

Abstract:
Heart failure characterized by cardiac remodeling is a global problem. Angiotensin II (Ang II) induces cardiac inflammation and oxidative stress, which also is implicated in the pathophysiology of adverse collagen accumulation–induced remodeling. Kaempferol (KPF), a kind of flavonoid compounds, is capable of anti-inflammatory and antioxidant activities. However, the target of KPF still remains blurred. In this study, we investigated the effect of KPF on Ang II-induced collagen accumulation and explored the underlying mechanisms. Our results suggested that KPF prevented Ang II-induced cardiac fibrosis and dysfunction, in mice challenged with subcutaneous injection of Ang II. In culture cells, KPF significantly reduced Ang II-induced collagen accumulation. Furthermore, KPF remarkably decreased inflammation and oxidative stress in Ang II-stimulated cardiac fibroblasts by modulating NF-κB/mitogen‐activated protein kinase and AMPK/Nrf2 pathways.
Key Words: kaempferol, angiotensin II, inflammation, oxidative stress, cardiac fibroblasts
INTRODUCTION
Heart failure is closely associated with high morbidity and accounts for a significant portion of medical care costs.1 Tissue remodeling plays a critical role during the pathological development of heart failure. Understanding the mechanisms underlying cardiac remodeling and investigating effective pharmacological intervention is important to the field of cardiovascular biology. Cardiac remodeling mainly results from abnormal collagen accumulation.2 Studies have demonstrated that angiotensin II (Ang II) promotes excessive deposition of collagen leading to cardiac remodeling as well as cardiac dysfunction and ultimately to heart failure.3,4
Several mechanisms have been implicated in the pathogenesis of heart failure, including cardiac inflammation,5,6 increased oxidative stress,7 mitochondrial dysfunction, cardiac cell apoptosis, and interstitial fibrosis. Inflammation and oxidative stress are 2 interactive factors and apparently located upstream of the other factors.8 Accordingly, therapies of anti-inflammatory and antioxidative seem to be potential approaches in treating with heart failure.
Kaempferol (KPF, 3, 4′, 5, 7 -tetrahydroxyflavone, Fig. 1A) is a flavonoid compound widely found in fruits and vegetables.9 The anti-inflammatory and antioxidant activities of KPF are well documented.10,11 Recent report indicates that high flavonols in diet, especially KPF, closely related with reduced serum inflammatory cytokine interleukin-6 (IL-6).12 Decreased expression of glutathione-S-transferase as well as glutathione in liver tissues can be reversed by KPF in the alcohol- and thermally oxidized polyunsaturated fatty acid-induced oxidative stress rat.13 KPF has shown much potential in cancer fight by several different mechanisms such as inhibition impacts of angiogenesis, inflammation, and metastasis.14
FIGURE 1.
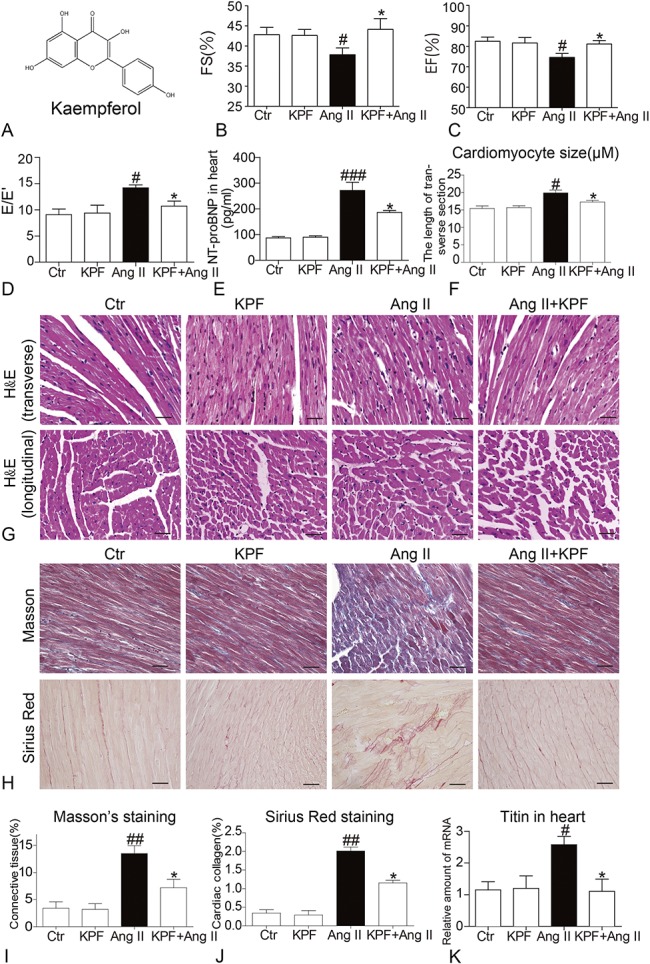
KPF reduces cardiac dysfunction and prevents Ang II-induced cardiac remodeling. A, The structure of kaempferol. Ang II-induced cardiac remodeling mice were developed, and KPF was administrated as described in the “Methods” section. Cardiac function of the experimental mice measured by echocardiography (n = 6 or 7 per group). B, Fractional shortening (FS). C, Eject fraction (EF). D, Diastolic function E/E'. E, ELISA determination of NT-proBNP in heart tissues. F, The represent images were acquired and quantified for myocyte cross-section length of 100 cells chosen from H&E. Heart tissues were subjected to H&E (G), Masson, and Sirius red (H) staining assay as described in the “Methods” section. The represent images were acquired and quantified for Masson's (I) and Sirius red (J) staining, respectively. K, Then, heart tissues were harvested and processed for the RT-qPCR analysis of titin (# vs. Ctr group; * vs. Ang II group; # and *P < 0.05, ##P < 0.01, ###P < 0.001).
With renewed interest in the pharmaceutical potential using natural products with minimal side effects, we explored the effect of KPF on Ang II-stimulated cardiac fibroblasts and further investigated the underlying mechanism. Our results demonstrated that KPF could attenuate Ang II-induced remodeling and diastolic dysfunction in vivo and reduce collagen accumulation, inflammation, and oxidative stress in cardiac fibroblasts. The beneficial actions of KPF are closely associated with its ability to increase AMPK/Nrf2 expression and inhibit NF-κB/mitogen‐activated protein kinase (MAPK) activation.
MATERIAL AND METHODS
Chemicals
Kaempferol (KPF) was purchased from Sigma-Aldrich (St. Louis, MO). Before used to the biological experiments, compounds were purified by recrystallization or silica gel chromatography to reach the purity higher than 97.0%. Compounds were dissolved in dimethylsulfoxide for in vitro experiments and were dissolved in 1% sodium carboxyl methyl cellulose (CMC-Na) for in vivo experiments.
Animal Experiments
All animal care and experimental procedures were performed in accordance with “The Detailed Rules and Regulations of Medical Animal Experiments Administration and Implementation”. Protocols for animal studies were approved by the Wenzhou Medical University Animal Policy and Welfare Committee. Eight-week-old male C57BL/6 mice weighing 18–22 g were obtained from the Wenzhou Medical University Animal Center (Wenzhou, China). Animals were housed at a constant room temperature with a 12:12-hour light–dark cycle and fed with a standard rodent diet. Ang II-mediated cardiac remodeling was induced by single subcutaneous injections of Ang II at 1.4 mg·kg−1·d−1 for 4 weeks in pH = 7.2 phosphate buffer. Ang II-induced cardiac remodeling mice were treated orally with KPF at 10 mg/kg every other day for 4 weeks, while the age-matched control groups were treated with vehicle alone. After 4 weeks of treatment, mice were anesthetized by intraperitoneal injection of 1% pentobarbital sodium (40 mg/kg) and killed; heart tissues were harvested. Heart tissues were immediately fixed in 4% paraformaldehyde for histopathological analysis.
Cardiac Function
Cardiac function was determined noninvasively by transthoracic echocardiography (Philips Electronics, Amsterdam, the Netherlands) in anesthetized mice 1 day before killing. Ejection fraction was calculated from left ventricle end-diastolic volume (LVEDV) and left ventricle end-systolic volume (LVESV) using the equation of (LVEDV − LVESV)/LVEDV × 100%. Fractional shortening (FS) was calculated using the equation: FS = [(LVIDd − LVIDs)/LVIDd] × 100%.
Heart Histology
Heart tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Five-micrometer thick sections were stained with hematoxylin and eosin (H&E) for histological analysis and Sirius red and Masson to evaluate the fibrosis content. Specimens were observed under a light microscope (×400 amplification; Nikon Tokyo, Japan).
Cell Culture and Treatment
Isolation and culture of neonatal rat primary cardiac fibroblasts was performed as described previously.7 For cell isolation, neonatal Sprague–Dawley rats' hearts were cut into pieces, washed repetitively in Hanks' Balanced Salt Solution without Ca2+, followed by 5 rounds of 8-minute digestions with 0.08% trypsin. After digestion, the cells were pelleted and suspended in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. After a 45-minute incubation, cells that were unattached were removed, and the attached cells were collected. Third to fourth passage cardiac fibroblasts were used for all experiments. The purity of these cultured cardiac fibroblasts was greater than 95% on the basis of positive staining for vimentin and negative staining for smooth muscle actin and von Willebrand factor (vWF; Sigma). Cardiac fibroblasts were treated with 10−8 M Ang II for 6–24 hours. In parallel experiments, cells were pretreated with 2.5- or 10-mM kaempferol for 30 minutes followed by treatment with Ang II (or left untreated) in the presence of kaempferol. This concentration of kaempferol in previous studies was shown to result in maximal inhibition of reactive oxygen species (ROS) release. All experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee. P65 or AMPK gene silencing in cells was achieved by transfecting cells with siRNA (5′-CUGGAUGACAUCUUAAACUTT-3-3′ for P65, 5′-GGGGAACAAACACGAGAGUUGGUUUAATT-3′ for AMPK) using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA). Knockdown was verified by Western blotting.
MTT Assay
After treatments, cells were washed for 3 times, changed medium into 1-mg/mL 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2‐H‐tetrazolium bromide, thiazolyl blue tetrazolium bromide (MTT) solution (100 μL/well, Sigma), and then incubated at 37°C for 4 hours. Cell viability was determined by measuring the absorbance by a microplate reader (SYNERGYTM 4; BioTek, Winooski, VT) at 570 nm.
Western Blot Analysis
The common procedure for Western blot analysis was described in previous publication.7 Antibodies for collagen I (1:300, cat. no. sc-25974), collagen III (1:300, cat. no. sc-271249), TGF-β (1:300, cat. no. sc-146), IκBα (1:300, cat. no. sc-1643), p-ERK (1:300, cat. no. sc-81492), ERK (1:300, cat. no. sc-514302), p-P38 (1:300, cat. no. sc-7973), P38 (1:300, cat. no. sc-7972), glyceraldehyde‐3‐phosphate dehydrogenase (1:300, cat. no. sc-32233), Nrf2 (1:300, cat. no. sc-722), and the secondary horseradish peroxidase–conjugated antibody (1:2000, anti-mouse IG-HRP cat. no. sc-3744; anti-rabbit IG-HRP cat. no. sc2313) were obtained from Santa Cruz Technology (Santa Cruz, CA). Antibodies for p-AMPK (1:1000, cat. no. 2535), AMPK (1:1000, cat. no. 2532) were obtained from Cell Signaling Technology (Danvers, MA). In all Western bolt analysis, glyceraldehyde‐3‐phosphate dehydrogenase was used as a loading control protein.
Real-Time Quantitative PCR
The common procedure for real‐time quantitative PCR (RT‐PCR) was described in previous publication.8 Primers for genes including connective tissue growth factor, collagen1, tumor necrosis factor (TNF)-α, IL-6, VACM-1, HO-1, NQO-1, and β-actin were synthesized in Invitrogen (Shanghai, China). The primer sequences used were shown in Table 1. The relative amount of each gene was normalized to the amount of β-actin.
TABLE 1.
Primers Used for Real-Time qPCR Assay

Determination of TNF-α and NT-proBNP by Enzyme-Linked Immunosorbent Assay
The TNF-α level in medium of cardiac fibroblasts was determined with an enzyme-linked immunosorbent assay (ELISA) kit (Bioscience, San Diego, CA) according to the manufacturer's instructions. The total amount of TNF-α in the cell medium was normalized to the total amount of protein in the viable cell pellets. The NT-proBNP level in heart tissues was determined with an ELISA kit (LifeSpan BioSciences, Inc, Seattle, WA, LS-F30872) according to the manufacturer's instructions.
H2O2 and O2 Staining
To analyze the ROS generation, various subtypes of ROS such as superoxide (O2−) and hydrogen peroxide (H2O2) were detected using 2-μM dihydroethidium and 2-μM DAF-2DA, respectively, as described previously.13 The fluorescence intensity for 10,000 events was acquired using fluorescence activated cell sorting, and cellular images were captured under the Nikon fluorescence microscope (×400 amplification; Nikon).
Statistical Analysis
Each in vitro experiment was performed in a group size of n > 3 independent samples. Representative images from 3 independent experiments were shown. Data were presented as mean ± SEMs. The statistical significance of differences between groups was obtained by analysis of variance followed by the multiple comparison test with Bonferroni correction in GraphPad Pro 5.0 (GraphPad, San Diego, CA). Differences were considered to be significant at P < 0.05.
RESULTS
KPF Significantly Attenuated Ang II-Induced Cardiac Dysfunction and Remodeling In Vivo
To explore the pharmacological effect of KPF on cardiac dysfunction and remodeling in vivo, Ang II-induced cardiac remodeling mice were treated with KPF 10 mg/kg for 4 weeks. Cardiac function was detected by noninvasive transthoracic echocardiograph 1 day before killing. As shown in Figures 1B, C, KPF significantly improved Ang II-induced decreasing of cardiac systolic function such as ejection fraction and fractional shortening. We also measured the diastolic function as detected by E/E′ and NT-proBNP. KPF significantly attenuated Ang II-induced increasing of E/E′ (Fig. 1D) and NT-proBNP (Fig. 1E). Histological analysis of heart tissues showed disorganized myofibers, deranged cellular structures, and increased myofibers cross-sectional area (transverse section) in Ang II mice, but improved in mice treated with KPF (Figs. 1F, G). Masson's trichrome stain for connective tissue and Sirius red for collagen further validated the antifibrotic effects of KPF (Figs. 1H–J). Titin, a giant protein that functions as a molecular spring which is responsible for the passive elasticity of muscle, plays a pivotal role in ventricular remodeling. As shown in Figure 1K, KPF decreases Ang II-induced changes of cardiac titin level by RT-qPCR analysis. These data suggested that mice with KPF were resistant to Ang II-induced cardiac dysfunction and remodeling.
KPF Attenuated Ang II-Induced Collagen Accumulation in Cardiac Fibroblasts
We initially determined the effect of KPF on cell viability. Cells were incubated with 10-mM KPF for 24 hours, and then, proliferation of cell was examined by MTT assay. As shown in Figure 2A, KPF had no obvious effect on viability of cardiac fibroblasts. Collagen accumulation leads to cardiac remodeling, which impenetrates in the process of heart failure.15 To investigate the effects of KPF on cardiac collagen biosynthesis and metabolism, we examined the levels of myocardial fibrosis–related proteins, including collagen (Col) I, Col III, and transforming growth factor (TGF)-β. As shown in Figure 2B, E, Ang II-stimulated cardiac fibroblasts increased expression of Col I, Col III, and TGF-β. KPF treatment inhibited Ang II-induced increased of these profibrosis genes (Figs. 2B–E). The mRNA data also revealed that KPF decreases Ang II-induced changes of cardiac Col I and connective tissue growth factor levels by real-time qPCR analysis (Figs. 2F, G). These results show that KPF protected against Ang II-induced cardiac fibrosis in a dose-dependent manner.
FIGURE 2.
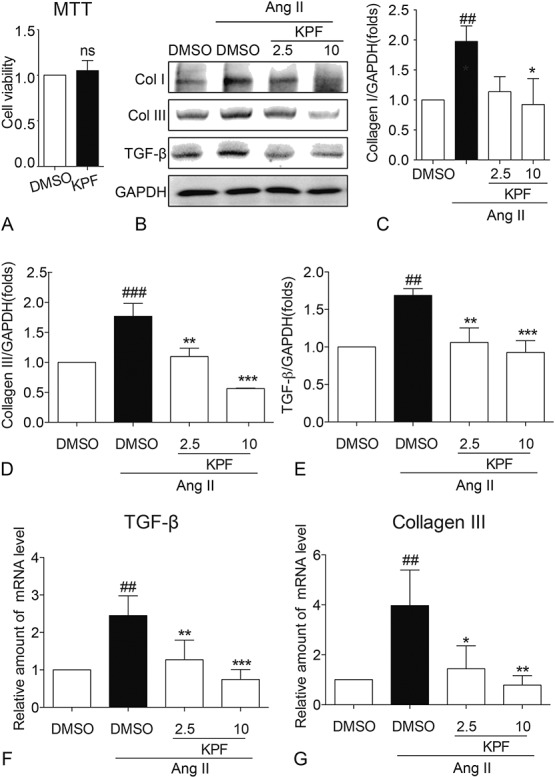
Kaempferol reduced the expression of Col I, Col III, and TGF in Ang II-stimulated cardiac fibroblasts. Cardiac fibroblasts were incubated with 10-mM KPF for 24 hours, and MTT assay was performed to detected cell viability (A). Three independent experiments were performed in duplicate. Cardiac fibroblasts were pretreated with 2.5-mM and 10-mM KPF for 24 hours and then incubated with 10−8 M Ang II for 24 hours. After that, cells were lysated, and the protein extracts were processed for Western blot analysis with GAPDH as a loading control (B). The normalized optical density of Col I (C), Col III (D), and TGF-β (E) in mean ± SE was from 3 independent experiments, respectively. Then cells were harvested and processed for the real-time qPCR analysis of CTGF (F) and Col I (G) gene expression (ns, no significant, # vs. DMSO group; * vs. DMSO + Ang II group; *P < 0.05, ## and **P < 0.01, ### and ***P < 0.001). CTGF, connective tissue growth factor; DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
KPF Attenuated Ang II-Induced Inflammation in Cardiac Fibroblasts by Inhibiting Activation of NF-κB/MAPK
Cardiac inflammation is ubiquitous in myocardial tissues, and it fuels the profibrotic process and therefore initiates adverse accumulation of collagen.16,17 Accordingly, we examined whether KPF altered Ang II-induced proinflammatory cytokine release. RT-qPCR showed increased mRNA levels of proinflammatory cytokines including TNF-α, interleukin-6 (IL-6), and cell adhesion molecule, vascular endothelial cell adhesion molecule (VCAM)-1 (Figs. 3A–C). Cell adhesion molecules are strongly associated with the macrophage infiltration clustering in inflammatory response. This increase was associated with increased level of TNF-α in medium (Fig. 3D). KPF treatment reduced level of TNF-α in medium and mRNA of TNF-α, IL-6, and VCAM-1(Fig. 3).
FIGURE 3.
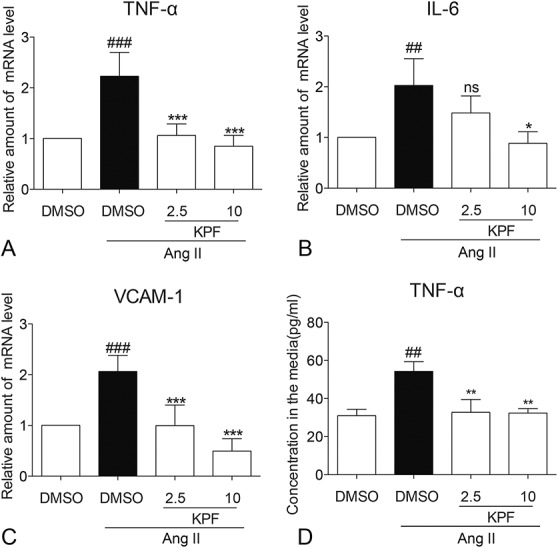
Kaempferol reduced TNF-α, IL-6, VCAM-1 mRNA expression, and content of TNF-α in Ang II-stimulated cardiac fibroblasts. Cardiac fibroblasts were pretreated with 2.5-mM and 10-mM KPF for 24 hours and then incubated with 10−8 M Ang II for 24 hours. Extracts from cardiac fibroblasts were conducted real-time qPCR analysis to determine TNF-α (A), IL-6 (B), and VCAM-1 (C) gene expression. The concentration of TNF-α (D) in medium of cardiac fibroblasts was detected by ELISA and normalized to the total amount of protein in the viable cell pellets (# vs. DMSO group; * vs. DMSO + Ang II group; ## and **P < 0.01; * P < 0.05, ### and ***P < 0.001, ns = not significant). DMSO, dimethylsulfoxide.
It has been found that NF-κB and MAPKs play a critical role in mediating inflammatory response.18 The degradation of IκB, a key step in the activation of NF-κB signaling pathway, exposes the nuclear localization sequence of NF-κB and enters the nuclear to initiate transcription.19 Figures 4A, B show that Ang II incubation for 1 hour remarkably increased IκB-α degradation, which was reversed by pretreating with KPF. MAPKs consist of extracellular signal-regulated kinase (ERK), MAPK p38 kinase, and c-Jun N-terminal kinase (JNK).20 Here, we identified that the levels of p-ERK and p-P38 were markedly elevated in Ang II-stimulated cardiac fibroblasts (Figs. 4A, C, D). However, pretreatment with KPF inhibited phosphorylation of ERK and P38 in a dose-dependent manner (Figs. 4A, C, D). To verify that KPF attenuates Ang II-induced inflammation by targeting NF-κB, we transfected cardiac fibroblasts with NF-κB P65 targeting siRNA (Fig. 4E). Silencing NF-κB P65 inhibited TNF-α and IL-6 release in Ang II-induced cardiac fibroblasts, and KPF was not able to reduce the content of TNF-α and IL-6 in the P65-knockdown cardiac fibroblasts (Figs. 4F, G). These results suggest that KPF attenuates Ang II-induced inflammation in cardiac fibroblasts by ameliorating activation of NF-κB/MAPK.
FIGURE 4.
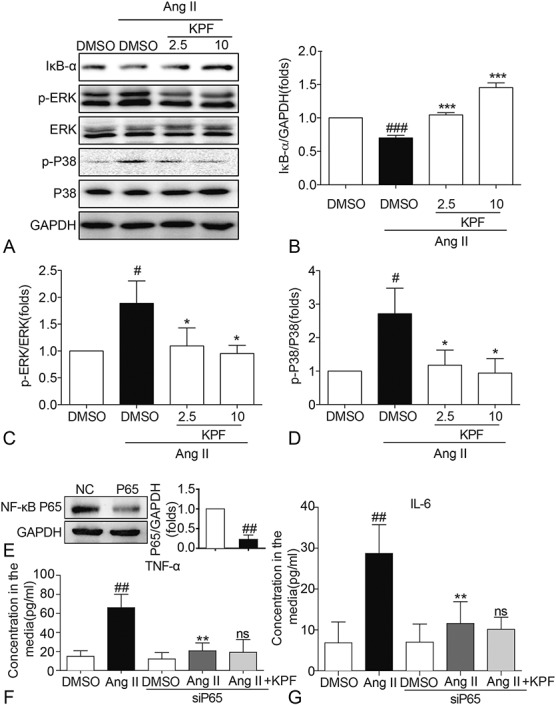
Kaempferol reduced the expression of IκB-α, p-ERK, and p-P38 in Ang II-stimulated cardiac fibroblasts. Cardiac fibroblasts pretreated with 2.5-mM and 10-mM KPF for 24 hours were incubated with 10−8 M Ang II for 24 hours. Western blot analysis was conducted on extracts from cardiac fibroblasts with GAPDH as a loading control (A). The normalized optical density of IκB-α (B), p-ERK (C), and p-P38 (D) in mean ± SE was from 3 independent experiments, respectively. E, Western blot analysis of P65 after siRNA transfection in cardiac fibroblasts. F, G, Effect of P65 silencing and KPF on Ang II-induced TNF-α (F) and IL-6 (G) mRNA levels (# vs. DMSO group; * vs. DMSO + Ang II group; ns, no significant, vs. siP65 + Ang II group; # and *P < 0.05, ## and **P < 0.01, ### and ***P < 0.001). DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
KPF Attenuated Ang II-Induced Oxidative Stress in Cardiac Fibroblasts by Activating AMPK/Nrf2 Pathway
Signaling pathways regulated by oxidative stress are increasingly recognized as an important contributor to the pathophysiology of cardiac remodeling.21 ROS refers to a group of small reactive molecules that include superoxide (O2−), hydrogen peroxide (H2O2), hydroxyl (OH−), and hypochlorite (OCl−).22 We examined the direct effects of KPF on Ang II-induced oxidative stress in cardiac fibroblasts. As shown in Figure 5, Ang II increased the production of H2O2 (Figs. 5A, B) and O2− (Figs. 5C, D), which was dose-dependently prevented by KPF in cardiac fibroblasts.
FIGURE 5.
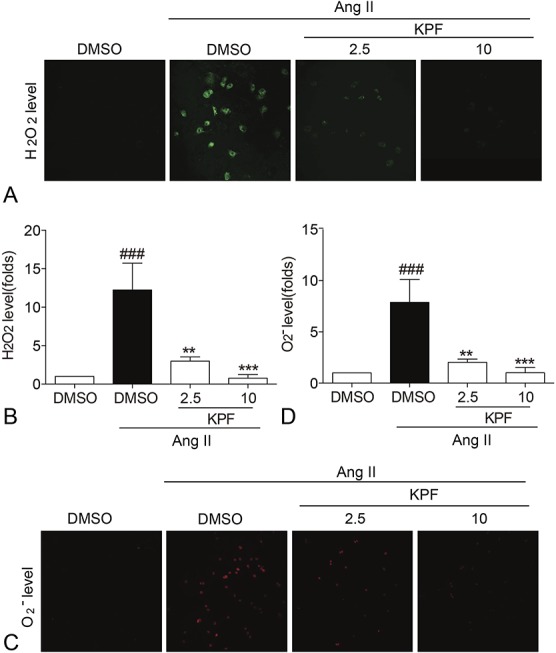
Kaempferol reduced the production of H2O2 and O2− in Ang II-stimulated cardiac fibroblasts. Cardiac fibroblasts pretreated with 2.5-mM and 10-mM KPF for 24 hours were incubated with 10−8 M Ang II for 24 hours, then DHE and DAF-2DA (2 μM) probes were loaded. Representative staining images and for H2O2 (A) and O2− (C) levels were quantified with mean fluorescence intensity (MFI) value normalized with DMSO group (B, D) (# vs. DMSO group; * vs. DMSO + Ang II group; **P < 0.01, ### and ***P < 0.001). DHE, dihydroethidium; DMSO, dimethylsulfoxide.
It is reported that the activation of AMPK can upregulate Nrf2 protein expression.23 Nrf2, a cellular defense molecule against oxidative or electrophilic stress, binds to the antioxidant-responsive elements to activate the expression of antioxidant enzymes.24,25 To further investigate the potential mechanism involved in KPF-mediated oxidative stress regulation, we examined the AMPK and Nrf2 pathways. Our data show that AMPK and Nrf-2 are all induced when treating with KPF (Figs. 6A–C). NADPH quinine oxidoreductase (NQO-1) and heme oxygenase-1 (HO-1) were Nrf-2–driven antioxidant response genes. mRNA levels of NQO-1 and HO-1 followed the same pattern as the Nrf-2 level in cardiac fibroblasts (Figs. 6D, E). In addition, to verify that KPF regulates Nrf2 expression through AMPK signal, we transfected cardiac fibroblasts with AMPK targeting siRNA (Fig. 6F). AMPK gene silencing downregulated Nrf2 protein levels in Ang II-induced cardiac fibroblasts; however, KPF was not able to induce Nrf2 protein levels in the AMPK-knockdown cardiac fibroblasts (Figs. 6G, H).Consequently, KPF attenuates Ang II-induced oxidative stress in cardiac fibroblasts by activating AMPK/Nrf2 pathway.
FIGURE 6.
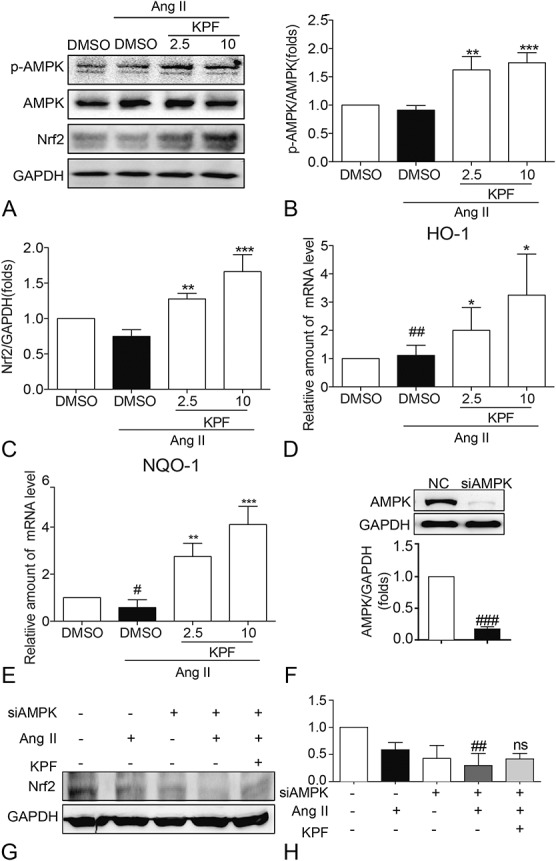
Kaempferol reduced the expression of p-AMPK, Nrf2, HO-1, and NQO-1 in Ang II-stimulated cardiac fibroblasts. Cardiac fibroblasts pretreated with 2.5-mM and 10-mM KPF for 24 hours were incubated with 10−8 M Ang II for 24 hours. Cells were harvested and processed for p-AMPK and Nrf2 detection by Western blot analysis with GAPDH as a loading control (A). The normalized optical density of p-AMPK (B) and Nrf2 (C) in mean ± SE was from 3 independent experiments, respectively. Total RNAs extracted from cardiac fibroblasts were processed for real-time qPCR assay for HO-1 (D) and NQO-1 (E). F, Western blot analysis of AMPK after siRNA transfection in cardiac fibroblasts. G and H, Effect of AMPK silencing and KPF on Ang II-induced Nrf2 protein expression (# vs. DMSO group; * vs. DMSO + Ang II group; ns, no significant, vs. siAMPK + Ang II group; # and *P < 0.05, ## and **P < 0.01, ### and ***P < 0.001). DMSO, dimethylsulfoxide; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase.
DISCUSSION
Heart failure is a global problem which refers to a syndrome that cardiac circulation is disordered caused by dysfunction of the heart's systolic and/or diastolic.33 Almost all the cardiac diseases ultimately lead to heart failure through cardiac remodeling and thus altering cardiac function. Ang II-mediated stimulation of fibroblast growth and collagen synthesis is believed to be an important component of the cardiac remodeling process in heart failure.34 Although accumulating knowledge has obtained during the past decades, we still have limited therapy for cardiac remodeling and heart failure and to investigating new medical agents are pressing. Natural products have strong advantages in the identification of bioactive lead compounds and their development into drugs for treating diseases. A great deal of native compounds has been shown to possess anti-inflammatory and antioxidant properties such as flavonoid and polyphenolic compounds.10,31,32 KPF is a kind of flavonols having non-negligible potential in preventing inflammation and oxidation stress.9,11,14 However, the mechanism of KPF effect is not fully explained. Hence, we designed this study to explore the therapeutic targets and regulatory functions.
An increasing body of evidence has emphasized that inflammation and oxidative stress are pivotal in the pathophysiology of cardiac remodeling.27–29 Redundant ROS and proinflammatory cytokines fuel the profibrotic process and therefore initiate adverse remodeling.35 Consistent with previous reports, Ang II-stimulated cardiac fibroblasts expressed apparently increased proinflammatory cytokines, TNF-α, IL-6, and cell adhesion molecule, VCAM-1 (Fig. 3). NF-κB and MAPKs are 2 important signals in mediating inflammatory response.18 The dissociation of IκB from the inactive cytoplasmic complex leads to the translocation of the active subunit NF-κB p65 from the cytosolic to nuclear fractions, which triggers inflammatory gene expression.19
KPF treatment has been associated with a positive outcome in many chronic inflammatory diseases.36 It is reported that KPF inactivates NF-κB, resulting in the decreased expression of TNF-α, IL-1, and IL-6.37 We found that KPF enhanced the level of IκB-α in Ang II-stimulated cardiac fibroblasts suggesting KPF suppressed NF-κB activation dependent inflammation (Figs. 4A, B). In addition, all the 3 subfamilies of MAPKs, especially ERK and p38, have been reported to be activated in response to inflammation. In this study, hyperphosphorylation of ERK and p38 was attenuated by KPF in Ang II-stimulated cardiac fibroblasts (Figs. 4A, C, D). To determine the potential mechanisms and target of KPF behind this protection, we transfected cardiac fibroblasts with P65 siRNA (Fig. 4E). KPF was not able to reduce the release of TNF-α and IL-6 in the P65-silencing cardiac fibroblasts (Figs. 4F, G). These indicate that KPF inhibits Ang II-induced cardiac inflammation through inactivation of NF-κB and MAPKs.
Several reports have shown that oxidative stress is one of risk factors of cardiac remodeling which indicates that oxidative stress may be a potential mechanism whereby Ang II leads to remodeling.21,38 Here, we found a remarkable augmentation of ROS when cardiac fibroblasts were treated with Ang II (Fig. 5). Nrf2 is an indispensable positive regulator of many antioxidant enzymes, which can be activated by AMPK.26 HO-1 and NQO-1 are the target genes of Nrf2 involving in expressing phase II detoxifying/antioxidant enzymes.39 Studies have indicated the action of KPF on triggering Nrf2 signaling in vitro.40 Our results demonstrated that the activation of KPF in AMPK/Nrf2 signaling (Figs. 6A–C). We further found that the expression of downstream genes of Nrf2, HO-1, and NQO-1 is also significantly increased by KPF treatment (Figs. 6D, E). Decreased KPF activity in cells transfected with AMPK siRNA suggested that KPF engaged AMPK in inducing Nrf2 in Ang II-stimulated cardiac fibroblasts (Figs. 6G, H). Correspondingly, KPF attenuates Ang II-induced oxidation stress by activation of AMPK/Nrf2 signaling.
Inflammation and oxidative stress are 2 closely interrelated pathways implicating in the process of collagen biosynthesis.41 It has been recorded that Ang II stimulates collagen deposit in many in vitro studies,42,43 and corresponding with that, our results show cardiac fibroblasts exposed to Ang II exhibited collagen biosynthesis favor (Fig. 2) accompanied with augmented inflammatory and oxidative stress level (Figs. 3 and 5). In addition, histopathological alterations in the heart were evident in cardiac remodeling mice, and these were accompanied by interstitial fibrosis and heart dysfunction (Fig. 1).
KPF has emerged decent potential in cancer fighting by modulating cellular signal transduction pathways linked to apoptosis, angiogenesis, inflammation, and oxidative stress.14 In this study, we also found KPF relieved Ang II-induced collagen accumulation in cardiac fibroblasts whereby inhibiting inflammation and oxidative stress. These results strongly suggest that targeting NF-κB/MAPK and Nrf2/AMPK may be a good therapeutic strategy in treating cardiac remodeling and heart failure. Agents including KPF and some natural active compounds with both antioxidant and anti-inflammatory properties may attract more attention for the treatment of this disease.
ACKNOWLEDGMENTS
The authors thank W. Ge and L. Jiang. They are the guarantors of this work and had full access to all the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
The authors report no conflicts of interest.
Y. Du and J. Han contribute equally to this article. Y. Du, J. Han, and W. Ge performed the research. Y. Du, J. Han, and L. Jiang designed the research study. L. Jiang and W. Ge contributed essential reagents or tools. Y. Du, H. Zhang, J. Xu, and J. Han analyzed the data. Y. Du, J. Han, and W. Ge wrote the article.
REFERENCES
- 1.Metra M, Teerlink JR. Heart failure. N Engl J Med. 2017;349:1002–1004. [Google Scholar]
- 2.Takano H, Hasegawa H, Nagai T, et al. Implication of cardiac remodeling in heart failure: mechanisms and therapeutic strategies. Intern Med. 2003;42:465–469. [DOI] [PubMed] [Google Scholar]
- 3.Brilla CG, Rupp H, Funck R, et al. The renin-angiotensin-aldosterone system and myocardial collagen matrix remodelling in congestive heart failure. Eur Heart J. 1995;16(suppl O):107–109. [DOI] [PubMed] [Google Scholar]
- 4.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. 1991;83:1849–1865. [DOI] [PubMed] [Google Scholar]
- 5.Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. 2014;70:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mooremorris T, Guimarãescamboa N, Yutzey KE, et al. Cardiac fibroblasts: from development to heart failure. J Mol Med. 2015;93:823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu M, Zheng Y, Sun HX, et al. Inhibitory effects of enalaprilat on rat cardiac fibroblast proliferation via ROS/P38MAPK/TGF-β1 signaling pathway. Molecules. 2012;17:2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang LT, Huang ZQ, Huang WJ, et al. Inhibition of epidermal growth factor receptor attenuates atherosclerosis via decreasing inflammation and oxidative stress. Sci Rep. 2017;8:45917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calderón-Montaño JM, Burgos-Morón E, Pérez-Guerrero C, et al. A review on the dietary flavonoid kaempferol. Mini Rev Med Chem. 2011;11:298. [DOI] [PubMed] [Google Scholar]
- 10.Kim HP, Son KH, Chang HW, et al. Anti-inflammatory plant flavonoids and cellular action mechanisms. J Pharmacol Sci. 2004;96:229. [DOI] [PubMed] [Google Scholar]
- 11.Samhan-Arias AK, MartíN-Romero, Gutiérrez-Merino C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for reactive oxygen species production at the plasma membrane in the commitment to apoptosis. Free Radic Biol Med. 2004;37:48–61. [DOI] [PubMed] [Google Scholar]
- 12.Bobe G, Albert PS, Sansbury LB, et al. Interleukin-6 as a potential indicator for prevention of high-risk adenoma recurrence by dietary flavonols in the polyp prevention trial. Cancer Prev Res (Phila). 2010;3:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shakya G, Manjini S, Hoda M, et al. Hepatoprotective role of kaempferol during alcohol- and ΔPUFA-induced oxidative stress. J J Basic Clin Physiol Pharmacol. 2014;25:73–79. [DOI] [PubMed] [Google Scholar]
- 14.Chen AY, Chen YC. A review of the dietary flavonoid, kaempferol on human health and cancer chemoprevention. Food Chem. 2013;138:2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurdi M, Booz GW. New take on the role of angiotensin II in cardiac hypertrophy and fibrosis. Hypertension. 2011;57:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yndestad A, Damås JK, Øie E, et al. Role of inflammation in the progression of heart failure. Curr Cardiol Rep. 2007;9:236. [DOI] [PubMed] [Google Scholar]
- 17.Lindner D, Zietsch C, Tank J, et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res Cardiol. 2014;109:428. [DOI] [PubMed] [Google Scholar]
- 18.Stan D, Calin M, Manduteanu I, et al. High glucose induces enhanced expression of resistin in human U937 monocyte-like cell line by MAPK- and NF-kB-dependent mechanisms; the modulating effect of insulin. Cell Tissue Res. 2011;343:379–387. [DOI] [PubMed] [Google Scholar]
- 19.Viatour P, Merville MP, Bours V, et al. Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. [DOI] [PubMed] [Google Scholar]
- 20.Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy—from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 2005;1754:253. [DOI] [PubMed] [Google Scholar]
- 21.Fiaschi T, Magherini F, Gamberi T, et al. Hyperglycemia and angiotensin II cooperate to enhance collagen I deposition by cardiac fibroblasts through a ROS-STAT3-dependent mechanism. Biochim Biophys Acta 2014;1843:2603. [DOI] [PubMed] [Google Scholar]
- 22.Hafstad AD, Nabeebaccus AA, Shah AM. Novel aspects of ROS signaling in heart failure. Archiv Für Kreislaufforschung. 2013;108:359. [DOI] [PubMed] [Google Scholar]
- 23.Zimmermann K, Baldinger J, Mayerhofer B, et al. Activated AMPK boosts the Nrf2/HO-1 signaling axis—a role for the unfolded protein response. Free Radic Biol Med. 2015;88:417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol. 2011;85:241–272. [DOI] [PubMed] [Google Scholar]
- 25.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W, Wu M, Tang L, et al. Novel curcumin analogue 14p protects against myocardial ischemia reperfusion injury through Nrf2-activating anti-oxidative activity. Toxicol Appl Pharmacol. 2015;282:175–183. [DOI] [PubMed] [Google Scholar]
- 27.Udelson JE, Konstam MA. Ventricular remodeling: fundamental to the progression (and regression) of heart failure. J Am Coll Cardiol. 2011;57:1477–1479. [DOI] [PubMed] [Google Scholar]
- 28.Briasoulis A, Androulakis E, Christophides T, et al. The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail Rev. 2016;21:169–176. [DOI] [PubMed] [Google Scholar]
- 29.Nediani C, Raimondi L, Borchi E, et al. NO/ROS generation and nitroso/redox imbalance in heart failure: from molecular mechanisms to therapeutic implications. Antioxid Redox Signal. 2011;14:289–331. [DOI] [PubMed] [Google Scholar]
- 30.Westermann D, Lindner D, Kasner M, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4:44. [DOI] [PubMed] [Google Scholar]
- 31.Naderi GA, Asgary S, Sarraf-Zadegan N, et al. Anti-oxidant effect of flavonoids on the susceptibility of LDL oxidation. Mol Cell Biochem. 2003;33:193–196. [PubMed] [Google Scholar]
- 32.Salah N, Miller NJ, Paganga G, et al. Polyphenolic flavanols as scavengers of aqueous phase radicals and as chain-breaking antioxidants. Arch Biochem Biophys. 1995;322:339. [DOI] [PubMed] [Google Scholar]
- 33.Braunwald E. The war against heart failure: the Lancet lecture. Lancet 2015;385:812–824. [DOI] [PubMed] [Google Scholar]
- 34.Jia L, Li Y, Xiao C, et al. Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci. 2012;17:221. [DOI] [PubMed] [Google Scholar]
- 35.Sciarretta S, Paneni F, Palano F, et al. Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clin Sci (Lond). 2009;116:467–477. [DOI] [PubMed] [Google Scholar]
- 36.Rajendran P, Rengarajan T, Nandakumar N, et al. Kaempferol, a potential cytostatic and cure for inflammatory disorders. Eur J Med Chem. 2014;86:103–112. [DOI] [PubMed] [Google Scholar]
- 37.Tang XL, Liu JX, Dong W, et al. Protective effect of kaempferol on LPS plus ATP-induced inflammatory response in cardiac fibroblasts. Inflammation 2015;38:94–101. [DOI] [PubMed] [Google Scholar]
- 38.Harrison DG, Cai H, Landmesser U, et al. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst. 2003;4:51. [DOI] [PubMed] [Google Scholar]
- 39.Yang CM, Huang SM, Liu CL, et al. Apo-8'-lycopenal induces expression of HO-1 and NQO-1 via the ERK/p38-Nrf2-ARE pathway in human HepG2 cells. J Agric Food Chem. 2012;60:1576–1585. [DOI] [PubMed] [Google Scholar]
- 40.Carreres Á. The berry constituents quercetin, kaempferol, and pterostilbene synergistically attenuate reactive oxygen species: involvement of the Nrf2-ARE signaling pathway. Food Chem Toxicol. 2014;72:303–311. [DOI] [PubMed] [Google Scholar]
- 41.Lijnen P, Papparella I, Petrov V, et al. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens. 2006;24:757–766. [DOI] [PubMed] [Google Scholar]
- 42.Bersi MR, Bellini C, Wu J, et al. Excessive adventitial remodeling leads to early aortic maladaptation in angiotensin-induced hypertension. Hypertension 2016;67:890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato H, Suzuki H, Tajima S, et al. Angiotensin II stimulates collagen synthesis in cultured vascular smooth muscle cells. J Hypertens. 1991;9:17–22. [PubMed] [Google Scholar]