CAR activation can be achieved by inclusion of domains in the CAR construct that come together and render the CAR functionally active only in the presence of an administered drug. Thus, in the absence of drug, CAR is functionally inactive. (D) CARs recognizing adaptor molecules. Generation of tumor-specific scFvs tagged with unique molecules can be used as adaptor molecules to mediate recognition of tumor by CARs specific to the unique tag. (E) CARs sensitive to antigen density. Because tumor cells often express antigens at a higher density than that of normal tissue, CARs can be generated to be sensitive only to high-density antigen by reducing the affinity of the scFv, thereby sparing normal tissue with low antigen density from CAR recognition.
Targeting CAR+ T cells to tumor site
Homing via introduced chemokine receptors
Co-expression of homing molecules with CAR, such as CCR2, CCR4, and CXCR2, can direct migration of genetically modified T cells to tumor sites (Fig. 2A).16-19 Engineering T cells with chemokine receptors matched to chemokines produced by the tumor, such as CXCR2 to migrate to melanoma, CCR2 to migrate to mesothelioma and neuroblastoma, and CCR4 to migrate to Hodgkin lymphoma results in enhanced tumor infiltration and antitumor activity.16-19 However, these models demonstrated significant T cell numbers in normal tissues (e.g., lung and liver) shortly after T-cell infusion, with chemokine-redirected T cells significantly accumulating at the tumor site around 6 days post infusion. Therefore, this strategy will not limit short-term normal tissue toxicity mediated by CAR T cells before their homing to tumor sites. Likewise, chemokines secreted by tumors can also be secreted in normal tissue, such as during tissue trauma and healing. Therefore, combining these treatments with other treatment modalities, such as surgery, chemotherapy, and radiation therapy, may risk attracting T cells to normal cells, with subsequent damage resulting in toxicity.
Conditional CAR expression in hypoxia
Development of a CAR preferentially expressed in hypoxic conditions, common in many tumors, has been achieved by fusing a CAR to an oxygen-dependent degradation domain (ODDD) derived from HIF-1α (Fig. 2B).20 A cMet-specific CAR fused to ODDD was expressed in low oxygen tension (1% O2) but degraded in normoxia (20% O2), resulting in c-Met-specific lysis in hypoxic, but not normoxic, conditions.21 Because the degradation of the CAR in T cells migrating from hypoxia to normoxia mediated by ODDD degradation may take minutes to hours, it is feasible for on-target, off-tissue toxicity to occur before CAR degradation. In addition, although the centers of many tumors are hypoxic, well-vascularized peripheral tumor regions may have sufficient oxygen concentration to degrade CARs, potentially limiting the antitumor activity of CAR T cells.22
Limiting CAR+ T-cell persistence
Suicide genes
Suicide genes that eliminate infused T cells that are exhibiting toxicity upon administration of a prodrug have been used (Fig. 3A, left). Initially, T cells were modified with herpes simplex virus-derived thymidine kinase (HSV-TK) to ablate T cells upon treatment with ganciclovir. However, HSV-TK proved to be immunogenic, and anti-transgene immune responses negatively affected T-cell persistence.23,24 A novel inducible caspase 9 (iCasp9) has been developed in which a chemical inducer of dimerization (CID), AP1903, can be administered to patients to cause iCasp9 activation, which triggers apoptosis. This results in >90% elimination of T cells within 30 min of CID administration and reversal of T-cell-related toxicity. The inducing agent is also biologically inert, thereby enhancing safety.25, 26 Although suicide genes effectively and rapidly reverse toxicity, ablation of T cells to mitigate normal tissue toxicity also mitigates the long-term potential for antitumor efficacy.
Alternatively, cell surface expression of a target recognizable by a clinically available antibody can be used to ablate T cells (Fig. 3A, right). Expression of CD20 in T cells rendered them sensitive to in vitro complement-mediated cytotoxicity, resulting in 86–97% of T cells specifically ablated in the presence of rituximab.27,28,29 Similarly, a truncated, inert form of the epidermal growth factor receptor (huEGFRt) specifically sensitized CAR T cells to in vitro cytotoxicity in the presence of cetuximab and activated peripheral blood mononuclear cells by antibody-dependent cell cytotoxicity (ADCC).30 Cetuximab treatment significantly delayed engraftment and eliminated murine T cells that expressed huEGFRt in 4–6 d. Because the CAR onset of T-cell mediated toxicity can be rapid, this elimination time mediated by antibody recognition of introduced targets may not be sufficient to reverse clinical manifestations of toxicity.
CAR expression by mRNA modification
An alternative strategy to self-limit persistence is to introduce CARs as mRNA species. CARs targeting CD19, mesothelin, HER2, EGFR, GD2, and CD33 have been transiently expressed by introduction of in vitro transcribed mRNA into human primary T cells (Fig. 3B).31-39 Expression of CARs by mRNA modification appears to offer a less cumbersome process for regulatory approval, because there is no genome integration, potentially shortening the preclinical development period for CAR T-cell therapy against a new TAA. Furthermore, generation of CAR-modified T cells by mRNA transfer is quicker than DNA-modification using viral and nonviral integration systems. Improving the speed of the regulatory approval processes and ex vivo manufacture time may quicken the pace of translation from bench-to-bedside and back and potentially improve potency and fine-tuning of these therapies for clinical application.
In preclinical studies of a mesothelin-specific CAR expressed via mRNA modification, reduced tumor burden and prolonged survival in large, vascularized mesothelioma model was observed after multiple injections.33 In a study targeting CD33, CARs stably-expressed via DNA modification mediated significant myelotoxicity.38 However, the transient expression of CARs via mRNA modification resulted in the reduction of myelotoxicity in a mouse model of acute myeloid leukemia (AML). Similarly, mRNA-modification of T cells to express an EGFR-specific CAR demonstrated desired transient CAR expression and the corresponding specificity for EGFR decreased with CAR expression.37 The half-life of CAR expression was shortened by cytokine and antigenic stimuli, mitigating cytotoxicity to normal EGFR-expressing kidney cells.
The transient nature of CAR expression by mRNA modification also limits antitumor activity, increasing the potential for relapse. This has been demonstrated in a disseminated model of neuroblastoma in which T cells expressing a GD2-specific CAR introduced by mRNA exhibited reduced antitumor efficacy and tumor infiltration relative to stably modified CAR T cells. This may be due to a lack of antigen-driven migration or a proliferative difference, both of which could arise from accelerated loss of the CAR after antigenic stimulation.37, 39 Extending the half-life of CAR expression by delivery of a higher dose of mRNA or by use of synthetic mRNA with stabilizing elements may enhance antitumor activity.33, 36 Duration of CAR expression impacts both the potential to reduce normal tissue toxicity and antitumor activity, and must be carefully balanced to maximize therapeutic efficacy while minimizing normal tissue toxicity. In addition, lymphodepletion immediately before infusion and weighted split-dosing, with one larger, front-loaded dose followed by smaller maintenance doses given weekly has been shown to enhance antitumor activity of RNA-modified CAR T cells in models of leukemia and disseminated ovarian cancer, approaching the efficacy observed from a single treatment with stably modified CAR T cells.35, 40
Early clinical results have demonstrated feasibility and safety of treatment with mRNA-modified mesothelin-specific CAR T cells, with infusions being well-tolerated.41 Although reduced antitumor activity of mRNA-modified CAR T cells may be overcome by multiple injections, this also poses risks for the development of immune responses to foreign CAR moieties and the development of anaphylaxis to CAR T cells, which was observed in one patient receiving mRNA-modified mesothelin-specific CAR T cells that was attributed to an intermittent dosing schedule.42 Transiently-modified CAR T cells have limited potential to mediate long-term normal tissue toxicity, but the potential for potent normal tissue activity exists from the moment of infusion, before CAR expression declines. Serious adverse events from T-cell therapy can progress rapidly from the onset of clinical symptoms; therefore, a strategy to protect normal tissue from the moment of CAR+ T-cell infusion is optimal.7, 8
Inducible CAR expression
Inducible CAR expression in the local tumor environment can be achieved via the dual synNotch receptor/CAR system, dubbed AND-gate T cells (Fig. 3C).43 In this system, the first receptor, a synNotch receptor recognizing a TAA designated “A” on the tumor, is constitutively expressed and activates the transcription of a second, traditional CAR under inducible control of synNotch response elements in the promoter region, which recognizes TAA “B” on tumor cells, thereby, activating cytolytic machinery in T cells. In a bilateral, subcutaneous tumor model, tumors expressing both antigen A and antigen B, but not antigen B alone, were significantly targeted by AND-gate T cells, indicating a lack of bystander cytotoxicity after priming in the tumor microenvironment. CAR-expressing AND-gate T cells infiltrated and accumulated in dual antigen-expressing tumors, but much less so in single antigen B-expressing tumors. This was attributed to the short half-life of the CAR, about 8 h, relative to the time required for T cells to migrate, and the increased receptor-engagement of AND-gate T cells in tissues expressing both antigens. Identifying two targets uniquely co-expressed on tumors and absent on normal tissue creates a profile to distinguish malignant from normal tissue and limits on-target, off-tissue toxicity. Because AND-gate T cells require dual antigen expression for CAR activation and tumor clearance, this approach is best suited for antigens with complete, homogeneous tumor expression, because elimination of either antigen will result in deactivation of CAR function and incomplete tumor clearance.
Inducible expression of cytokines such as IL-12 has been demonstrated in genetically engineered cells via a drug-activated inducible promoter.44, 45 This Rheo-Switch Therapeutic System® (RTS®) allows for tight regulation of protein expression by administration of an oral ligand, veledimex, to activate the promoter. Preclinical and clinical studies have demonstrated efficacy and biologic activity of the RTS. Control of CAR expression by this drug-inducible promoter regulation could be used to conditionally express the CAR, and effectively discontinue CAR expression in the event of observed toxicity (Fig. 3D). This is a theoretical approach and represents a future area of investigation.
Clinical applicability of ligand-induced CARs requires that (i) the promoter is specifically activated by the drug, (ii) no immunoreceptor is expressed in the absence of the drug, and (iii) the kinetics of CAR expression and recycling are characterized. To reduce on-target, off-tissue toxicity, withdrawal of the oral activator ligand must reduce expression of the CAR quickly enough to resolve symptoms of clinical toxicity before they become severe. A benefit to this approach is that reduced CAR expression by withdrawal of the oral ligand is reversible, and therapy could potentially be resumed once the toxicity has subsided, in contrast to ablating CAR-expressing T cells with suicide genes or transient expression of CAR in mRNA-modified T cells.
Restricting CAR+ T-cell activation
Dissociated signaling domains
Dual-specific, complementary receptors with two specificities can selectively activate T cells when two targeted TAAs are co-expressed on the tumor, by dissociating signaling domains. In this system, a traditional CAR that recognizes TAA “A” signals via CD3ζ, while a second receptor (termed a chimeric co-stimulation receptor [CCR]), recognizing TAA “B,” activates co-stimulation endodomains (Fig. 4A). Thus, complete T-cell activation and effector functions are only attained with simultaneous engagement of CAR and CCR by co-expression of the two antigens.46-48 This approach has been piloted with CAR and CCR with specificities redirected toward HER2 and MUC1 for breast cancer, prostate-specific membrane antigen (PSMA) and PSCA for prostate cancer, and mesothelin and α-folate receptor for ovarian cancer. However, studies have demonstrated that low-level T-cell activation and lytic function can occur against single antigen-expressing targets via CAR expression in the absence of CCR activation.46, 47 One strategy to overcome this limitation is to develop a CAR with suboptimal affinity, such that it barely renders T cells functional when activated by a single antigen, and activation is only rescued by ligation of CCR.48 The requirement for two immunoreceptors to be co-expressed on T cells requires consideration of the spatial stoichiometry of the two TAAs on malignant cells for efficient T-cell activation. In addition, loss of either antigen increases the potential for the emergence of antigen-escape variants.
Inhibitory CARs to normal tissue antigen
An inhibitory CAR (iCAR) with specificity for an antigen found on normal cells but not tumor cells, and coupled to programmed death receptor 1 (PD-1) signaling endodomains, is capable of inhibiting T-cell-mediated killing and cytokine production after binding of normal tissue antigen (Fig. 4B).49 iCAR T-cell inhibition is reversible, and T cells are subsequently capable of productive responses upon encountering TAAs. This strategy's success also depends on consideration of temporal and spatial stoichiometric expression of CAR/iCAR and their respective antigens. Therefore, normal tissue toxicity could still occur if iCAR or antigen expression is insufficient in the presence of overwhelming CAR/TAA expression.
Pharmacologic control of CAR activation
Inducible CAR activation has been achieved by using a pair of well-defined heterodimerizing proteins, the FK506-binding protein (FKBP) and FKBP-rapamycin binding (FRB) domains, which are brought together in the presence of the small molecule rapamycin or its less immunosuppressive analog AP21967 (Fig. 4C).50,51 Two studies have engineered CARs including these domains to result in conditional activation in the presence of dimerizing small molecules. In one engineering strategy, the CAR was dissociated into two separate proteins: an antigen-binding domain that recognizes TAA and an intracellular signaling domain containing 41BB and CD3ζ.50 These two receptors dimerized to produce an activated CAR complex at engineered FKBP and FRB domains in the presence of AP21967. Antigen-specific activity of CAR T cells, including production of IL-2 and IFNγ, proliferation, and in vitro and in vivo cytotoxicity required both antigen and small molecule presence and was titratable.
In a second engineering strategy, the FKBP and FRB domains were included within the extracellular hinge region of a CAR, which destabilizes cell-surface expression by distancing the scFv from the cell membrane.51 Addition of rapamycin or its analog AP21967 binds and brings together the FKBP and FRB domains, shortening the CAR extracellular domain and “switching on” CAR expression. CAR expression increased 15-fold after rapamycin addition at clinically relevant concentrations. Lytic activity in response to CD19+ Daudi cells was titratable by the amount of small molecule activator and was insignificant in the activator's absence. However, Daudi cells have relatively low CD19 expression relative to Nalm-6 or Raji cell lines.52 Whereas CAR expression was reduced in the absence of rapamycin or its analog AP21967, it was not completely absent, raising the possibility that basal CAR expression would contribute to toxicity. Indeed, T-cell activation is impacted by the density of CAR and target antigen expression, such that a low density of CAR may activate T cells by highly expressed antigen, but not low levels of antigen.53 Thus, it is possible that the low levels of CAR expression in the absence of rapamycin or AP2967 are still capable of on-target, off-tissue toxicity in response to high levels of antigen expression.
Sustained locoregional delivery of the small molecule could be used to exercise spatial control over CAR activity and reduce normal tissue toxicity. Because the rapalog AP21967 has a relatively short half-life (4 hours), cessation of administration at the onset of clinical symptoms of serious adverse events should result in rapid clearance and reversal of CAR-mediated toxicity. In addition, tacrolimus binds the FKBP, but not FRB, domain, and can be used to antagonize CAR expression by competing with rapamycin.51 Therefore, this approach for controlling CAR expression may have an added clinical benefit of being able to switch-off CAR expression in the event of adverse toxicity.
CARs recognizing adaptor molecules
Another method to limit the spectrum of CAR binding includes the use of adaptor molecules to mediate recognition between universal CARs and TAAs (Fig. 4D). Antibodies specific to antigen are modified to express a switch, fluorescein isothiocyanate (FITC) or a yeast-derived peptide neo-epitope (PNE) sequence, and infused with switch-specific (i.e., FITC-specific or PNE-specific) CAR+ T cells.54,55 Optimization of the adaptor antibody demonstrated that switch conjugates located proximal to antigen binding domains improved CD19-specific CAR activation and antitumor function, but that distal conjugation was optimal for CD22-specific CARs, suggesting that optimal switch conjugation needs to be empirically evaluated for each antibody/CAR pair. In vivo evaluation demonstrated potent antitumor activity that was dependent on the presence of antigen and adaptor molecule dose and comparable with traditional CD19-specific CARs. Titration of the antibody could be used to mitigate acute toxicity, and halting the administration of the adaptor resulted in normal B-cell repopulation. However, it is as yet unclear whether the elimination of the switch will result in rapid reversal of severe, acute toxicity in a clinical setting. In addition, because the adaptor molecule and genetically modified T cells are administered systemically, it does not protect against short-term normal tissue toxicity. There are added expenses associated with co-infusing antibody and CAR+ T cells, which will need to be addressed for widespread clinical application.
CARs sensitive to antigen density
Some TAAs are expressed on tumors at a higher density than their physiologic level of expression on normal tissue(s). Recent studies have exploited this difference to generate CARs capable of distinguishing malignant from normal cells. Reducing the affinity of scFv within a CAR allows the design to discriminate between target cells based on antigen density, such that lower affinity CARs bind preferentially to targets with high TAA density but not those with low density (Fig. 4E). These data have been demonstrated in CAR species targeting HER2, α-folate receptor, β-folate receptor, and EGFR.56-59 scFvs with reduced affinity can be engineered using screening programs and site-directed mutagenesis to the complement-determining regions (CDRs). One advantage of tuning CAR affinity to reduce targeting of normal cells is that T-cell effector function is not dampened in response to docking with high-density TAAs on tumors. However, affinity-tuned CAR T cells may also have reduced capacity to mediate cytotoxicity against low-density TAA-expressing tumors, which may increase the potential for outgrowth of tumor-escape variants.
In vivo modeling of on-target, off-tissue toxicity
Preclinical CAR T cell efficacy is often evaluated in orthotopic murine model systems in which human T cells are modified to target human xenografts in immune deficient murine hosts. Inherent weaknesses of this model include the inability to model the interactions of CAR T cells with the systemic and local immune system and inability to predict on-target, off-tissue toxicity since scFvs for human antigens often do not cross react with murine counterparts. Thus, alternative models are desirable to model CAR T cell activity in vivo.
Immune competent murine models evaluating CD19-specific CAR T cell function have shown long-term B cell aplasia that was also observed in human clinical trials, indicating their utility in detecting normal tissue toxicity.60 In a fully murine model of glioblastoma expressing EGFRvIII, EGFRvIII-specific CAR T cells were able to cure mice and were protected from rechallenge with EGFRvIIIneg tumor, suggesting potential epitope spreading.61 However, in human clinical trials of EGFRvIII-specific CAR T cells, the patients recurred with EGFRvIII negative tumors, indicating immune competent murine models may still be insufficient for human modeling. Optimization of dosing regimen and implication of systemic immune activation in this model highlight benefits of immunocompetent murine models; however, building the murine CAR (including scFv specific for murine counterpart, and intracellular signaling components) is laborious and may not recapitulate biology of the human CAR. Furthermore, distribution of TAAs on normal tissue varies from species to species, and targeting the murine epitope may not recapitulate toxicity in human tissues. Normal tissue toxicity of a ROR1-specific CAR was screened in an immunocompetent primate model since primates have high homology to human.62 Importantly, these models are often cost-prohibitive as the numbers of subjects needed are not sufficient to make meaningful conclusions regarding the risks of toxicity.
Investigation of additional models to measure on-target, off-tissue toxicity of CAR T cells is warranted. Orthotopic models in mice transgenic for human antigen may provide a simple, cost-effective method for evaluating on-target, off-tissue toxicity, although location and density of human antigen will need to be assayed for physiological relevance. Alternatively, treatment of spontaneous tumors in companion canines using syngeneic CAR T cells may be a middle-ground between immunocompetent murine models with disparate physiology and expensive primate models, since canine T cells can be manufactured using methods similar to those used to modify human T cells.63
Conclusion
Determining the approach best suited to reduce normal tissue toxicity by a particular CAR/antigen pair requires understanding and leverage of the biology of the specific tumor against itself. For example, the presence of unique factors in the tumor microenvironment, such as hypoxia or secreted chemokines, can be used to concentrate effector T cells at the tumor site. Understanding the pattern of expression of the targeted TAA on normal and tumor tissue is critical. Antigens widely expressed on normal tissues require mechanisms with tight control of CAR expression or T cell activation and should be designed with back-up secondary control mechanisms, such as suicide gene expression. Such a combination might include an inducible CAR, AND-gate CARs requiring dual antigen expression, or CARs recognizing adaptor molecules with highly effective and fast-acting suicide signals via iCasp9. Targeting multiple antigens uniquely co-expressed on tumors provides an advantageous strategy to restrict activation at normal tissue sites only expressing one antigen. In these instances, understanding the pattern of expression of both antigens relative to each other is key. Two antigens expressed homogenously in a tumor are ideal targets for AND-gate T cells or CARs with dissociated signaling domains as uneven expression of one antigen over another may lead to incomplete CAR activation and tumor clearance. Use of inhibitory CARs to dampen activation in response to normal tissue should take the stoichiometry of activating and inhibitory antigens on normal tissue into account, as a higher density of antigen for inhibitory receptor would be ideal to ensure a robust signal to overcome the activating CAR signal. Finally, strategies in which toxicity is greatly reduced to normal tissue, but may potentially still occur, such as affinity-modulated CARs, could be strategically combined with approaches to restrict their expression/activation to the tumor microenvironment, such as homing via chemokine receptors or utilized in an AND-gate approach to further reduce the likelihood of normal tissue toxicity. These advances in the bioengineering of T cells provide a pathway to expand T cell therapies to TAAs with more widespread normal tissue expression, widening application of CARs to additional tumors.
Disclosure of potential conflicts of interest
On May 7, 2015, Dr. Cooper was appointed as the Chief Executive Officer at ZIOPHARM Oncology. Dr. Cooper is now a Visiting Scientist at MD Anderson. Some of the technology reviewed in this article was licensed by MD Anderson for commercial application to ZIOPHARM Oncology, Inc., and Intrexon Corporation in exchange for equity interests in these companies.
Acknowledgments
We thank David M. Wildrick, Ph.D. for editorial assistance.
References
- 1.Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol 2016; 13:370-83; PMID:27000958; http://dx.doi.org/ 10.1038/nrclinonc.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turtle CJ, Riddell SR, Maloney DG. CD19-targeted chimeric antigen receptor-modified T cell immunotherapy for B cell malignancies. Clin Pharmacol Ther 2016; 100(3):252-8; PMID:27170467; 10.1002/cpt.392 [DOI] [PubMed] [Google Scholar]
- 3.Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015; 125:4017-23; PMID:25999455; http://dx.doi.org/ 10.1182/blood-2014-12-580068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadelain M. CAR therapy: the CD19 paradigm. J Clin Investig 2015; 125:3392-400; PMID:26325036; http://dx.doi.org/ 10.1172/JCI80010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V et al.. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7:303ra139; http://dx.doi.org/ 10.1126/scitranslmed.aac5415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507-17; PMID:25317870; http://dx.doi.org/ 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al.. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18; PMID:23527958; http://dx.doi.org/ 10.1056/NEJMoa1215134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725-33; PMID:21830940; http://dx.doi.org/ 10.1056/NEJMoa1103849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, den Bakker M, Oosterwijk E, Debets R et al.. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21:904-12; PMID:23423337; http://dx.doi.org/ 10.1038/mt.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18:843-51; PMID:20179677; http://dx.doi.org/ 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A et al.. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T Cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol 2015; 33:1688-96; PMID:25800760; http://dx.doi.org/ 10.1200/JCO.2014.58.0225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han WD. EGFR-targeted chimeric antigen receptor-modified T cells immunotherapy for patients with EGFR-expressing advanced or relapsed/refractory solid tumors. In: Proceedings of the 5th World Congress on Cancer Therapy, 2015; Atlanta, GA. [Google Scholar]
- 13.Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF et al.. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res 2015; 21:3149-59; PMID:25850950; http://dx.doi.org/ 10.1158/1078-0432.CCR-14-1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC et al.. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 2015; 21(18):4062-72; 10.1158/1078-0432.CCR-15-0428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J et al.. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med 2014; 6:261ra151; PMID:25378643; http://dx.doi.org/21610146 10.1126/scitranslmed.3010162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, Powell DJ Jr, Riley JL, June CH, Albelda SM. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res 2011; 17:4719-30; PMID:21610146; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, Whittington M, Yang Y, Overwijk WW, Lizée G et al.. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res 2010; 16:5458-68; PMID:20889916; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009; 113:6392-402; PMID:19377047; http://dx.doi.org/ 10.1182/blood-2009-03-209650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother 2010; 33:780-8; PMID:20842059; http://dx.doi.org/ 10.1097/CJI.0b013e3181ee6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol Cell Biol 2005; 25:6415-26; PMID:16024780; http://dx.doi.org/ 10.1128/MCB.25.15.6415-6426.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ang SO, Olivares S, Deniger DC, Lee DA, Champlin RE, Cooper LJ. Conditional activation of T cells to specifically target c-Met under hypoxia. Mol Ther 2009; 17:S25-6. [Google Scholar]
- 22.Vartanian A, Singh SK, Agnihotri S, Jalali S, Burrell K, Aldape KD, Zadeh G. GBM's multifaceted landscape: highlighting regional and microenvironmental heterogeneity. Neuro-oncology 2014; 16(9):1167-75; PMID:24642524; 10.1093/neuonc/nou035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 2006; 107:2294-302; PMID:16282341; http://dx.doi.org/ 10.1182/blood-2005-08-3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rettig MP, Ritchey JK, Prior JL, Haug JS, Piwnica-Worms D, DiPersio JF. Kinetics of in vivo elimination of suicide gene-expressing T cells affects engraftment, graft-versus-host disease, and graft-versus-leukemia after allogeneic bone marrow transplantation. J Immunol 2004; 173:3620-30; PMID:15356106; http://dx.doi.org/ 10.4049/jimmunol.173.6.3620 [DOI] [PubMed] [Google Scholar]
- 25.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B et al.. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365:1673-83; PMID:22047558; http://dx.doi.org/ 10.1056/NEJMoa1106152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, Brouns SA, Spencer DM, Till BG, Jensen MC et al.. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One 2013; 8:e82742; PMID:24358223; http://dx.doi.org/ 10.1371/journal.pone.0082742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serafini M, Manganini M, Borleri G, Bonamino M, Imberti L, Biondi A, Golay J, Rambaldi A, Introna M. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther 2004; 15:63-76; PMID:14965378; http://dx.doi.org/ 10.1089/10430340460732463 [DOI] [PubMed] [Google Scholar]
- 28.Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JH, Heemskerk MH. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica 2009; 94:1316-20; PMID:19734426; http://dx.doi.org/ 10.3324/haematol.2008.001677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, Marin V, Marafioti T, Chakraverty R, Linch D et al.. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014; 124:1277-87; PMID:24970931; http://dx.doi.org/ 10.1182/blood-2014-01-545020 [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011; 118:1255-63; PMID:21653320; http://dx.doi.org/ 10.1182/blood-2011-02-337360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabinovich PM, Komarovskaya ME, Ye ZJ, Imai C, Campana D, Bahceci E, Weissman SM. Synthetic messenger RNA as a tool for gene therapy. Hum Gene Ther 2006; 17:1027-35; PMID:17007566; http://dx.doi.org/ 10.1089/hum.2006.17.1027 [DOI] [PubMed] [Google Scholar]
- 32.Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY, Kim TG. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther 2009; 16:489-97; PMID:19096447; http://dx.doi.org/ 10.1038/cgt.2008.98 [DOI] [PubMed] [Google Scholar]
- 33.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL et al.. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 2010; 70:9053-61; PMID:20926399; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther 2011; 22:1575-86; PMID:21838572; http://dx.doi.org/ 10.1089/hum.2011.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 2013; 24:717-27; PMID:23883116; http://dx.doi.org/ 10.1089/hum.2013.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabinovich PM, Komarovskaya ME, Wrzesinski SH, Alderman JL, Budak-Alpdogan T, Karpikov A, Guo H, Flavell RA, Cheung NK, Weissman SM et al.. Chimeric receptor mRNA transfection as a tool to generate antineoplastic lymphocytes. Hum Gene Ther 2009; 20:51-61; PMID:19025415; http://dx.doi.org/ 10.1089/hum.2008.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caruso HG, Torikai H, Zhang L, Maiti S, Dai J, Do KA, Singh H, Huls H, Lee DA, Champlin RE et al.. Redirecting T-cell specificity to EGFR using mRNA to self-limit expression of chimeric antigen receptor. J Immunother 2016; 39:205-17; PMID:27163741; http://dx.doi.org/ 10.1097/CJI.0000000000000126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, Scholler J, Song D, Porter DL, Carroll M et al.. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015; 29:1637-47; PMID:25721896; http://dx.doi.org/ 10.1038/leu.2015.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA, Barrett DM, Zhao Y. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res 2014; 2:1059-70; PMID:25104548; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schutsky K, Song DG, Lynn R, Smith JB, Poussin M, Figini M, Zhao Y, Powell DJ. Rigorous optimization and validation of potent RNA CAR T cell therapy for the treatment of common epithelial cancers expressing folate receptor. Oncotarget 2015; 6(30):28911-28; PMID:26359629; http://dx.doi.org10.18632/oncotarget.5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM et al.. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2014; 2:112-20; PMID:24579088; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 2013; 1:26-31; PMID:24777247; http://dx.doi.org/26830879 10.1158/2326-6066.CIR-13-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 2016; 164:770-9; PMID:26830879; http://dx.doi.org/ 10.1016/j.cell.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrett JA, Cai H, Miao J, Sun L, Murugesan S, Chan T, Chakiath M, Krishnan S, Einstein R, Lebel F, Cooper LJN. A Synthetic Biology Rheoswitch Therapeutic System® for the Controlled Local Expression of IL-12 as an Immunotherapy for the Treatment of Cancer. Cell Biol. 2016; 5:2; 10.4172/2324-9293.1000126 [DOI] [Google Scholar]
- 45.Huang C, Ramakrishnan R, Trkulja M, Ren X, Gabrilovich DI. Therapeutic effect of intratumoral administration of DCs with conditional expression of combination of different cytokines. Cancer Immunol Immunother 2012; 61:573-9; PMID:22223258; http://dx.doi.org/ 10.1007/s00262-011-1198-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 2012; 32:1059-70; PMID:22526592; http://dx.doi.org/ 10.1007/s10875-012-9689-9 [DOI] [PubMed] [Google Scholar]
- 47.Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH et al.. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused anti-tumor activity with reduced potential for toxicity. Cancer Immunol Res 2013; 1(1):43-53; PMID:24409448; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 2013; 31:71-5; PMID:23242161; http://dx.doi.org/ 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 2013; 5:215ra172; PMID: 24337479; http://dx.doi.org/26750734 10.1126/scitranslmed.3006597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015; 350(6258):aab4077; PMID:26405231; 26750734 10.1126/science.aab4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, Duchateau P. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep 2016; 6:18950; PMID:26750734; http://dx.doi.org/ 10.1038/srep18950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM et al.. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013; 121:1165-74; PMID:23243285; http://dx.doi.org/ 10.1182/blood-2012-06-438002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther 2000; 7:35-42; PMID:10680014; http://dx.doi.org/ 10.1038/sj.gt.3301051 [DOI] [PubMed] [Google Scholar]
- 54.Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB et al.. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci USA 2016; 113:E450-8; PMID:26759368; http://dx.doi.org/ 10.1073/pnas.1524193113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O et al.. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci USA 2016; 113:E459-68; PMID:26759369; http://dx.doi.org/ 10.1073/pnas.1524155113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B et al.. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015; 75:3596-607; PMID:26330166; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song DG, Ye Q, Poussin M, Liu L, Figini M, Powell DJ Jr. A fully human chimeric antigen receptor with potent activity against cancer cells but reduced risk for off-tumor toxicity. Oncotarget 2015; 6(25):21533-46; PMID:26101914; http://dx.doi.org/0.18632/oncotarget.407126330164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, Powell DJ. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia 2016; 30:1355-64; PMID:26898190; 26330164 10.1038/leu.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H et al.. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 2015; 75:3505-18; PMID:26330164; http://dx.doi.org/ 10.1158/0008-5472.CAN-15-0139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B cell aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One 2013; 8:e61338; PMID:23585892; http://dx.doi.org/ 10.1371/journal.pone.0061338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, Schmittling RJ, Nair SK, Reap EA, Norberg PK et al.. EGFRvIII mCAR-modified T cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res 2013; 20(4):972-84; PMID:24352643; 10.1158/1078-0432.CCR-13-0709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ, Kosasih PL, Rader C, Riddell SR. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res 2015; 3:206-16; PMID:25355068; http://dx.doi.org/ 10.1158/2326-6066.CIR-14-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Connor CM, Sheppard S, Hartline CA, Huls H, Johnson M, Palla SL, Maiti S, Ma W, Davis RE, Craig S et al.. Adoptive T-cell therapy improves treatment of canine non-Hodgkin lymphoma post chemotherapy. Sci Rep 2012; 2:249; PMID:22355761; http://dx.doi.org/ 10.1038/srep00249 [DOI] [PMC free article] [PubMed] [Google Scholar]