

ABSTRACT
Histone methyltransferase KMT2D has diverse functions and distinct mechanisms in different cancers. Although we have previously found KMT2D serves as an oncogene that promotes tumor growth and metastasis in prostate cancer (PCa), the functions and mechanisms of KMT2D are complicated and most remain undefined. Here, the function of KMT2D regarding DNA damage in PCa and the underlying mechanisms of KMT2D in epigenetic regulation were explored in a series of studies. Knockdown of KMT2D sensitized cells to DNA damage through the disturbance of antioxidative gene expression and increased levels of intracellular reactive oxygen species, which led to cell apoptosis and senescence. The loss of KMT2D reduced the abundance of enhancer activity markers H3K4me1 and H3K27ac, which blocked the DNA binding of FOXO3, a critical mediator of the cellular response to oxidative stress, and suppressed antioxidative gene transcription. Moreover, KMT2D deletion in PCa cells also increased their sensitivity to genotoxic anticancer drugs and a PARP inhibitor, which suggested that lower levels of KMT2D may mediate the response of PCa to particular treatments. These findings further highlighted the important role of KMT2D in PCa progression and suggested that targeting KMT2D might be therapeutically beneficial for advanced PCa treatment.
KEYWORDS: Histone methyltransferase, histone lysine methyltransferase 2D, DNA damage, forkhead box O3, reactive oxygen species, prostate cancer
Introduction
Prostate cancer (PCa) is one of most common malignancies and the predominant cause of cancer-related death in men worldwide [1]. Though radical prostatectomy and androgen deprivation therapy provide palliative benefits, recalcitrant disease recurs and progresses to a lethal stage [2]. None of the current treatments are curative and the median survival time is 2–3 y [3]. Recently, with the technology of next-generation sequencing, multiple genetic alterations such as gene mutations, copy number variants, and gene rearrangements have been revealed in localized and metastatic PCa [4–6]. However, the biological consequences of these genetic alterations have not been fully elucidated in either cell culture or animal model systems [7].
Emerging evidence suggests that the function and expression of certain epigenetic modifiers, such as TET2 [8], BAZ2A [9], and EZH2 [10], are closely related to PCa tumorigenesis, metastasis, and prognosis. Histone lysine methyltransferase 2D (KMT2D/MLL2) is an epigenetic modifier that directs the mono-methylation of H3K4 residues (histone H3 lysine 4-monomethylation, H3K4me1). We previously found that KMT2D is both highly mutated and overexpressed in Chinese patients with PCa and correlates with poor disease prognosis [11,12]. KMT2D epigenetically upregulates the expression of leukemia inhibitory factor receptor (LIFR) and Kruppel-like factor-4 (KLF4), which activates PI3K/Akt and EMT oncogenic pathways, thereby promoting PCa outgrowth and metastasis [11]. Moreover, accumulating evidence reveals that KMT2D correlates with tumorigenesis and anti-tumor drug resistance. The genetic ablation of KMT2D in B cells perturbs tumor-expression of the tumor-suppressor genes that control B cell-activating pathways and contributes to lymphoma development [13]. In breast cancer, PI3Kα inhibition enhances KMT2D activity and triggers endoplasmic reticulum (ER)-dependent transcription that limits therapeutic efficacy [14]. Given the divergent functions of KMT2D in other cancers, the pathophysiological function of KMT2D in PCa is likely to be complicated and needs to be comprehensively investigated.
DNA damage and subsequent genomic instability give rise to gene mutations and chromosomal damage, which are facilitating characteristics that help generate the hallmarks of cancer [15,16]. On the other hand, extensive or unpaired DNA damage is toxic to cancer cells. DNA damage may cause cell cycle arrest and cell elimination, inducing apoptosis or necrotic cell death [17]. Thus, DNA damage also can function as a tumor suppressor [18]. Endogenous agents can induce DNA damage. Among them, reactive oxygen species (ROS) is responsible for a major portion of the damage. Altered metabolism, deficiency in antioxidative ability, hypoxia, and mitochondria dysfunction all increase intracellular ROS levels and further promote DNA damage, leading to premature cellular senescence and programmed cell death [19]. Santos et al. reported that KMT2D knockout increases ROS-mediated DNA damage in hematopoietic stem cells (HSCs) via transcriptional regulation of antioxidative response genes, which enhances myelopoiesis and myeloid differentiation of leukemic blasts [20]. Therefore, KMT2D might serve as a protector against the ROS-mediated DNA damage for cancer cells. However, whether KMT2D correlates with DNA damage and antioxidative response in PCa remains unknown.
In the current study, we showed that the loss of KMT2D promoted ROS-mediated DNA damage in PCa and led to both cell apoptosis and senescence, which blocks cancer development and progression. We found that the expression of enhanced activity markers H3K4me1 and H3K27ac were decreased after KMT2D was silenced. The low expression of enhancer activity markers disturbed the transcription regulation of forkhead box O3 (FOXO3) and reduced cell antioxidative gene expression. Moreover, the increased DNA damage and greater deficiency of antioxidants resulted in the KMT2D-deficient cells being more sensitive to genotoxic anticancer drugs. Therefore, KMT2D status might be useful to stratify human PCa for effective treatment.
Results
KMT2D depletion led to DNA damage in PCa
To explore whether KMT2D depletion was associated with DNA damage in PCa, we established a PC-3 cell line with KMT2D stably silenced by transfection with lentiviruses that expressed two KMT2D-specific short hairpin RNAs (shRNAs; #1 and #2). In addition, another PCa cell line, DU145, was also chosen due to the relatively high abundance of KMT2D mRNA abundance [11] and was transfected with KMT2D small interfering RNA (siRNA) ‘smartpool,’ which is a mixture of four siRNAs. After transfection, both the transcriptional and translational levels of KMT2D in PC-3 and DU145 cells were significantly reduced compared with that of the control group (Supplementary Figure S1(a,b)). Also, the H3K4me1 protein level was consistently decreased (Supplementary Figure S1(c)).
KMT2D-silenced cells and control cells were used in a variety of assays to explore the correlation between KMT2D and DNA damage. First, single cell gel electrophoresis was performed. The results revealed that the DNA damage ratio, reflected by the tail moment, was significantly increased in KMT2D-knockdown PC-3 and DU145 cells (range, 2.43–4.55-fold, P < 0.001; Figure 1(a)). Second, the fluorescence of the DNA damage sensor γH2AX was detected significantly increased by confocal microscopy after KMT2D knockdown (Figure 1(b)). Third, through flow cytometry, the DNA damaged cells were quantified (Figure 1(c)). As a result of KMT2D depletion, the percentage of γH2A.X cells was significantly elevated in PC-3 and DU145 cells (range, 1.44–2.03-fold, P < 0.001; Figure 1(c)).
Figure 1.
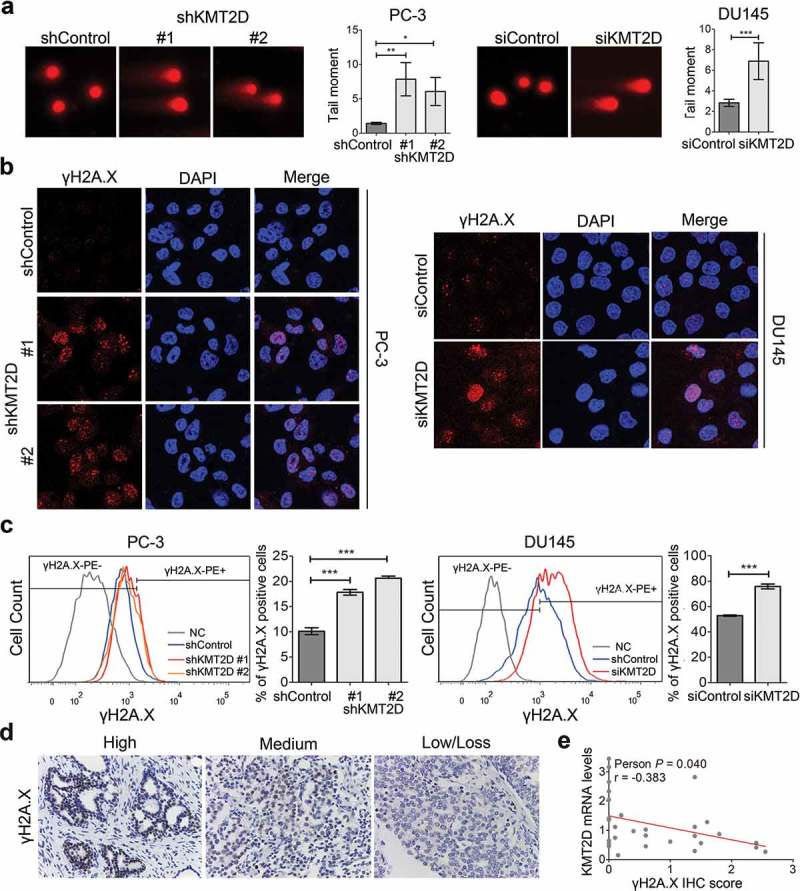
Knockdown of KMT2D led to DNA damage. (a) Single cell alkaline gel electrophoretic analysis of PC-3 and DU145 cells. The tail moment was quantified using CometScore software. (b) Immunofluorescence staining analysis of γH2AX (red) in PC-3 and DU145 cells. (c) γH2A.X-positive cells were evaluated by flow cytometry using Alexa Fluor 647 anti-γH2A.X in PC-3 and DU145 cells. * P < 0.05, ** P < 0.01, *** P < 0.001, compared with that of control cells. (d) Representative images of immunohistochemistry staining of PCa specimens (n = 29). (e) KMT2D mRNA levels negatively correlated with γH2A.X IHC scores in PCa tissues. All values are means ± SD. Data are representative of three independent experiments.
To further validate the correlation between KMT2D and DNA damage, we analyzed specimens from 29 patients with PCa. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) was used to evaluate the expression of KMT2D and immunohistochemical staining of γH2A.X was performed to mark DNA damage. Representative IHC images of the different staining of γH2A.X are shown in Figure 1(d). As expected, our analysis showed that KMT2D expression was inversely correlated with γH2A.X (Person r = −0.383, P = 0.040; Figure 1(e), Supplementary Table S1). Thus, the findings provided compelling evidence that KMT2D loss results in DNA damage in PCa.
Increased intracellular ROS level was associated with DNA damage
Elevated intracellular ROS level is a major cause of DNA damage. Therefore, we measured the level of ROS in PCa cells. CellROX was used as a probe and the ROS levels in PC-3 and DU145 cells were analyzed by flow cytometry. We observed that the intracellular ROS levels were significantly increased after KMT2D knockdown compared with that in the control cells (range 1.45–2.61-fold, P < 0.001; Figure 2(a)). The level of 8-hydroxy-2ʹ-deoxyguanosine (8-OHdG), an indicator of common oxidized base damage, was significantly higher in KMT2D-silenced PC-3 and DU145 cells based on analysis by enzyme-linked immunosorbent assay (ELISA; range 1.29–3.87-fold, P < 0.05; Figure 2(b)).
Figure 2.
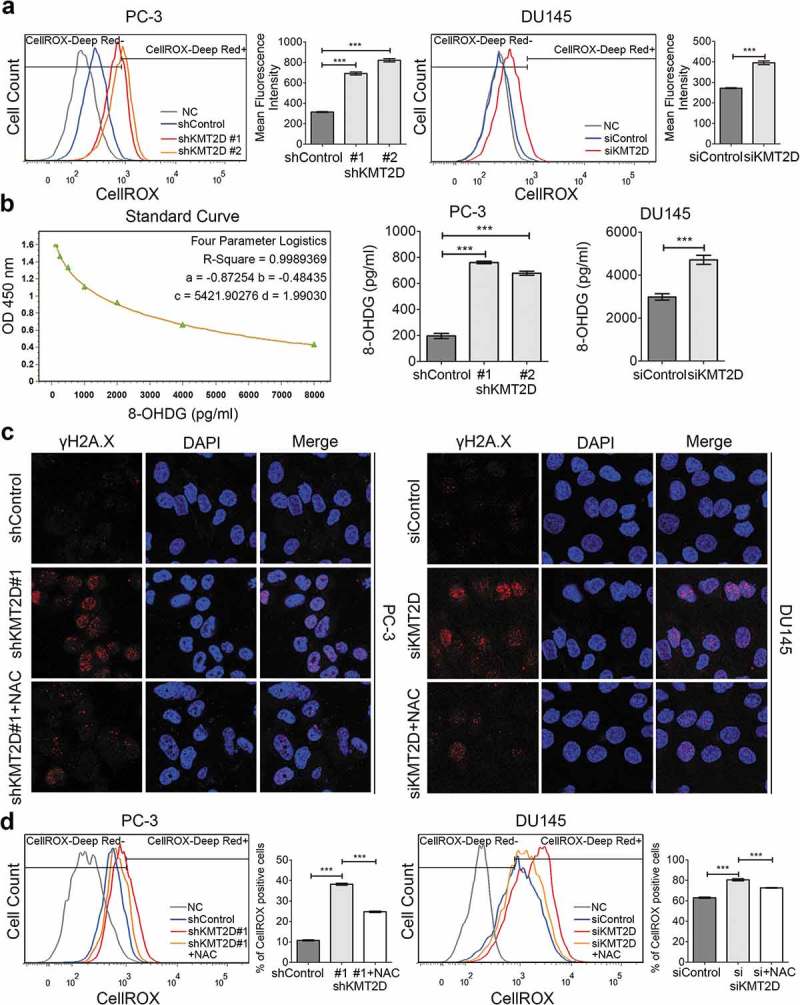
Increased intracellular ROS level contributed to DNA damage in the absence of KMT2D. (a) ROS levels in PC-3 and DU145 cells were detected by flow cytometry using CellROX. *** P < 0.001, compared with that of control cells. (b) The 8-OHdG concentrations of DNA were evaluated by ELISA in PC-3 and DU145 cells. * P < 0.05, *** P < 0.001, compared with that of control cells. (c) Representative images of γH2AX foci (red) of KMT2D silenced PC-3 and DU145 cells treated with NAC. (d) The percentage of γH2AX positive cells were quantified by flow cytometry. All values are means ± SD. Data are representative of three independent experiments.
To confirm whether the increased ROS levels contributed to DNA damage in KMT2D-deficient cells, the antioxidant N-acetylcysteine (NAC) was used. Immunofluorescence staining showed the percentage of γH2A.X-positive cells decreased due to NAC treatment in KMT2D deficiency cells (Figure 2(c)). Fluorescence-activated cell sorting (FACS) analysis showed similar results in that NAC treatment significantly reversed the DNA damage induced by the loss of KMT2D expression in PC-3 and DU145 cells (P < 0.001; Figure 2(d)). These findings suggested that the increase in ROS is responsible for the elevation of DNA damage in PCa with low KMT2D expression.
ROS-mediated DNA damage prompted PCa cell apoptosis and senescence
ROS-mediated DNA damage can trigger cell-cycle arrest, premature cellular senescence, or apoptosis and thereby suppress tumor progression. We reasoned that the ROS-mediated DNA damage caused by KMT2D loss might also result in cytotoxicity for PCa cells. Fluorescence antibodies specific for γH2A.X and cleaved poly ADP ribose polymerase (PARP) were used to immunostain DNA damaged and apoptotic cells, respectively, and were then detected by flow cytometry. The results showed that the percentage of apoptotic cells significantly increased in KMT2D-silenced cells (range 9.74–14.66-fold, P < 0.001; Figure 3(a)). Meanwhile, almost all the apoptotic cells were also γH2A.X positive (P < 0.001; Figure 3(b)), which suggested that the ROS-mediated DNA damage was responsible for the increased apoptosis after KMT2D knockdown. The flow cytometry results were further confirmed by western blot (Supplementary Figure S2).
Figure 3.
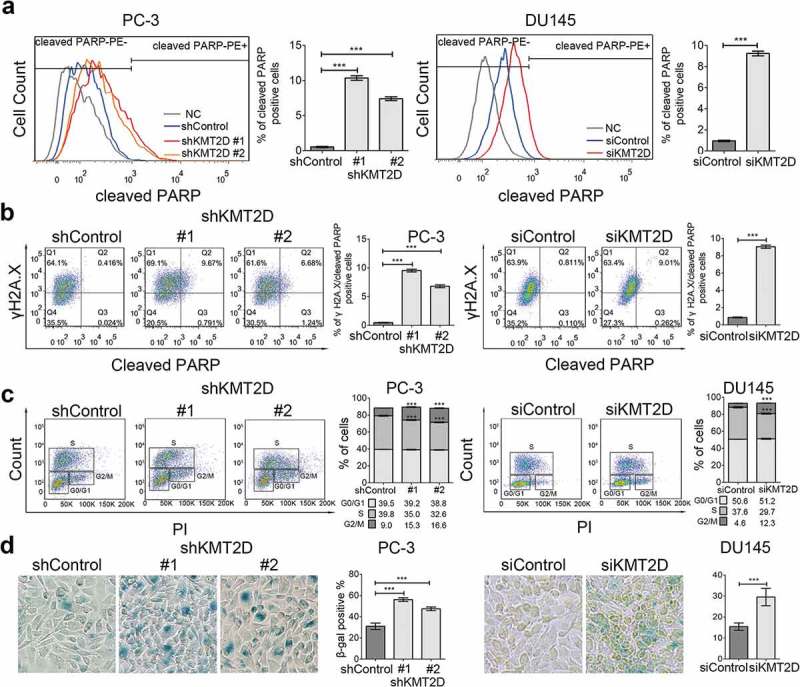
ROS-mediated DNA damage induced PCa cell apoptosis and senescence. (a) Cell apoptosis was evaluated with flow cytometry using PE anti-cleaved PARP in PC-3 and DU145 cells. *** P < 0.001, compared with that of control cells. (b) Representative FACS analysis of cleaved PARP and γH2A.X double positive cells in PC-3 and DU145 cells. *** P < 0.001, compared with that of control cells. (c) Cell cycle analysis of the PC-3 and DU145 cells using BrdU incorporation. Quadrangular gates represent cell cycle stages of G1/G0, S, and G2/M. *** P < 0.001, compared with that of control cells. (d) Representative images of SA-β-Galactosidase staining (blue-green) of PC-3 and DU145 cells. All values are means ± SD. Data are representative of three independent experiments.
In addition, we analyzed cell cycle effects using flow cytometry and BrdU incorporation. We observed that KMT2D-depletion increased the number of PC-3 and DU145 cells in the G2/M phase and decreased the number of cells in the S phase (P < 0.001; Figure 3(c)). Furthermore, an SA-β-Gal assay showed that KMT2D knockdown increased the percentage of senescent cells (β-Gal-positive cells) in PC-3 and DU145 cells (range 1.53–1.91, P < 0.001; Figure 3(d)). Hence, ROS-mediated DNA damage also triggered DNA damage response signaling to block the cell cycle and prompted cell senescence in PCa.
KMT2D knockdown attenuated antioxidative gene expression and FOXO3 DNA binding
To understand how KMT2D loss induced ROS-mediated DNA damage, we performed gene expression profiling on stable KMT2D knockdown and control PC-3 cells using an RT-PCR array. The results showed that the expression of mostly oxidative stress-specific genes was decreased after KMT2D depletion, including the genes encoding glutathione peroxidases, peroxiredoxins, and superoxide dismutases (Figure 4(a)). These gene-expression alterations were confirmed by qRT-PCR analysis of PC-3 and DU145 cells. Four representative antioxidative genes, GPX1, PRDX2, SOD2, and CAT, were significantly suppressed after KMT2D expression was silenced (P < 0.01; Figure 4(b)). These findings suggested that KMT2D played an important role in sustaining transcriptional programs programs associated with the antioxidative response in PCa.
Figure 4.

KMT2D was required for antioxidative gene expression and FOXO3 DNA binding. (a) A heatmap showing the fold changes of 84 oxidative stress genes compared with that of the control group. (b) Analysis of PC-3 and DU145 cells by qRT-PCR showed the decreased expression of four representative antioxidative genes, GPX1, PRDX2, SOD2, and CAT. ** P < 0.01, *** P < 0.001, compared with that of the control cells. (c) Immunofluorescence staining analysis of PC-3 and DU145 cells using anti-FOXO3 (red); nuclear staining with DAPI. (d) western blot analysis of FOXO3 in PC-3 and DU145 cells. (e) ChIP-PCR for FOXO3 to representative antioxidative genes in PC-3 cells transfected with shKMT2D. ** P < 0.01, *** P < 0.001, compared with that in the control cells. (f) ROS levels in PC-3 cells transfected with shKMT2D and FOXO3 vectors were detected by flow cytometry using CellROX. *** P < 0.001, compared with that in the control cells. All values are means ± SD. Data are representative of three independent experiments.
We next aimed to identify how KMT2D orchestrated the expression of the antioxidative genes. It has been previously shown that KMT2D plays a critical role in the function of FOXO3 in breast cancer and glioblastoma multiforme [21,22]. Additionally, FOXO3 was recently found to be activated in the presence of oxidative stress, which induces the expression of antioxidant defense enzymes to reduce ROS levels and oxidative stress [23]. Thus, we hypothesized that FOXO3 participated in the defective oxidative stress response after KMT2D knockdown. To test this hypothesis, we first evaluated the nuclear levels of FOXO3 using immunofluorescence staining. Our results indicated that the abundance of FOXO3 was suppressed in KMT2D-silenced PC-3 and DU145 cells compared to that in control cells (Figure 4(c)). This result was confirmed by western blot (Figure 4(d)). More importantly, through ChIP-PCR, KMT2D suppression also led to a considerable decrease in the amount of FOXO3 occupying the forkhead motif near the representative antioxidative genes (Figure 4(e), Supplementary Figure S3). In addition, we overexpressed FOXO3 in stable KMT2D silenced PC-3 cells (Figure S1(d)). Flow cytometric analysis revealed that FOXO3 overexpression only partially rescued the upregulated intracellular levels of ROS caused by KMT2D loss (P < 0.001; Figure 4(f)). These results suggested that KMT2D is indispensable for the DNA binding and function of FOXO3 in PCa.
KMT2D deficiency decreased the pre-existing levels of enhancer activity markers
Eijkelenboom et al. have found that unlike other transcription factors, FOXO3 transcription regulation primarily proceeds through enhancer regions [24]. FOXO3 DNA binding prior to FOXO3 activation is largely determined by the pre-existing levels of enhancer activity markers [25]. Thus, genetic alterations that affect the levels of enhancer activity without affecting FOXO3 itself could influence FOXO3 DNA binding and affect the FOXO3-mediated regulation of target genes [25]. Moreover, as a crucial epigenetic modifier, recent studies have proven that upon impairment of KMT2D function, changes in the levels of H3K4me1 and H3K27ac are observed in enhancer regions, indicating a direct role for KMT2D in enhancer regulated processes [26]. Based on these findings, we proposed that KMT2D deficiency suppressed FOXO3 DNA binding through blocking the formation of the enhancer activity marker. To test this hypothesis, we evaluated the changes in enhancer activity markers after KMT2D knockdown. Since we previously confirmed that the H3K4me1 abundance is suppressed in KMT2D silenced cells [11], in the current study we only evaluated the enhancer activity marker H3K27ac. Consistent with the findings for H3K4me1, both immunofluorescent staining (Figure 5(a)) and western blot analysis (Figure 5(b)) showed that H3K27ac was significantly decreased in the nucleus after KMT2D knockdown (P < 0.001). Subsequently, we investigated patterns of enhancer activity markers on antioxidative genes using Cistrome DB, a comprehensive annotated resource of publicly available ChIP-seq and chromatin accessibility data [27]. As shown in Figure 5(c), the overlapping ChIP-seq peaks for both H3K4me1 and H3K27ac were found in the upstream region of the representative antioxidative genes GPX1, PRDX2, SOD2, and CAT. This raised the possibility that KMT2D might sustain the enhancer activity of these genes. Using ChIP-PCR, we found the KMT2D knockdown significantly dampened the abundance of H3K4me1 and H3K27ac in these peaks (P < 0.05; Figure 5(d)). In addition, from the Cistrome DB, we also obtained the human KMT2D ChIP-seq data from two different studies (GSE67314 and GSE71854). The KMT2D peaks were found in the upstream region of the four representative antioxidative genes GPX1, PRDX2, SOD2, and CAT (Supplementary Figures S4–S7) and closed to the H3K4me1 and H3K27ac peaks in PC-3 cells. The consistent binding sites of H3K4me1, H3K27ac and KMT2D further supported that KMT2D might have a direct role in antioxidative gene regulation. Together, our findings suggested that KMT2D is a critical epigenetic mediator of enhancer activity that directs FOXO3 binding and the antioxidative transcriptional response.
Figure 5.

Loss of KMT2D attenuated the enhancer activity markers in PCa. (a) Immunofluorescence staining analysis of H3K27ac (red) in PC-3 and DU145 cells. (b) Western blot analysis of H3K27ac in PC-3 and DU145 cells. (c) Tracks of H3K4me1 and H3K27ac ChIP-seq occupancy in PC-3 cells based on data from the Cistrome DB database. (d) ChIP-PCR for H3K4me1 and H3K27ac to representative antioxidative genes in PC-3 cells transfected with shKMT2D. * P < 0.05, ** P < 0.01, *** P < 0.001, compared with that in the control cells.
KMT2D depletion increased sensitivity to genotoxic anticancer drugs and PARP inhibitors
The dependence of PCa cells on KMT2D for survival and ROS-mediated DNA damage makes it a prime therapeutic target. We expected that low expression of KMT2D could increase the sensitivity of PCa cells to genotoxic anticancer drugs and provide potentially favorable prognosis in patients with low KMT2D expression. To test this hypothesis, we treated control and KMT2D-silenced cells with the common genotoxic anticancer drugs doxorubicin and carboplatin. We observed an increased sensitivity to the two drugs in KMT2D knockdown cells compared with that in control cells (Figure 6(a,b)). In addition, we determined whether KMT2D loss would result in a higher sensitivity to PARP inhibitors. PARP inhibitors are usually used in patients with defective homologous recombination (HR) such as BRCA mutations [28] and have been approved by the US Food and Drug Administration (FDA) as a form of monotherapy treatment for metastatic castration-resistant prostate cancer (mCRPC). Interestingly, KMT2D-deficient cells were also more sensitive to the PARP inhibitor olaparib compared with that of control cells (Figure 6(c)). Taken together, our study indicated that KMT2D loss sensitizes PCa cells to both genotoxic anticancer drugs and PARP inhibitors.
Figure 6.
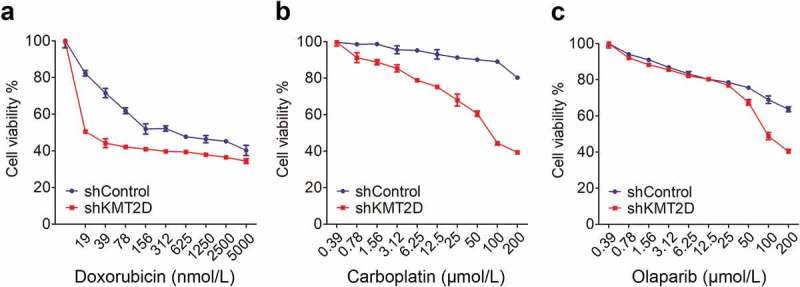
Depletion of KMT2D increased the sensitivity of PCa cells to genotoxic anticancer drugs and a PARP inhibitor. Cell viability was examined using MTT assays after 72 h of drug treatment. Data shown are mean OD values from triplicate wells normalized to that of the untreated wells.
Discussion
Large-scale human PCa gene sequencing analysis revealed that PCa is characterized by the presence of multiple genetic alterations. However, most of these abnormal lesions and the role they play in prostate cancer have not been functionally defined. We previously found KMT2D was recurrent mutation and high transcription, and first reported this histone methyltransferase serves as an oncogene in PCa [11,12]. In the current study, we investigated this gene and identified a new function of KMT2D in PCa. By generating KMT2D-depleted PCa cells, we demonstrated that the loss of KMT2D expression could lead to ROS-mediated DNA damage, which is known to prompt cell apoptosis and senescence through decreasing the pre-existing levels of enhancer activity markers and suppressing FOXO3 DNA binding, and thereby impaired the antioxidative response in PCa cells. Furthermore, we also reported that KMT2D-deficient PCa cells were sensitive to genotoxic anticancer drugs and a PARP inhibitor, which suggested that KMT2D status might be a potential predictor of treatment response to DNA-damaging agents.
As an epigenetic modifier, the function and underlying mechanisms of KMT2D in cancer are multifaceted and not fully understood. Data from the current study demonstrated that deletion or suppression of KMT2D induced a robust increase in DNA damage through the accumulation of ROS in PCa. In agreement with our findings, DNA damage and increased ROS levels are also observed in bone marrow HSCs expressing the MLL-AF9 oncogene and lacking KMT2D. In this situation, KMT2D is required for stem-cell activity and represents an aggressive form of acute myeloid leukaemia [20]. Kantidakis et al reported that KMT2D is critical for maintaining genomic stability. KMT2D-mutated cells display transcription stress that results in DNA damage and genomic instability, which are considered to be strong drivers in tumorigenesis [29]. In addition, KMT2D is a part of the KMT2 family and acts as catalytic subunits for mammalian complex proteins associated with Set1 (COMPASS)-like complexes by modifying the methylation of histone H3 lysine [30]. There are multiple reports showing that the depletion of the components of COMPASS-like complexes, such as WDR5, induce DNA damage. These findings suggest a connection between DNA damage resulting from a deficiency in KMT2D and its role in the COMPASS complex [31,32].
In the current study, we found that ROS-mediated DNA damage led to both cell apoptosis and senescence in PCa. The elevated levels of ROS increased the amount of DNA damage and genome instability, resulting in gene mutations and chromosomal damage, which are causal events in oncogenic transformation and tumor progression [18]. However, extensive oxidative DNA damage may abrogate the cellular capacity for repair and become lethal to the cell and thus block cancer progression [19]. Moreover, Lei K, et al. use the CRISPR-based KMT2D knockout cell models and find KMT2D is important for the propagation of PCa cells [33]. This result is consistent with our findings and further confirm the essential role of KMT2D in PCa progression.
There are multiple factors involved in the cell fate process between survival and death following DNA damage. Among them is the Akt kinase, which is an important pro-survival protein that suppresses apoptosis after DNA damage in a PI3K-dependent manner [34]. Numerous studies have confirmed that Akt suppression of apoptosis leads to increased cell survival following DNA damage [35]. We previously showed that KMT2D loss results in the inactivation of the PI3K/Akt signaling pathway and decreased anti-apoptotic protein expression [11], which might contribute to PCa cell apoptosis following ROS-mediated DNA damage. Moreover, our findings also suggested that ROS-mediated DNA damage in KMT2D-knockdown PCa cells may have triggered cell senescence in PCa, whereas cell proliferation was markedly suppressed by cell cycle arrest at the G2/M phase. Therefore, ROS-mediated DNA damage in PCa may be able to inhibit cancer development, which indicates that inhibition of KMT2D may be a viable strategy for cancer therapeutics.
The most interesting discovery from our current study is the development of a new model for the regulation of antioxidative gene expression by KMT2D and FOXO3. Various previous studies have revealed that KMT2D is a prominent H3K3 mono- methyltransferase of enhancer regions [36]. In addition, KMT2D is required for H3K27 acetyltransferase of CREB-binding protein (CBP) and/or p300 on enhancers, and for determining the formation of H3K27ac [37]. ChIP-seq analysis has shown that deletion of KMT2D prevents the accumulation of H3K4me1 and H3K27ac on enhancers, which results in severe defects in gene expression and cell differentiation [38]. Because H3K4me1 and H3K27ac are markers of enhancer activity, KMT2D is considered an important regulator of enhancer activity [30,39]. FOXO3 transcription factors are members of the forkhead box family of transcription factors consisting of FOXO1, FOXO3, FOXO4, and FOXO6. Previous findings in human cancers suggest that FOXO3 transcription factors are both sensors of oxidative stress signals and effectors of the antioxidative response [23]. Emerging evidence indicates that in breast cancer and glioblastoma multiforme, KMT2D is indispensable to the function of FOXO3. However, the connection between KMT2D and FOXO3 is unclear. In our current study, we found that KMT2D deficiency decreased the pre-existing levels of enhancer activity markers and suppressed FOXO3 DNA binding in PCa, suggesting epigenetic regulation contributes to the FOXO3 transcriptional response. Our results are consistent with the findings by Eijkelenboom et al. that the preexisting chromatin state can direct FOXO3 gene regulation and regulate cell-specific homeostasis [25], further supporting the conclusion that KMT2D may be a relevant determinant to modulate FOXO function and transcriptional output.
In addition, previous studies have reported that the FOXO family genes could be stimulated by FOXO3 in a positive feedback loop [40]. FOXO3 can bind to its own and activate transcription [41]. In PCa, KMT2D knockdown might also block this positive feedback and lead to FOXO3 reduction. This might explain why the level of FOXO3 was reduced upon the knockdown of KMT2D in our study. The similar regulation pattern of KMT2D was also identified in murine hepatic steatosis [42]. KMT2D directs overnutrition-induced murine steatosis trough ABL-PPARg2-KMT2D axis. In this pathway, the Pparg2 (encoding PPARg2) itself is a direct target gene of the ABL-PPARg2-KMT2D axis.
Further work is required to determine the extent of KMT2D loss in the enhancement of sensitivity to genotoxic drugs. Here, we noted that KMT2D silencing increased PCa sensitivity to the genotoxic drugs doxorubicin and carboplatin. Combination of carboplatin with docetaxel or cabazitaxel is one of the key chemotherapy strategies for mCRPC [43]. Moreover, KMT2D loss was found in our current study to be associated with increased sensitivity to the PARP inhibitor olaparib, implying that KMT2D might be involved in the DNA-damage-response process and the loss of KMT2D might attenuate HR [44]. How KMT2D associates with HR in PCa requires further study. Here, our study indicated that the low expression of KMT2D might attribute to an improved response to genotoxic anticancer drugs and PARP inhibitor treatments in patients with advanced PCa.
In summary, we reported that the loss of KMT2D led to ROS-mediated DNA damage and the suppression of tumor progression in PCa. We showed that KMT2D loss decreased the enhancer activity through the damping of histone modification of H3K4me1 and H3K27ac, which inhibited the binding of the antioxidative transcript factor FOXO3 and the transcription response of oxidative stress. Our data provide a possible approach to treat patients with PCa that harboring KMT2D deletion through using genotoxic anticancer drugs or PARP inhibitor to enhance their potential clinical benefit in this subclass of PCa.
Material and methods
Patients and tissue specimens
This study was approved by Medical Ethics Committee of Nanfang Hospital. Written consent was obtained from the patients in accordance with the Declaration of Helsinki. Twenty-nine patients with histologically confirmed prostate cancer at Nanfang Hospital between 2011 to 2015 were enrolled in the study. All samples have been confirmed as a diagnosis of PCa by two independent pathologists. Patients that received pre-operative hormone therapy, chemotherapy, or radiotherapy were excluded from the study.
Immunohistochemistry
Consecutive 4-μm sections were obtained from paraffin blocks of prostate tissue specimens from all 29 patients with PCa. The sections were deparaffinized and rehydrated using routine methods. The antibodies against γH2A.X (#ab26530, 1:1000; Abcam) were used as primary antibodies. Antigen retrieval and immunostaining were performed as described previously [11]. The staining intensity and staining percentage for each tissue were used for quantification as previous study [45]. Intensity was graded 1 to 3, with 1 being no or low staining, 2 medium staining, and 3 strong staining. The final IHC score was calculated by multiplying values of staining intensity and staining percentage and used for correlation analysis. All sections stained for the target proteins were evaluated by two independent pathologists.
Cell culture
Human PCa cell lines PC-3 and DU145 were purchased from Cellcook Biological Technology Co., Ltd. (Guangzhou, China). All cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA) and maintained at 37°C in a humidified atmosphere of 5% carbon dioxide. All cell lines were authenticated by short tandem repeat (STR) profiling.
Quantitative real-time PCR
Total RNA from the 29 PCa specimens was isolated using an RNeasy FFPE Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. RNA from the cell lines was isolated using TRIzol Reagent (Invitrogen, CA, USA). The extracted RNA was quantified using a Qubit fluorimeter (Thermo Fisher Scientific). Then, 1 μg RNA from each specimen was used to generate complementary DNA (cDNA) by reverse transcription (RT) using a QuantiTect Reverse Transcription Kit (Qiagen). The RT products were mixed with nuclease-free water, gene-specific primers (Supplementary Table S2), and reagents from a SYBR Premix Ex Taq II Kit (Takara, Shiga, Japan) according to the manufacturer’s instructions. Quantitative PCR was performed using a CFX384 Touch Real-Time PCR system (Bio-Rad, Hercules, CA, USA). Data was analyzed using a 2−ΔΔCt method [46].
Western blot analysis
Whole protein lysates of PC-3 and DU145 cells were prepared by RIPA buffer containing 1% phenylmethylsulfonyl fluoride (PMSF) and phosphatase inhibitor cocktail (Roche). As described previously [13], the blots were probed with primary antibodies specific for KMT2D (# sc-68671, 1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), FOXO3 (#ab53287, 1:1000; Abcam), H3K27ac (ab4729, 1:1000; Abcam), and GAPDH (#2118, 1:1000; Cell Signaling Technology). The protein bands were visualized using an ECL chemiluminescence detection reagent (Santa Clara, CA, United States) and quantified using Quantity One software (Bio-Rad, Hercules, CA, USA).
RNA interference and FOXO3 overexpression
Lentiviral shRNAs were used to knockdown KMT2D expression in PC-3 cells. Cells were incubated with lentivirus particles and 5 μg/ml polybrene for 24 h and then washed with medium. The transfected cells were selected with 0.5 μg/ml puromycin for 2 wk. The positive vector sequences and negative control vector sequences are shown in Supplementary Table S3. For siRNA transfection, a KMT2D siRNA ‘smartpool’ mixture of four siRNAs and a negative control siRNA were purchased from GE Dharmacon (Lafayette, USA). A final concentration of 25 pmol of each siRNA combined with 1 μl DharmaFECT 2 Transfection Reagent (GE Dharmacon, Lafayette, USA) was used to transfect the DU145 cells (1 × 105). For overexpression of FOXO3, human FOXO3 cDNA was PCR amplified and cloned into the pGCMV vector. Then PC-3 cells were transfected with the FOXO3 vector or negative control vector using Lipofectamine 2000 DNA Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Western blotting and qRT-PCR were used to verify the protein and mRNA expression, respectively, of KMT2D and FOXO3.
Single-cell gel electrophoresis
PC-3 and DU145 cells (1 × 103) were resuspended in 0.7% low-temperature agarose and applied onto glass slides pre-coated with 0.8% agarose. Additional low-temperature agarose was used to cover the layer of agarose containing the cells. The glass slides were then incubated with alkaline cell lysis buffer for 1 h at 4 °C and subjected to electrophoresis. The DNA was stained with ethidium bromide (1 μg/ml) and the fluorescent signals were detected using a fluorescence microscope (Leica, Jena, Germany).
Immunofluorescence staining
PC-3 and DU145 cells were fixed, permeabilized, and blocked. Cells were stained overnight with primary antibodies to γH2A.X (#ab26530, 1:500; Abcam), FOXO3a (#ab53287, 1:100; Abcam), or H3K27ac (#ab177178, 1:1000; Abcam), The cells were subsequently incubated with a secondary fluorescent antibody conjugated with phycoerythrin (PE; 1:200; Santa Cruz Biotechnology) and 4ʹ,6-diamidino-2-phenylindole (DAPI; 5 μg/ml). Fluorescent signals were detected using a confocal fluorescence microscope (Leica).
Flow cytometry analysis
To detect ROS, PC-3, and DU145, cells were incubated with CellROX Deep Red Reagent (Invitrogen) according to the to the manufacturer’s instructions for 30 min at 37℃ and analyzed by flow cytometry (BD Biosciences, San Diego, CA, USA).
To detect DNA damage and cell apoptosis, the PC-3 and DU145 cells were fixed and permeabilized using a BD Cytofix/Cytoperm Kit (BD Biosciences, Auckland, New Zealand) as described by the manufacturer. Alexa Fluor 647 anti-γH2A.X (#560447; BD Biosciences, Auckland, New Zealand) and PE anti-cleaved PARP (# 564130; BD Biosciences, Auckland, New Zealand) were added to the cells for 20 min at room temperature followed by flow cytometry.
To detect cell cycle distribution, PC-3 and DU145 cells were incubated with BrdU solution (BD Biosciences, Auckland, New Zealand) for 30 min at 37°C. The cells were then fixed and permeabilized using a BD Cytofix/Cytoperm Kit and treated with DNase to expose the incorporated BrdU. PerCP-Cy5.5 Anti-BrdU (# 560809; BD Biosciences, Auckland, New Zealand) was added to the cells for 20 min at room temperature. The cells were resuspended and stained with propidium iodide (PI; BD Biosciences, Auckland, New Zealand) and analyzed by flow cytometry.
8-OHdG ELISA test
DNA was isolated from PC-3 and DU145 cells using a DNeasy Blood & Tissue Kit (Qiagen). The 8-OHdG concentrations were measured using a DNA Damage Competitive ELISA Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The absorbance of each well was measured at 450 nm using a microplate reader (PerkinElmer, MA, USA). For each experiment, an 8-OHdG standard curve was constructed (62.5–8,000 pg/mL) and a curve-fitting software program (Curve Expert 1.4) was used to quantify the 8-OHdG concentrations.
Senescence-associated (SA)-β-galactosidase cytochemical staining
PC-3 and DU145 cells were plated into 12-well plates. After transfection with siRNAs for 72 h, the cells were fixed in 4% paraformaldehyde and analyzed using a SA-β-Gal kit (Cell Signaling Technology).
ChIP assays
ChIP assays using PC-3 cells were performed as described [11] using a Pierce Agarose ChIP Kit (Thermo Fisher Scientific). The antibodies used for the ChIP assays were specific for H3K4me1 (#ab8895; Abcam), H3K27ac (#ab177178; Abcam), and FOXO3a (#ab12162; Abcam). The ChIP-DNA analysis was then performed using qRT-PCR to evaluate the levels of H3K4me1, H3K27ac, and FOXO3a in the target loci. The primers used in this study are listed in Supplementary Table S2.
Cell chemotherapy response assay
PC-3 and DU145 cells (8 × 103) were seeded into 96-well plates. After incubation with common the genotoxic anticancer drugs doxorubicin and carboplatin or with PARP inhibitor olaparib for 48 h, cell viability was determined by quantifying the reduction of the tetrazolium dye MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to its insoluble formazan using standard MTT assays. The formazan was solubilized with 150 μl of dimethyl sulfoxide (DMSO) and quantified spectrophotometrically using a microplate reader (PerkinElmer). To generate log–dose-response curves, the percentage of cell viability was calculated using the maximal optical density (OD) as being 100% viability.
Statistical analysis
The experiment results were statistically analyzed using Student’s t-test (two groups) or one-way ANOVA followed by the LSD post-hoc test (for more than two groups) or Mann-Whitney U test (nonparametric analysis). *P < 0.05, **P < 0.01, and ***P < 0.001 considered significant. All the experiments had a miniumum of three trials performed. The values are presented as the means ± standard deviation (SD) and were calculated using GraphPad Prism software (GraphPad Software, CA, USA).
Funding Statement
This work was mainly supported by National Natural Science Foundation of China (21773199, 61427807 and 81872092); National Key R&D Program of China (2017YFC1103403); Sanming Project of Medicine in Shenzhen (SZSM201612019); The PhD Start-up Fund of Natural Science Foundation of Guangdong Province of China (2016A030310287).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data can be accessed here.
References
- [1].Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015. March;65(2):87–108. PubMed PMID: 25651787. [DOI] [PubMed] [Google Scholar]
- [2].Mahajan K, Malla P, Lawrence HR, et al. ACK1/TNK2 regulates histone H4 Tyr88-phosphorylation and AR gene expression in castration-resistant prostate cancer. Cancer Cell. 2017;31(6):790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].James ND, Spears MR, Clarke NW, et al. Survival with newly diagnosed metastatic prostate cancer in the “docetaxel era”: data from 917 patients in the control arm of the STAMPEDE trial (MRC PR08, CRUK/06/019). Eur Urol. 2015;67(6):1028–1038. [DOI] [PubMed] [Google Scholar]
- [4].Barbieri CE, Bangma CH, Bjartell A, et al. The mutational landscape of prostate cancer. Eur Urol. 2013;64(4):567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44(6):685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castrate resistant prostate cancer. Nature. 2012;487(7406):239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yang Y, Blee AM, Wang D, et al. Loss of FOXO1 cooperates with TMPRSS2-ERG overexpression to promote prostate tumorigenesis and cell invasion. Cancer Res. 2017;77(23):canres.0686.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Takayama K, Misawa A, Suzuki T, et al. TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression. Nat Commun. 2015. September 25;(6):8219.PubMed PMID: 26404510. [DOI] [PubMed] [Google Scholar]
- [9].Gu L, Frommel SC, Oakes CC, et al. BAZ2A (TIP5) is involved in epigenetic alterations in prostate cancer and its overexpression predicts disease recurrence. Nat Genet. 2015. January;47(1):22–30. PubMed PMID: 25485837. [DOI] [PubMed] [Google Scholar]
- [10].Taniguchi H, Jacinto FV, Villanueva A, et al. Silencing of Kruppel-like factor 2 by the histone methyltransferase EZH2 in human cancer. Oncogene. 2012. April 12;31(15):1988–1994. PubMed PMID: 21892211; PubMed Central PMCID: PMC3325596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lv S, Ji L, Chen B, et al. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene. 2018 Mar;37(10):1354–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lv S-D, Wang H-Y, Yu X-P, et al. Integrative molecular characterization of Chinese prostate cancer specimens. Asian J Androl. 2019 May 28. doi: 10.4103/aja.aja_36_19. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ortega-Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015. October;21(10):1199–1208. PubMed PMID: 26366710; PubMed Central PMCID: PMCPmc4676270. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Toska E, Osmanbeyoglu HU, Castel P, et al. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355(6331):1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Andor N, Maley CC, Ji HP.. Genomic instability in cancer: teetering on the limit of tolerance. Cancer Res. 2017;77(9):2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hanahan D, Weinberg RA.. Hallmarks of cancer: the next generation. cell. 2011. March 4;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- [17].Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. [DOI] [PubMed] [Google Scholar]
- [18].Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2015;16(1):20. [DOI] [PubMed] [Google Scholar]
- [19].Ding Y, Wang H, Niu J, et al. Induction of ROS overload by alantolactone prompts oxidative DNA damage and apoptosis in colorectal cancer cells. Int J Mol Sci. 2016;17(4):558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Santos MA, Faryabi RB, Ergen AV, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514(7520):107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Matkar S, Sharma P, Gao S, et al. An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc axis. Cancer Cell. 2015;28(4):472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qian Z, Ren L, Wu D, et al. Overexpression of FoxO3a is associated with glioblastoma progression and predicts poor patient prognosis. Int J Cancer. 2017;140(12):2792–2804. [DOI] [PubMed] [Google Scholar]
- [23].Myatt SS, Brosens JJ, Lam EW. Sense and sensitivity: FOXO and ROS in cancer development and treatment. Antioxid Redox Signal. 2011;14(4):675–687. [DOI] [PubMed] [Google Scholar]
- [24].Eijkelenboom A, Mokry M, De WE, et al. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol. 2013;9(1):638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eijkelenboom A, Mokry M, Smits LM, et al. FOXO3 selectively amplifies enhancer activity to establish target gene regulation. Cell Rep. 2013. December 26;5(6):1664–1678. PubMed PMID: 24360957. [DOI] [PubMed] [Google Scholar]
- [26].Herz HM, Hu D, Shilatifard A. Enhancer malfunction in cancer. Mol Cell. 2014;53(6):859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mei S, Qin Q, Wu Q, et al. Cistrome data browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017;45(Databaseissue):D658–D662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. [DOI] [PubMed] [Google Scholar]
- [29].Kantidakis T, Saponaro M, Mitter R, et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016;30(4):408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ford DJ, Dingwall AK. The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet. 2015;208(5):178. [DOI] [PubMed] [Google Scholar]
- [31].Ali A, Veeranki SN, Chinchole A, et al. MLL/WDR5 complex regulates Kif2A localization to ensure chromosome congression and proper spindle assembly during mitosis. Dev Cell. 2017;41(6):605–622. [DOI] [PubMed] [Google Scholar]
- [32].Neilsen BK, Chakraborty B, Mccall JL, et al. WDR5 supports colon cancer cells by promoting methylation of H3K4 and suppressing DNA damage. Bmc Cancer. 2018;18(1):673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lei K, Sun R, Chen LH, et al. Mutant allele quantification reveals a genetic basis for TP53 mutation-driven castration resistance in prostate cancer cells. Sci Rep. 2018;8(1):12507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267(5206):2003–2006. [DOI] [PubMed] [Google Scholar]
- [35].Wendel HG, De SE, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428(6980):332–337. [DOI] [PubMed] [Google Scholar]
- [36].Froimchuk E, Jang Y, Ge K. Histone H3 lysine 4 methyltransferase KMT2D. Gene. 2017;627:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lai B, Lee JE, Jang Y, et al. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017;45(11):6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee JE, Wang C, Xu S, et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013;2(2):e01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rada-Iglesias A. Is H3K4me1 at enhancers correlative or causative? Nat Genet. 2018;50(1):4. [DOI] [PubMed] [Google Scholar]
- [40].Essaghir A, Dif N, Marbehant CY, et al. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem. 2009;284(16):10334–10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kannike K, Sepp M, Zuccato C, et al. Forkhead transcription factor FOXO3a levels are increased in Huntington disease because of overactivated positive autofeedback loop. J Biol Chem. 2014;289(47):32845–32857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim D-H, Kim J, Kwon J-S, et al. Critical roles of the histone methyltransferase MLL4/KMT2D in murine hepatic steatosis directed by ABL1 and PPARγ2. Cell Rep. 2016;17(6):1671–1682. [DOI] [PubMed] [Google Scholar]
- [43].Quinn DI, Sandler HM, Horvath LG, et al. The evolution of chemotherapy for the treatment of prostate cancer. Ann Oncol. 2017 Nov 1;28(11):2658–2669. [DOI] [PubMed] [Google Scholar]
- [44].Mateo J, Boysen G, Barbieri CE, et al. DNA repair in prostate cancer: biology and clinical implications. Eur Urol. 2017 Mar;71(3):417–425. [DOI] [PubMed] [Google Scholar]
- [45].Yang Y, Blee AM, Wang D, et al. Loss of FOXO1 cooperates with TMPRSS2–ERG overexpression to promote prostate tumorigenesis and cell invasion. Cancer Res. 2017;77(23):6524–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. PubMed PMID: 18546601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.