

Abstract
Blood is an ideal body fluid for the discovery or monitoring of diagnostic and prognostic protein biomarkers. However, discovering robust biomarkers requires the analysis of large numbers of samples to appropriately represent interindividual variability. To address this analytical challenge, we established a high-throughput and cost-effective proteomics workflow for accurate and comprehensive proteomics at an analytical depth applicable for clinical studies. For validation, we processed one μL each from 62 plasma samples in 96-well plates and analyzed the product by quantitative data-independent acquisition (DIA) liquid chromatography/mass spectrometry (LC/MS); the data were queried using feature quantification with Spectronaut. To show the applicability of our workflow to serum, we analyzed a unique set of samples from 48 chronic pancreatitis patients, pre- and post Total Pancreatectomy with Islet AutoTransplantation (TPIAT) surgery. We identified 16 serum proteins with a statistically significant abundance alteration, representing a molecular signature distinct from that of chronic pancreatitis. In summary, we established a cost-efficient high throughput workflow for comprehensive proteomics using PVDF-membrane-based digestion that is robust, automatable, and applicable to small plasma- and serum volumes, e.g. fingerstick. Application of this plasma/serum proteomics workflow resulted in the first mapping of the molecular implications of TPIAT on the serum proteome.
Keywords: plasma, serum, TPIAT, biomarker, data independent acquisition
Graphical Abstract
INTRODUCTION
Blood is a readily accessible and obtainable body fluid in contact with most of human cells and tissues. Accordingly, concentrations of key proteins, cytokines, salts, and metabolites in plasma and serum are routinely monitored for diagnostic, prognostic, and/or predictive purposes.1 However, multiple pathologies and personalized/precision medicine strategies necessitate a comprehensive assessment of multiple protein markers that could assist in disease diagnosis or to monitor disease progression. Targeted protein analysis assays, while being highly sensitive, only allow for measuring a short predefined list of protein candidates.2 In contrast, mass spectrometry (MS)-based proteomics does not require any predefined list of proteins of interest, and allows for a hypothesis-generating, data-driven protein identification and quantification. Thus, MS has become the method of choice for protein biomarker discovery.3 As the analysis is not restricted to specific proteins, additional hypotheses may be retrospectively tested after a sample has been comprehensively analyzed by MS. This is of particular importance in initial discovery-focused research where novel findings often generate new hypotheses, but where re-analysis of samples often is impossible due to limited resources or limited sample access. Additionally, comprehensive data-driven plasma analysis could have far-reaching implications in clinical settings as it would allow practitioners to establish diagnoses not easily identified from patient symptoms or standard samples. However promising, MS-based proteomics analysis of plasma and serum remains highly challenging, mainly due to the vast protein concentration range which spans ~11 to 12 orders of magnitude.4-6 Such a dynamic range is similar to the difference between the diameter of a US quarter coin and the closest distance between Earth and Mars. To remedy this complication, it is possible to deplete the most abundant proteins from plasma and serum prior to analysis to increase the number of detectable proteins. A recent study by Keshishian H et al. (2015) identified >5300 different plasma proteins using extensive depletion and fractionation prior to thorough LC-MS analysis.8 The study demonstrated the extreme complexity encountered in plasma and the potential for discovering novel diagnostic markers. However, the study methodology includes separating the plasma sample into 30 fractions and submitting each fraction to 3.5 hours of MS-analysis. Consequently, each analysis takes >105 h, making the strategy incompatible with the analysis of hundreds of samples or implementation in a clinical setting. Even when combining this approach with multiplexing such as TMT-labeling, the instrument time per sample is still enormous. Another consideration is the significant financial cost associated with the applied abundant protein depletion system (100 times usage Seppro Supermix column ~$14650, and IgY14~$23950 USD = $386 USD per samples), which will be a limitation for many large-scale proteomics projects. Finally, the use of depletion strategies for generating signatures of e.g. the human immune system can be detrimental as numerous highly abundant proteins in plasma, which are among the proteins to be depleted, are immunologically highly relevant, including IgGs, IgMs and IgAs, and at least 18 components of the complement pathway. Additionally, the most abundant protein, albumin, is a protein carrier whose depletion also removes many albumin-bound proteins, including clinically useful biomarker from the plasma and serum samples of interest.7
Despite the fact that there are thousands of proteins in plasma down to pg/L concentrations, characterizing the higher abundance proteins at >0.1 mg/L is often sufficient for many research studies, and translational and clinical projects. This is particularly the case if the objective of such study is to obtain information about significant changes in the organismal state that affect e.g. complement system proteins and/or apolipoproteins. If need be, the generated hypotheses can subsequently be refined or expanded on a subset of the samples using targeted or more elaborate analytical strategies to detect low abundant proteins such as cytokines such as interleukins. Therefore, to facilitate robust and cost-effective large-scale plasma proteomics studies with hundreds of samples, we set-out to develop an automatable and cost-effective workflow for robustly mapping the classic serum and plasma proteome without high abundant protein depletion. We based the workflow on a 96-well plates sample preparation strategy and subsequent data-independent acquisition (DIA)-based MS method. Using the DIA workflow, we identified significantly more plasma proteins when compared to conventional data-dependent acquisition (DDA)-based MS workflows, without compromising quality of protein quantitation. The workflow was validated on 62 plasma samples and even though we targeted the classic plasma proteome with our improved plasma proteomics workflow, we detected almost half of all Food and Drug Administration (FDA)-approved blood-based biomarkers. To further demonstrated that our workflow is applicable to plasma and serum, we analyzed 96 serum samples obtained pre and post total pancreatectomy with islet autotransplantation (TPIAT). TPIAT is a specialized surgical procedure performed to relieve the pain of severe chronic pancreatitis (CP). Approximately 1000 patients have undergone the TPIAT surgery since its inception 40 years ago. In this procedure, the pancreas is entirely resected and only the islets are infused back into the liver of the recipient, in order to minimize the burden of post-surgical diabetes.9 This study represents the first mapping of the molecular effects of TPIAT.
MATERIALS AND METHODS
Plasma samples for platform development and validation
At the Hospital of Southern Denmark, Denmark, blood samples were obtained in EDTA tubes (Sarstedt, Nürnberg, Germany) from 62 Danish adults for the study (age: 48.9±18.5 years; sex: 51.6% males; BMI: 25.4±11.1) (Supplementary Table S2). Samples were centrifuged at 1500 g for 20 min, plasma collected, and stored at −80° until processing. The participants had provided oral and written informed consent and the biobank has been approved by Danish Data Protection Agency (2008-58-0035) and the regional Ethical Committee (S-20120176). Ten μL of each sample was pooled for generating a spectral library needed for the DIA method. The highly abundant proteins were depleted from some of the aliquots of the pooled sample using Seppro IgY14 spin columns (Sigma-Aldrich), according to the manufacturer’s instructions.
Serum samples to characterize the impact of TPIAT
At the University of Minnesota Medical Center, 48 chronic pancreatitis patients were enrolled in the study based on criterion eligibility for TPIAT.9 Blood samples were collected fasting, within the week prior to TPIAT and fasting at a one-year later follow-up. The samples were collected into 5 mL serum tubes and incubated at room temperature for 30 min until the clot was formed. The sample was then centrifuged at 2,000 × g for 10 min using a swinging bucket rotor. The upper layer of serum was then transferred to cryotubes and stored at −80°C until time of processing. The surgical procedure of TPIAT was performed as previously described.9 Briefly the entire pancreas was resected, along with partial duodenectomy, roux-en-Y duodenojejunostomy, choledochojenunostomy, and splenectomy. Islets were isolated by distending the pancreas with collagenase and neutral protease, followed by mechanical digestion using the semi-automated method of Ricordi et al.10 Isolated islets were returned to the operating room and infused intraportally. The participants had provided oral and written informed consent and samples were collected under IRB# 1006M83756.
96-Well Sample Processing/Digestion and Cleanup
We optimized the in-house developed MStern blotting protocol, originally designed for 96-well preparation of urine to accommodate plasma and serum samples.11,12 All solvent transfer and addition steps employed a multichannel pipette. Initially, all samples were randomized13, and 100 μL urea buffer (8 M urea, 50 mM ammonium bicarbonate (ABC) at pH 8.5) and 1 μL plasma or serum (i.e. ~70 μg protein) was added to a 96-well plate. Cysteine residues were reduced by addition of 30 μL 0.05 M dithiothreitol in urea buffer. The samples were incubated for 20 min, and the reduced cysteine residues were alkylated by addition of 30 μL 0.25 M iodoacetamide in urea buffer. After incubation for 20 min in the dark, 30 μL (i.e. ~13 μg protein) of the samples were transferred to a 96-well plate with a polyvinylidene fluoride (PVDF) membrane at the bottom (MSIPS4510, Millipore), which had been activated with 150 μL 70% ethanol, and primed with 200 μL urea buffer. A vacuum manifold was used to facilitate the liquid transfer through the 96-well PVDF membrane plate. The filter-bound proteins were washed with 200 μL 50 mM ABC. Protein digestion was performed with sequencing grade trypsin (V5111, Promega, nominal enzyme to protein ratio 1:25), by adding 100 μL digestion buffer (0.4 μg trypsin, 5% acetonitrile (ACN), 5% trifluoroethanol, and 50 mM ABC, pH 8.5) to the 96-well PVDF membrane plate. After incubation for 2 h at 37 °C in a humidified incubator, the remaining digestion buffer was evacuated from the 96-well PVDF membrane plate, and peptides were eluted twice with 150 μL 40% ACN, 0.1% formic acid (FA). The flow through and the eluents were pooled in a 96-well plate and centrifuged to dryness in a vacuum centrifuge. The peptides were resuspended in 100 μL 0.1% FA, and transferred to a 96-well MACROSPIN C18 plate (TARGA, Nest Group), which had been activated with 100 μL 70% ACN, 0.1% FA, and equilibrated with 100 μL 0.1% FA. All transfer steps were done by centrifugation at 2000x g for 2 min. The samples were washed with 100 μL 0.1% FA, and peptides were eluted with 100 μL 40% ACN, 0.1% FA, and 100 μL 70% ACN, 0.1% FA. The eluents were combined, dried down in a vacuum centrifuge and stored at −20°C until analysis.
HPLC-based High pH Reversed Phase Peptide Prefractionation
For HPLC-based high pH reversed phase peptide prefractionation, 400 μg peptide material was resuspended in 80 μL buffer A (20 mM NH4OH, pH 10), and loaded onto a XBridge column (3.5 μm C18, 2.1×100 mm, Waters) with buffer A at 0.3 mL/min, using an Agilent 1260 HPLC system (Agilent, Santa Clara, USA) connected to a sample collector. The peptides were eluted from the column on a 60 min gradient on a 0-42% ramp gradient with buffer B (20 mM NH4OH in 90% ACN, pH adjusted to 10 with FA) into 32 fractions, and combined into 20 pools.
Tip-based High pH Reversed Phase Peptide Prefractionation
For tip-based high pH reversed phase peptide prefractionation, 150 μg peptide material was resuspended in 500 μL buffer A (20 mM NH4OH, pH 10), and loaded onto OASIS C18 columns (Waters Corporation, Milford, MA). The peptides were washed with 800 μL buffer A, and step-wise eluted with 500 μL buffer B (20 mM 20 mM NH4OH in 90% ACN, pH adjusted to 10 with FA) in buffer A with ratios 10%, 12%, 14%, 16%, 18%, 20%, 22%, 70%. The eluted peptides were dried down in a vacuum centrifuge and stored at −20°C until analysis.
DDA Sample Acquisition
For the generation of a spectral library for the DIA methodology, iRT peptides (Biognosys, Schlieren, Switzerland) were spiked into the samples according to manufacturer instructions. 0.5 μg peptide material was loaded onto a capflow PicoChip column (150 μm × 10 cm Acquity BEH C18 1.7 μm 130 Å, New Objective, Woburn, MA) with 2 μL min solvent A (0.1% FA) using a micro-autosampler AS2 and nanoflow HPLC pump module (Eksigent/Sciex, Framingham, USA). The proteolytic peptides were eluted from the column using 2% solvent B (0.1% FA in ACN) in solvent A, which was increased from 2-30% on a 40 min ramp gradient and to 35% on a 5 min ramp gradient, at a flowrate of 1000 nL/min. The gradient length was in some cases varied to investigate the impact on identifications. The PicoChip containing an emitter for nanospray ionization was kept at 50°C, and mounted directly at the inlet to a 1st generation Orbitrap Q Exactive mass spectrometer (Thermo Scientific, Bremen, Germany). The mass spectrometer was operated in positive DDA top 12 mode with the following MS1 scan settings: mass-to-charge (m/z) range 375-1400, resolution 70000 @ m/z 200, AGC target 3e6, max IT 60 ms. MS2 scan settings: resolution 17500 @ m/z 200, AGC target 1e5, max IT 100 ms, isolation window m/z 1.6, NCE 27, underfill ratio 1% (intensity threshold 1e4), charge state exclusion unassigned, 1, >6, peptide match preferred, exclude isotopes on, dynamic exclusion 40 s.14
Spectral Library Construction
The .RAW-files were directly processed with MaxQuant v1.5.4.1. using the UniProt Human reference proteome (downloaded April 17th 2016, 70634 protein entries) with the iRT peptide sequences added.15-17 Standard settings were employed in MaxQuant, including a first search with 20 ppm mass tolerance followed by data calibration, and a main search with 4.5 ppm mass tolerance. A maximum of three tryptic missed cleavages were allowed. Additionally, the following modifications were found to be abundant with the applied MStern blotting protocol, and were included in the search: carbamidomethylated cysteine residues (fixed), acetylation of the N-terminal of proteins (variable), oxidation of methionine (variable), deamidation of asparagine and glutamine residues (variable).6,18 Identified proteins and peptide spectral matches were filtered to <1% false discovery rate (FDR) using a forward/reverse database search strategy in MaxQuant. For the DDA analysis of the pooled plasma sample, the match between runs feature was enabled in MaxQuant using standard parameters. This allows the transfer of MS2 identifications across LC-MS samples based on accurate m/z and retention time. LFQ was performed with a minimum ratio count of one.5,15 A spectral library was generated in Spectronaut v10 (Biognosys) using standard parameters including 0.01 peptide spectral match FDR and a Best N-filer with min three and max six fragment ions per peptide.19 The spectral library data has been deposited to the ProteomeXchange consortium via the PRIDE partner repository, and is available with the dataset identifier PXD008163.20,21
DIA Sample Acquisition
HRM calibration peptides (Biognosys, Schlieren, Switzerland) were spiked into the DIA samples according to manufacturer instructions. The samples were analyzed on the same LC-MS system as the DDA runs, using identical LC parameters (45 min gradient, total runtime 59 min). The mass-range m/z 375 – 1200, covering 95% of the identified peptide m/z, was divided into 15 variable windows based on density (Supplementary table S3), and the following parameters were used: resolution 35000 @ m/z 200, AGC target 3e6, maximum IT 120 ms, fixed first mass m/z 200, NCE 27. The DIA-scans preceded a MS1 Full scan with identical parameters, yielding a total cycle-time of 2.4 s.
DIA Data Analysis
All DIA data was analyzed directly in Spectronaut v10 (Biognosys).19 Standard settings were employed, which included dynamic peak detection, automatic precision non-linear iRT calibration, interference correction, and cross run normalization (total peak area) enabled. Spectronaut utilizes the spiked-in HRM peptides for m/z and retention time calibration. All results were filtered by a q-value of 0.01 (equal to a FDR of 1% on peptide level). All other settings were set to default.
Statistical Analysis
To evaluate the reproducibility of the DDA and DIA method, a pooled plasma sample was analyzed in triplicate by capflow-LC-MS. For statistical analysis, the data was imported into Perseus v1.5.5.3.22,23 A peptide is defined as a unique amino acid sequence, charge, and modification. Coefficient of variance (CV) was calculated for proteins and peptides identified in all three replicates. In the analysis of 62 plasma samples, proteins with less than a total of 21 valid values (33%) were removed. Male and female specific plasma proteins were identified by independent two-samples t-test with multiple testing correction by permutation-based FDR using standard parameters in Perseus (FDR < 0.05, s0 = 0.1). Prior to t-testing, missing values were imputed using numbers drawn from a normal distribution, using standard parameters in Perseus (0.3 width, 1.8 downshift) to simulate signals from low-abundant proteins.14
Similarly, the identified proteins in the TPIAT serum proteins (FDR < 1%), were further filtered for potential contaminants, and proteins with valid values in < 70% of either the post- or pre TPIAT group were removed. Proteins with a TPIAT specific change of abundance were identified by paired two-samples t-test with multiple testing correction by permutation-based FDR using standard parameters in Perseus (FDR < 0.05, s0 = 0.1).
RESULTS AND DISCUSSION
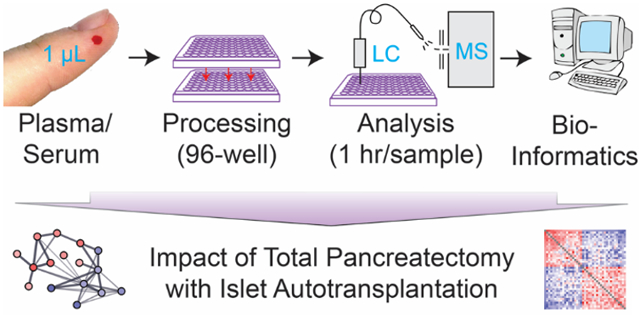
We set-out to develop an automatable and cost-effective workflow for robustly mapping the classic serum and plasma proteome, usable for data-driven high-throughput plasma proteomic studies where depletion of the most abundant proteins and/or fractionation is infeasible and/or undesirable. Such a workflow is of particular importance for large scale plasma proteomic studies with hundreds of samples. Given the documented advantages of the MStern blotting approach for the 96-well plate-based processing of urine samples11,24, we optimized the workflow for applicability to plasma and serum, and established a DIA strategy for the subsequent LC/MS analysis similar to what we and other have described for body fluid proteomics.11 Once optimized, we validated the workflow using 62 plasma samples for validation, and further demonstrate the applicability of the workflow on 96 serum samples, through an analysis of the systemic impact of TPIAT on the serum proteome, which has not previously been characterized.
96-Well PVDF membrane plate digestion strategy
We prepared all samples for proteomics analysis using a serum/plasma adaptation to our MStern blotting sample processing strategy using 96-well PVDF membrane plates.11 The strategy requires <1 μL of plasma/serum and enables preparation of 96 samples in parallel in <3 hours. While the optimized protocol as described in the Materials and Methods sections works well for plasma and serum, a C18 cleanup step is strongly recommend for larger number of samples to minimize the risk of clogging the analytical reversed phase column of the LC/MS set-up. When using a 96-well C18 desalting plate, the sample clean-up is easily integrated into the 96-well plate-based MStern blotting workflow.
Multichannel pipettes are used throughout the protocol which reduces the risk of pipetting errors when working with many samples indicating that the workflow easily automatable. Additionally, the protocol is highly cost effective and the estimated sample preparation cost for 96 samples is approximately $122 USD or $1.3 USD/sample (PVDF-membrane plate $34, trypsin $48, chemicals and materials $40). Including the optional 96 well C18-plate purification ($224), the total cost becomes $346 USD ($3.6 USD/sample). Comparably, the estimated cost for 96 samples with e.g. the iST approach from the Mann group, the commercial 96x kit from Preomics runs at $3200 USD or ~$33 USD/sample.25
Spectral Library Generation
For the generation of a spectral library for the DIA methodology, a fraction of the purified peptide product was pooled and analyzed by DDA LC-MS. We performed high pH C18 fractionation which has been reported to outperform the fractionation by OFFGEL isoelectric focusing (peptide), SDS-PAGE (proteins), and strong cation exchange chromatography (peptide) for plasma in terms of identified proteins.26 Cumulated, we identified 21788 peptide precursors corresponding to 1000 proteins (both at <1% FDR) in plasma (Table 1), which constitutes the spectral library for the DIA method used for this study. Based on our findings, high pH C18 fractionation is an efficient scheme for generating spectral libraries. Depletion of the 14 most abundant plasma proteins (Top14) only resulted in a moderate increase in the number of identifications, with 14.5% of the identified proteins and 26.4% of the identified peptide precursors in the spectral library were unique to the depleted plasma samples. The depletion of a broader range of high-abundance protein and/or more extensive fractionation scheme would likely have enabled the identification of additional plasma proteins.8 However, as the goal was to establish a reliable spectral library to facilitate DIA detection of the classic and most abundant tissue leakage plasma proteins in non-depleted samples, we reasoned that additional proteins identified after more extensive fractionation would likely be outside the instrument’s detection limit when conducting performing high throughput DIA analysis on non-depleted plasma.
Table 1.
Precursor and protein identifications in the developed plasma spectral library.
| Protein Depleted |
Fractionation | #Precursors | #Proteins | LC-MS time [h] |
|---|---|---|---|---|
| No | None | 7381 | 511 | 56 |
| No | HPLC-based 20 fractions | 12579 | 660 | 48 |
| No | Tip-based 8 fractions | 7277 | 476 | 36 |
| Top14 | None | 10703 | 483 | 30 |
| Top14 | Tip-based 8 fractions | 12358 | 688 | 75 |
| Cumulated | 21788 | 1,000 | 212 |
Top14: Depletion of the 14 most abundant plasma proteins prior to proteolytic digestion. Fractionation: Peptides were high pH C18 reversed phase fractionated prior to LC-MS analysis using a HPLC system or pipette compatible tips.
Highly Reproducible Identification and Quantitation with DIA
Reproducibility is a prerequisite for any (bio)analytical technique. Therefore, to assess the reproducibility of the DIA-based peptide and protein quantitation, we analyzed a pooled plasma sample in triplicates using DIA and DDA. Although there was a percentagewise slightly larger overlap in peptide identifications with DDA compared to DIA (87.0% vs. 76.8%,), the absolute number of overlapping peptide identifications for the DIA dataset vastly exceeded the number of overlapping proteins in the DDA data set: 5092 (DIA) vs. 2533 (DDA). When computationally assembling the peptides into protein identifications, the DIA method was superior both in number of identified proteins and in percentage of proteins identified in all three replicates (424 vs. 203, and 82.5% vs. 79.9%).
Similarly, we also calculated the coefficient of variation (CV) for peptides and proteins identified and quantified in all three replicates (Figure 1A). The median %CV of the peptide precursors identified with DIA was close to half compared to DDA (8.8% vs 16.0%) while having more than twice the number of identifications. In case of the normalized protein quantitation, the median %CV was slightly lower for the DDA data compared to the DIA method (9.6% vs 9.2%). However, 109% (221) more proteins were quantifiable with the DIA method compared to the DDA method with a minimal impact on the median %CV.
Figure 1.

Triplicate analysis of a pooled plasma sample. A) Coefficient of variance (CV) for features found in all three replicates with median CV, number of features, and color-coded point density (light blue, highest density; green, lowest density). B) Proteins ranked on increasing CV. C) Concentration-range of the plasma proteins as represented by intensity-based absolute quantitation (iBAQ) values.
We analyzed the proportion of proteins with < 10% and 20% CV, as the latter is a commonly applied cutoff for in vitro diagnostic assays (Figure 1B).27 Reassuringly, our findings on the protein and peptide CVs are consistent with previous reports on DIA vs. DDA.24,28 Of the 424 DIA proteins, 51.6% were detected with CV < 10%, which is comparable to the 56.7% of the 203 DDA proteins with CV < 10%. However, the absolute number of proteins below the two CV-thresholds was for both cutoffs almost twice as large with the DIA methodology, indicating that the quantitative accuracy is not compromised with the higher number of plasma proteins.
Our goal was to establish a robust method for the reproducible detection and quantitation of the classic and more abundant tissue leakage plasma proteins. Thus, we estimated the concentration range of the identifiable plasma proteins with DIA and DDA. We used the list of 1000 proteins from the spectral library, to estimate the intensity-based absolute quantitation (iBAQ) value for each protein, which is an estimate of the absolute concentration of the proteins in the sample; MaxQuant was used to calculate the iBAQ values.29 Based on these iBAQ values, the dynamic range of the spectral library spanned 7.5 orders of magnitude, the DIA method 7.2 orders of magnitude, and the DDA method 6.3 orders of magnitude (Figure 1C), demonstrating the superior dynamic range and detection limit of the DIA method as compared to the conventional DDA approach. In summary, our plasma proteomics data confirm previous reports of increased quantitative accuracy and sensitivity of the DIA method compared to the DDA method.24,28,30
Workflow validation with 62 plasma samples
To evaluate the 96-well plate-based plasma and serum proteomics pipelines, we first analyzed plasma from 62 participants (32 males, 30 females). The samples were digested without depletion of any highly abundant proteins and analyzed using a 45 min microflow (1 μL/min) LC gradient (59 min analysis time per sample), without encountering clogging/high pressure issues. The mass spectrometer was operated using the optimized DIA plasma method and cumulated 664 plasma proteins were identified (Supplementary Table S4). On average, 439 plasma proteins were identified in each sample, of which 55%, i.e. 241 were identified in all 62 plasma samples (Supplementary Table S5). In comparison, Liu et at 2015 identified on average 425 proteins in plasma samples using a similar approach with SWATH MS, but using almost three times as much instrument time per sample.28
To investigate the dynamic range of the absolute plasma-concentrations of the quantifiable proteins in our study, we added published absolute concentrations for all proteins (257) with data available from The Plasma Proteome Database in mol/L (Figure 2) and g/L (Supplementary Figure S1).31 For proteins with several reported absolute concentrations, we calculated the average protein concentration.
Figure 2.

Absolute concentration of the quantifiable plasma proteins with the workflow, demonstrating a dynamic range covering over 7 orders of magnitude. Averaged data from The Plasma Proteome Database is reported.29
The proteins spans a concentration range covering 7.5 orders of magnitude (g/L and mol/L) in agreement with the analysis of the iBAQ-range of the identified proteins in the pooled plasma sample, and span from albumin at ~39 g/L (0.56 mM) to fatty acid synthase at ~4.8 μg/L (18 pM). The ranges include the complement system proteins e.g. complement C3 at ~1.2 g/L (6.6 μM) and complement factor D at ~2.2 mg/L (82 nM) and apolipoproteins e.g. apolipoprotein A1 at ~1.1 g/L (36 μM) and apolipoprotein C-IV at ~1.0 mg/L (69 nM). The developed methodology thereby covers the classical plasma proteins such as the complement system proteins and apolipoproteins at concentrations down to μg/L.
To investigate the extent to which we could quantify clinically validated biomarkers, we compared our plasma proteomics data with the list of 109 Food and Drug Administration (FDA)-approved biomarkers in plasma and serum from Anderson et al. (2010).32 In the 62 plasma samples, 50 (46%) of the FDA-approved biomarkers could be detected (Supplementary Table S6), i.e. ~20% more than Liu et al. 2015, who detected 42 (40%) FDR-approved biomarkers in their SWATH-based plasma proteomics study28. In the present study, 2/3 (i.e. 33) of the biomarkers were found in all 62 samples without a single missing value, and 38 within at least 90% of the samples. To analyze the quantitative nature of our DIA-based workflow, we further examined proteins with sex specific abundances (Figure 3). In concordance with Geyer and Mann et al.,5 we found only two significantly changed proteins, namely pregnancy zone protein (PZP) and sex hormone-binding protein (SHBG), both of which were found in lower plasma concentration in samples from the male participants (two-samples t-tests, FDR < 0.05). Verifying these known differences, provides further validation to the applied methodology.
Figure 3.

Proteins with a sex-specific abundance. Pregnancy zone protein (PZP, o) and sex hormone-binding protein (SHBG, o) was significantly decreased in male plasma compared to female. + indicates not significantly changed proteins. The black curve represents the statistical significance cutoff (FDR < 0.05)
Extension to serum proteomics – studying the effect of TPIAT on the serum proteome
Upon demonstrating the robustness and cost effectiveness of our 96-well PVDF membrane plate-based plasma proteomics platform that requires less than 1 μL of plasma, we investigated the applicability of our platform to serum as a next step. To this end, we processed and analyzed a unique set of samples from chronic pancreatitis (CP) patients before and after total pancreatectomy with islet auto transplantation (TPIAT). CP is responsible for > 56,000 hospitalizations annually in the USA.33 It is a painful and disabling condition, which can arise secondary to a prolonged pancreatic inflammation and fibrosis, and results in several clinical features, including abdominal pain, diabetes, malabsorption, and steatorrhoea.34 The majority of the patients are treated medically, but some patients experience disabling pain leaving them dependent on opiates and with a reduced quality of life. For this subgroup of patients, treatment options are limited and include total pancreatectomy where the pancreas is removed, which can provide complete relief. However, the invasive procedure has several consequences for the patients, including insulin-dependent diabetes. Islet isolation and extraction of the islets from the pancreas and subsequent autologous transplantation into the liver can retain some production of insulin, C-peptide, and glucagon, thereby easing the daily diabetic control or even preventing the onset of diabetes.35,36 The TPIAT procedure was developed at the University of Minnesota in 1977, and so far only a few centers in the USA have established annual programs.34,37 Numerous clinical studies have been published, however, little is known about TPAT-induced changes at the molecular level, which might be explained by the limited sample availability as only ~1000 patients have undergone the TPIAT surgery since its inception 40 years ago. To this end, we decided to undertake a first-of-its-kind serum proteomics study on paired serum samples from 48 chronic pancreatitis patients collected pre- and post-TPIAT surgery to assess the systemic impact of TPIAT on the serum proteome. The 96 serum samples were processed using the same method as the plasma samples without any clogging issues or other problems, underscoring the robustness of the workflow.
Cumulated, we identified a total of 487 proteins in serum. Comparing the pre- vs post-TPIAT serum samples using paired two-samples t-tests, we identified 16 serum proteins with a statistically significant change of abundance (multiple testing correction controlled by FDR < 0.05, Table 2); 9 proteins were more abundant and 7 proteins less abundant post TPIAT (Figure 4).
Table 2.
Significantly changing serum-proteins post vs pre TPIAT.
| Fold change Log2(Post vs Pre) |
P-value | UPID | Protein Name | Gene Name |
|---|---|---|---|---|
| 2.04 | 1.30E-09 | P02775 | Platelet basic protein | PPBP |
| 1.99 | 1.18E-03 | P10720 | Platelet factor 4 variant | PF4V1 |
| 1.73 | 2.63E-03 | Q9GZX7 | Single-stranded DNA cytosine deaminase | AICDA |
| 1.56 | 1.20E-03 | P01703 | Ig lambda chain V-I region NEWM | IGLV1-40 |
| 1.56 | 9.80E-03 | P00441 | Superoxide dismutase [Cu-Zn] | SOD1 |
| 1.56 | 2.49E-04 | P07996 | Thrombospondin-1 | THBS1 |
| 1.53 | 3.44E-07 | P60709 | Actin, cytoplasmic 1 | ACTB |
| 1.49 | 7.03E-03 | P68871 | Hemoglobin subunit beta | HBB |
| 1.43 | 3.15E-03 | P80748 | Ig lambda chain V-III region LOI | IGLV3-21 |
| 0.82 | 7.67E-06 | O14791 | Apolipoprotein L1 | APOL1 |
| 0.75 | 1.97E-04 | P01871 | Ig mu chain C region | IGHM |
| 0.69 | 5.71E-05 | P02656 | Apolipoprotein C-III | APOC3 |
| 0.68 | 3.79E-04 | P00734 | Prothrombin | F2 |
| 0.67 | 2.45E-03 | P08519 | Apolipoprotein(a) | LPA |
| 0.66 | 3.58E-03 | P02753 | Retinol-binding protein 4 | RBP4 |
| 0.64 | 6.85E-03 | P55056 | Apolipoprotein C-IV | APOC4 |
List of proteins with statistically significantly changed abundance (FDR < 0.05) post vs pre TPIAT. UPID: UniProtKB protein identifier.
Figure 4.

Proteins with post vs pre TPIAT specific abundance. The black curve represents the statistical significance cutoff (FDR < 0.05). □: more abundant post; o: less abundant post, +: not significantly changed.
TPIAT induced molecular changes are distinct from those associated with pancreatitis
Taking a closer look at the list of proteins with statistically significant abundance differences, one of the most notable findings were reduced serum concentrations of several apolipoproteins, which bind to lipids in the blood and function as structural components of lipoprotein particles which are transported through the lymphatic and circulatory systems. Of the 7 proteins showing significant downregulation in serum post TPIAT, 4 were members of the apolipoprotein family: Apolipoprotein L1 (APOL1), Apolipoprotein C-III (APOC3), Apolipoprotein C-IV (APOC4), and Apolipoprotein (a) (LPA). Additionally, Apolipoprotein E (APOE) showed an almost statistically significant (p-value=0.001; q-value=0.053) post-TPIAT decrease. Apart from the five above mentioned apolipoproteins with decreased serum concentrations post TPIAT, five additional apolipoproteins were identified in our dataset (Apolipoprotein A-I, Apolipoprotein A-II, Apolipoprotein B-100, Apolipoprotein D, and Apolipoprotein M). However, none of these other 5 apolipoproteins demonstrated a TPIAT-induced abundance difference anywhere near statistical significance (p-values >0.25; q-values >0.5), dividing the identified apolipoproteins into two groups. Changes to the serum-concentration of several apolipoproteins in patients with CP has previously been described in two independent serum proteomics studies in the context of chronic pancreatitis.38,39
Further investigation of the changing vs. not changing apolipoproteins we noted that the apolipoproteins with statistically reduced abundances are part of the smaller HDL-particles and are associated with a faster clearance, as compared to the non-changing apolipoproteins (Dr. S. Singh (Brigham and Women’s Hospital), personal communication and reference 40). This finding might indicate that the TPIAT procedure induces changes in the HDL-particle distributions, possibly because of altered fat absorption due to post-TPIAT enzyme replacement therapy.
Because this is the first systems biology characterization of the impact of TPIAT, we took a closer look at the remaining 12 proteins that we had found to be of significant altered abundance post TPIAT. We noted that the vast majority of these proteins had been described in the context of pancreatic cancer and/or chronic pancreatitis. For instance, actin is well known to be upregulated in CP, probably due to increased fibrosis, platelet counts are down in CP41,42 and activation-induced cytidine deaminase (AICDA) has been described to be increased in pancreatic ductal adenocarcinomas (PDAC) and various pre-cancerous lesions.43 Similarly, differences in concentrations of IgG heavy chains,44,45 Cu-Zn superoxide dismutase (SOD1),46 thrombospondin-1 (THBS1),47 hemoglobin beta (HBB)48 and retinal binding protein 4 (RBP4) in sera from patients with CP and/or pancreatic exocrine insufficiency have been described. 38,48
When starting this project, we hypothesized that any described CP-induced changes would revert back to normal levels upon pancreatectomy. This hypothesis held up for a subset of a subset of proteins found to be of significantly different abundance post TPIAT. This subset included the platelet proteins (PPBP and PF4V1), thrombosponding-1, Cu-Zn Superoxide dismutase, and hemoglobin beta; the blood levels of these five proteins had been described as being suppressed in CP vs. normal, i.e. by removing the diseased organ and thus the assumed cause of the suppression their levels reverted back to more normal levels. However, to our surprise, we found as many proteins whose abundance differences did not show the expected directionality, i.e. proteins that had been described of being e.g. upregulated in CP, showed unexpectedly further increases post-TPIAT (or vice versa). Members of this protein subgroup included retinol binding protein 4 (RBP4), two IgG heavy chains, activation-induced cytidine deaminase (AICDA), and actin (ACTB). For example, it is expected that IgGs and actin are increased in CP vs. normal due to the CP-induced inflammation and fibrosis. Although we expected decreased levels of these proteins after removal of the inflamed, fibrosed organ, we observed increased serum actin and IgG levels post-TPIAT. As the post-TPIAT samples had been collected at least 12 months post-surgery, an acute inflammatory response to the surgical procedure can be excluded. To investigate whether these two post-TPIAT upregulated IgGs were not just outliers, we took a closer look at all identified immunoglobulins (see Supplementary Figure S1). The volcano plot for all the immunoglobulins shows some of the identified immunoglobulins decrease in abundance levels post-TPIAT; however, overall the identified heavy and light chains showed an increase post TPIAT providing evidence for a more, not less, active immune system more than one year post TPIAT.
Although it was reassuring that the majority of the serum proteins observed to be of altered abundance post TPIAT had been described before in the pancreas disease context, rationalizing the con- or discordance of the observed and previously described abundance level changes were less obvious. Our findings clearly indicate that the assumption that all CP-induced changes are reverted back to normal levels after removal of the pancreas, i.e. the diseased organ, is too simplistic. Instead, differentiation into pancreatitis-induced and pancreas activity-induced abundance differences is necessary: while the former will be reverted back to normal after TPIAT, it is expected that the latter changes will be amplified post TPIAT as the removal of the pancreas results is equivalent to the complete loss of pancreatic activity.
Thus, more detailed work will be necessary to better understand the observed TPIAT-induced abundance changes for a subset of the identified and quantified serum proteins and to pinpoint the pancreas and pancreatic activity-related serum proteome changes. Such follow-up experiment will include the deconvolution of the effect of systematic TPIAT-associated confounders including changes in pain management, induced pancreatic exocrine insufficiency and concomitant enzyme replacement therapy, and diabetes.
CONCLUSIONS
In this paper, we present a robust and cost-effective plasma- and serum proteomics workflow, which allows for the parallel preparation of 96 samples in the course of four hours for less than $4 USD per sample. The workflow allows for monitoring larger organismal changes such as the immune system or mapping the profile of apolipoprotein and associated/co-regulated proteins. Using filter plates with hydrophobic PVDF membranes and DIA for the subsequent data acquisition, < 1 μL of plasma or serum is required which makes the workflow compatible with finger stick-based sampling. The use of microflow LC columns in the 150 to 200 μm inner diameter range, enabled the analysis of one plasma sample in <1 hour while still maintaining a 45 min LC gradient, and all samples in the study were analyzed without column clogging issues demonstrating the robustness of the microflow setup. Moving from conventional DDA to DIA allowed us to more than double the number of identified and quantified proteins, without compromising quantitation accuracy as determined by median CV. The workflow does not rely on chemical labelling or depletion of abundant proteins and still provides insights into the classic and tissue leakage proteome covering 7+ orders of magnitude dynamic range. The robustness of our proteomics platform was demonstrated by analyzing 62 plasma and 96 serum samples. The analysis of the 62 plasma samples served as proof of concept, identifying and quantifying 453 proteins on average in each sample, including 46% of FDA approved blood protein biomarkers, in effect removing the need for pre-selection of biomarker candidates in plasma. Although the samples for the presented proof of concept study were readily processed without any liquid handling robot, automation would be easily implementable. Subsequently, we conducted a first of its kind serum proteomics analysis of samples taken from 48 chronic pancreatitis patients before and after total pancreatectomy with Islet Auto-Transplantation (TPIAT), a surgery devised 40 years ago and since then performed on only ~1,000 patients. Most of the serum proteins, that show statistically significant abundance differences after TPIAT had been described in the context of pancreatic diseases and malignancies. The directionality of the observed TPIAT-induced changes points towards the need to differentiate between pancreatitis and pancreatic function dependent changes, each with potential for diagnosis and/or pancreatic function monitoring, both of which will be the objective of follow-up studies. This first investigation of the molecular implications of TPIAT complement the 250+ publications describing TPIAT and its the macroscopic, physiological effects.
Supplementary Material
Supplementary table S1 (.pdf) Cover page.
Supplementary table S2 (.xls) Participant information.
Supplementary table S3 (.xls) DIA plasma method variable windows.
Supplementary table S4 (.xls) Cumulated list of plasma proteins in the 62 participants.
Supplementary table S5 (.xls) List of plasma proteins shared between all 62 participants.
Supplementary table S6 (.xls) List of identified FDA-approved biomarkers.
Supplementary Figure S1 (.png) Absolute concentration of the quantifiable plasma proteins with the workflow reported in g/L.
Supplementary Figure S2 (.png) Volcanoplot of TPIAT changes to the serum proteome with Igs.
ACKNOWLEDGMENT
OL and HS’s laboratories are supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Human Immunology Project Consortium (HIPC) Award Number U19AI118608. The research reported in this publication was also supported by the National Cancer Institute and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under award number U01DK108327 (HS, DC). MDB’s laboratory was supported by grants from the American Diabetes Association (1-11-CT-06) and the National Institutes of Health (K23 DK084315). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Furthermore, we would like to acknowledge the following grants that enabled the work described in this manuscript: TBB – funding from the Lundbeck Foundation (R181-2014-3372, and R275-2017-2219), The Carlsberg Foundation (CF14-0561), and A. P. Møller. AS – funding from the Obelske Family Foundation, the Svend Andersen Foundation, the Spar Nord Foundation and the Danish National Mass Spectrometry Platform for Functional Proteomics (PRO-MS). VA – funding from the Lundbeck Foundation (R126-2012-12194), Hospital of Southern Jutland, University of Southern Denmark, and “Knud og Edith Eriksens Mindefond”.
ABBREVIATIONS
- ABC
ammonium bicarbonate
- ACN
acetonitrile
- ACPA
autoantibody against citrullinated protein
- BMI
body mass index
- CP
chronic pancreatitis
- CV
coefficient of variation
- DDA
data dependent acquisition
- DIA
data independent acquisition
- FA
formic acid
- FDA
food and drug administration
- FDR
false discovery rate
- HPLC
high performance LC
- iBAQ
intensity-based absolute quantitation
- LC
liquid chromatography
- MS
mass spectrometry
- MS1
survey scan
- MS2
fragment ion scan
- m/z
mass-to-charge
- PVDF
polyvinylidene fluoride
- PZP
pregnancy zone protein
- SHBG
sex hormone-binding protein
- TPIAT
total pancreatectomy with islet auto-transplantation
Footnotes
The spectral library data has been deposited to the ProteomeXchange consortium via the PRIDE partner repository, and is available with the dataset identifier PXD008163.
Conflict of Interest Pertaining to the Presented Work:
VA receives compensation for consultancy and for being a member of an advisory board for Merck (MSD) and Janssen. The other authors declare no competing interests.
REFERENCES
- (1).Malekzadeh A; Twaalfhoven H; Wijnstok NJ; Killestein J; Blankenstein MA; Teunissen CE Comparison of Multiplex Platforms for Cytokine Assessments and Their Potential Use for Biomarker Profiling in Multiple Sclerosis. Cytokine 2017, 91, 145–152. [DOI] [PubMed] [Google Scholar]
- (2).Bennike T Biomarkers in Inflammatory Bowel Diseases: Current Status and Proteomics Identification Strategies. World J. Gastroenterol. 2014, 20 (12), 3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Mann M Origins of Mass Spectrometry-Based Proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17 (11), 678–678. [DOI] [PubMed] [Google Scholar]
- (4).Anderson NL The Human Plasma Proteome: History, Character, and Diagnostic Prospects. Mol. Cell. Proteomics 2002, 1 (11), 845–867. [DOI] [PubMed] [Google Scholar]
- (5).Geyer PE; Kulak NA; Pichler G; Holdt LM; Teupser D; Mann M Plasma Proteome Profiling to Assess Human Health and Disease. Cell Syst. 2016, 2 (3), 185–195. [DOI] [PubMed] [Google Scholar]
- (6).Bennike T; Ayturk U; Haslauer CM; Froehlich JW; Proffen BL; Barnaby O; Birkelund S; Murray MM; Warman ML; Stensballe A; et al. A Normative Study of the Synovial Fluid Proteome from Healthy Porcine Knee Joints. J. Proteome Res. 2014, 13 (10), 4377–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhou M; Lucas DA; Chan KC; Issaq HJ; Petricoin EF; Liotta LA; Veenstra TD; Conrads TP An Investigation into the Human Serum “Interactome.” ELECTROPHORESIS 2004, 25 (9), 1289–1298. [DOI] [PubMed] [Google Scholar]
- (8).Keshishian H; Burgess MW; Gillette MA; Mertins P; Clauser KR; Mani DR; Kuhn EW; Farrell LA; Gerszten RE; Carr SA Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell. Proteomics 2015, mcp.M114.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chinnakotla S; Beilman GJ; Dunn TB; Bellin MD; Freeman ML; Radosevich DM; Arain M; Amateau SK; Mallery JS; Schwarzenberg SJ; et al. Factors Predicting Outcomes After a Total Pancreatectomy and Islet Autotransplantation Lessons Learned From Over 500 Cases. Ann. Surg. 2015, 262 (4), 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ricordi C; Lacy PE; Finke EH; Olack BJ; Scharp DW Automated Method for Isolation of Human Pancreatic Islets. Diabetes 1988, 37 (4), 413–420. [DOI] [PubMed] [Google Scholar]
- (11).Berger ST; Ahmed S; Muntel J; Cuevas Polo N; Bachur R; Kentsis A; Steen J; Steen H MStern Blotting–High Throughput Polyvinylidene Fluoride (PVDF) Membrane-Based Proteomic Sample Preparation for 96-Well Plates. Mol. Cell. Proteomics 2015, 14 (10), 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bennike TB; Steen H High-Throughput Parallel Proteomic Sample Preparation Using 96-Well Polyvinylidene Fluoride (PVDF) Membranes and C18 Purification Plates In Serum/Plasma Proteomics; Greening DW, Simpson RJ, Eds.; Springer New York: New York, NY, 2017; Vol. 1619, pp 395–402. [DOI] [PubMed] [Google Scholar]
- (13).Urbaniak GC; Plous S Research Randomizer (Version 4.0) [Computer software] http://www.randomizer.org/ (accessed Jun 22, 2013). [Google Scholar]
- (14).Bennike TB; Ellingsen T; Glerup H; Bonderup OK; Carlsen TG; Meyer MK; Bøgsted M; Christiansen G; Birkelund S; Andersen V; et al. Proteome Analysis of Rheumatoid Arthritis Gut Mucosa. J. Proteome Res. 2017, 16 (1), 346–354. [DOI] [PubMed] [Google Scholar]
- (15).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics MCP 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann M Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10 (4), 1794–1805. [DOI] [PubMed] [Google Scholar]
- (17).Cox J; Mann M MaxQuant Enables High Peptide Identification Rates, Individualized p.p.b.-Range Mass Accuracies and Proteome-Wide Protein Quantification. Nat. Biotechnol. 2008, 26 (12), 1367–1372. [DOI] [PubMed] [Google Scholar]
- (18).Bennike TB; Kastaniegaard K; Padurariu S; Gaihede M; Birkelund S; Andersen V; Stensballe A ☆Comparing the Proteome of Snap Frozen, RNAlater Preserved, and Formalin-Fixed Paraffin-Embedded Human Tissue Samples. EuPA Open Proteomics 2016, 10, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Reiter L; Rinner O; Picotti P; Hüttenhain R; Beck M; Brusniak M-Y; Hengartner MO; Aebersold R MProphet: Automated Data Processing and Statistical Validation for Large-Scale SRM Experiments. Nat. Methods 2011, 8 (5), 430–435. [DOI] [PubMed] [Google Scholar]
- (20).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; et al. The ProteomeXchange Consortium in 2017: Supporting the Cultural Change in Proteomics Public Data Deposition. Nucleic Acids Res. 2017, 45 (Database issue), D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vizcaíno JA; Csordas A; del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; et al. 2016 Update of the PRIDE Database and Its Related Tools. Nucleic Acids Res. 2016, 44 (Database issue), D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13 (9), 731–740. [DOI] [PubMed] [Google Scholar]
- (23).Cox J; Mann M 1D and 2D Annotation Enrichment: A Statistical Method Integrating Quantitative Proteomics with Complementary High-Throughput Data. BMC Bioinformatics 2012, 13 Suppl 16, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Muntel J; Xuan Y; Berger ST; Reiter L; Bachur R; Kentsis A; Steen H Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. J. Proteome Res. 2015, 14 (11), 4752–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kulak NA; Pichler G; Paron I; Nagaraj N; Mann M Minimal, Encapsulated Proteomic-Sample Processing Applied to Copy-Number Estimation in Eukaryotic Cells. Nat. Methods 2014, 11 (3), 319–324. [DOI] [PubMed] [Google Scholar]
- (26).Cao Z; Tang H-Y; Wang H; Liu Q; Speicher DW Systematic Comparison of Fractionation Methods for In-Depth Analysis of Plasma Proteomes. J. Proteome Res. 2012, 11 (6), 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gao J Bioanalytical Method Validation for Studies on Pharmacokinetics, Bioavailability and Bioequivalence: Highlights of the FDA’s Guidance. Asian J Drug Metab Pharmacokinet 2004, 4 (1), 5–13. [Google Scholar]
- (28).Liu Y; Buil A; Collins BC; Gillet LC; Blum LC; Cheng L-Y; Vitek O; Mouritsen J; Lachance G; Spector TD; et al. Quantitative Variability of 342 Plasma Proteins in a Human Twin Population. Mol. Syst. Biol. 2015, 11 (2), 786–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Schwanhäusser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473 (7347), 337–342. [DOI] [PubMed] [Google Scholar]
- (30).Bjelosevic S; Pascovici D; Ping H; Karlaftis V; Zaw T; Song X; Molloy MP; Monagle P; Ignjatovic V Quantitative Age-Specific Variability of Plasma Proteins in Healthy Neonates, Children and Adults. Mol. Cell. Proteomics 2017, 16 (5), 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nanjappa V; Thomas JK; Marimuthu A; Muthusamy B; Radhakrishnan A; Sharma R; Ahmad Khan A; Balakrishnan L; Sahasrabuddhe NA; Kumar S; et al. Plasma Proteome Database as a Resource for Proteomics Research: 2014 Update. Nucleic Acids Res. 2014, 42 (D1), D959–D965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Anderson NL The Clinical Plasma Proteome: A Survey of Clinical Assays for Proteins in Plasma and Serum. Clin. Chem. 2010, 56 (2), 177–185. [DOI] [PubMed] [Google Scholar]
- (33).Everhart JE; Ruhl CE Burden of Digestive Diseases in the United States Part III: Liver, Biliary Tract, and Pancreas. Gastroenterology 2009, 136 (4), 1134–1144. [DOI] [PubMed] [Google Scholar]
- (34).Kesseli SJ; Smith KA; Gardner TB Total Pancreatectomy with Islet Autologous Transplantation: The Cure for Chronic Pancreatitis? Clin. Transl. Gastroenterol 2015, 6 (1), e73–e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wahoff DC; Papalois BE; Najarian JS; Kendall DM; Farney AC; Leone JP; Jessurun J; Dunn DL; Robertson RP; Sutherland DE Autologous Islet Transplantation to Prevent Diabetes after Pancreatic Resection. Ann. Surg. 1995, 222 (4), 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Clayton HA; Davies JE; Pollard CA; White SA; Musto PP; Dennison AR Pancreatectomy with Islet Autotransplantation for the Treatment of Severe Chronic Pancreatitis: The First 40 Patients at the Leicester General Hospital: Transplantation 2003, 76 (1), 92–98. [DOI] [PubMed] [Google Scholar]
- (37).Sutherland DE; Matas AJ; Najarian JS Pancreatic Islet Cell Transplantation. Surg. Clin. North Am. 1978, 58 (2), 365–382. [DOI] [PubMed] [Google Scholar]
- (38).Hartmann D; Felix K; Ehmann M; Schnölzer M; Fiedler S; Bogumil R; Büchler M; Friess H Protein Expression Profiling Reveals Distinctive Changes in Serum Proteins Associated with Chronic Pancreatitis. Pancreas 2007, 35 (4), 334–342. [DOI] [PubMed] [Google Scholar]
- (39).Chen J; Anderson M; Misek DE; Simeone DM; Lubman DM Characterization of Apolipoprotein and Apolipoprotein Precursors in Pancreatic Cancer Serum Samples via Two-Dimensional Liquid Chromatography and Mass Spectrometry. J. Chromatogr. A 2007, 1162 (2), 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Singh SA; Andraski AB; Pieper B; Goh W; Mendivil CO; Sacks FM; Aikawa M Multiple Apolipoprotein Kinetics Measured in Human HDL by High-Resolution/Accurate Mass Parallel Reaction Monitoring. J. Lipid Res. 2016, 57 (4), 714–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lei J-J; Zhou L; Liu Q; Xiong C; Xu C-F Can Mean Platelet Volume Play a Role in Evaluating the Severity of Acute Pancreatitis? World J. Gastroenterol. 2017, 23 (13), 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).SATO T; OHNO K; TAMAMOTO T; OISHI M; KANEMOTO H; FUKUSHIMA K; GOTO-KOSHINO Y; TAKAHASHI M; TSUJIMOTO H Assesment of Severity and Changes in C-Reactive Protein Concentration and Various Biomarkers in Dogs with Pancreatitis. J. Vet. Med. Sci. 2017, 79 (1), 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Sawai Y; Kodama Y; Shimizu T; Ota Y; Maruno T; Eso Y; Kurita A; Shiokawa M; Tsuji Y; Uza N; et al. Activation-Induced Cytidine Deaminase Contributes to Pancreatic Tumorigenesis by Inducing Tumor-Related Gene Mutations. Cancer Res. 2015, 75 (16), 3292–3301. [DOI] [PubMed] [Google Scholar]
- (44).Felix K; Hauck O; Fritz S; Hinz U; Schnölzer M; Kempf T; Warnken U; Michel A; Pawlita M; Werner J Serum Protein Signatures Differentiating Autoimmune Pancreatitis versus Pancreatic Cancer. PLoS ONE 2013, 8 (12), e82755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sandström A; Andersson R; Segersvärd R; Löhr M; Borrebaeck CAK; Wingren C Serum Proteome Profiling of Pancreatitis Using Recombinant Antibody Microarrays Reveals Disease-Associated Biomarker Signatures. PROTEOMICS - Clin. Appl. 2012, 6 (9–10), 486–496. [DOI] [PubMed] [Google Scholar]
- (46).Cullen JJ; Mitros FA; Oberley LW Expression of Antioxidant Enzymes in Diseases of the Human Pancreas: Another Link between Chronic Pancreatitis and Pancreatic Cancer. Pancreas 2003, 26 (1), 23–27. [DOI] [PubMed] [Google Scholar]
- (47).Jenkinson C; Elliott VL; Evans A; Oldfield L; Jenkins RE; O’Brien DP; Apostolidou S; Gentry-Maharaj A; Fourkala E-O; Jacobs IJ; et al. Decreased Serum Thrombospondin-1 Levels in Pancreatic Cancer Patients up to 24 Months Prior to Clinical Diagnosis: Association with Diabetes Mellitus. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22 (7), 1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lindkvist B; Domínguez-Muñoz JE; Luaces-Regueira M; Castiñeiras-Alvariño M; Nieto-Garcia L; Iglesias-Garcia J Serum Nutritional Markers for Prediction of Pancreatic Exocrine Insufficiency in Chronic Pancreatitis. Pancreatology 2012, 12 (4), 305–310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table S1 (.pdf) Cover page.
Supplementary table S2 (.xls) Participant information.
Supplementary table S3 (.xls) DIA plasma method variable windows.
Supplementary table S4 (.xls) Cumulated list of plasma proteins in the 62 participants.
Supplementary table S5 (.xls) List of plasma proteins shared between all 62 participants.
Supplementary table S6 (.xls) List of identified FDA-approved biomarkers.
Supplementary Figure S1 (.png) Absolute concentration of the quantifiable plasma proteins with the workflow reported in g/L.
Supplementary Figure S2 (.png) Volcanoplot of TPIAT changes to the serum proteome with Igs.