

ABSTRACT
Epigenetic mechanisms such as genomic imprinting have a fundamental role in embryo and fetal development. Hence, we here studied expression levels of epigenetic modifiers and imprinted genes in cases of ididopathic spontaneous abortion (SA). Thirty-five placental samples and 35 matched fetal tissues from second trimester SA were analysed; including 16 controls (placental and fetal infections as the known cause of spontaneous abortion) and 19 idiopathic SA cases. Transcript levels of epigenetic regulators and imprinted genes were measured by qRT-PCR and methylation at imprinted genes was studied by bisulfite genomic sequencing and MS-MLPA. Global DNA hydroxymethylation (5-hmC) levels were measured by an ELISA-based assay.
We observed an upregulation of TET2 and TET3 in placental samples from idiopathic SA cases; however, no significant difference in global 5-hmC levels was observed. On the contrary, in fetal tissues, TET3 was markedly downregulated in idiopathic SA, showing an opposite trend to that observed in placental tissue. IGF2 and CDKN1C were upregulated and MEST downregulated in placentas from idiopathic SA cases; concordantly, IGF2 was also upregulated in fetal tissues from idiopathic SA cases. Although not reaching statistical significance, an increase in methylation levels of MEST, KvDMR1 and H19 DMRs was observed in idiopathic SA cases, concordantly with the observed changes in expression. Our study reveals, for the first time, deregulation of epigenetic modifiers and imprinted genes in both placental and fetal tissues from idiopathic SA cases in the second trimester of pregnancy, indicating a critical role during pregnancy.
KEYWORDS: Spontaneous abortion, DNA methylation, DNA hydroxymethylation, TET enzymes, imprinted genes
Introduction
Spontaneous abortion (SA) has a high clinical and social impact and may be defined as a clinically recognized pregnancy loss before the 24th weeks of gestation [1,2]. Approximately 10% to 15% of these pregnancies terminate spontaneously and about 1–5% of couples suffer recurrent SA (two or more pregnancy losses) [2–4]. Pregnancy loss has a heterogeneous etiology, which results from several known risk factors, such as endocrine, immunological, environmental, infectious, thrombophilic and genetic factors [5,6]. Despite the different causes, genetic factors are considered the main contributor for sporadic and recurrent SA, given that fetal chromosomal abnormalities represent about 50–60% of the cases. However, approximately 40–50% of recurrent SA reveal a normal karyotype and its causes are still poorly understood [2,4].
Epigenetic marks regulate the expression of genes [7] and can constitute epimutations that could contribute to these cases. DNA methylation has been the most studied epigenetic modification and is associated to gene silencing when occurring in the promoter region of genes [8]. This epigenetic mark usually occurs in CpG dinucleotides and consists in the addition of a methyl group from the S-adenosylmethionine donor (SAM) to the fifth carbon of a cytosine, originating a 5-methylcytosine (5-mC) [9,10]. The enzymes that catalyze DNA methylation are well characterized and are designated DNA methyltransferases (DNMTs). Within this family, three enzymatically active types are identified, the maintenance methyltransferase DNMT1 and the de novo methyltransferases, DNMT3A and DNMT3B [9,11].
More recently, another family of enzymes – TET1, TET2 and TET3 – was identified, being able to oxidize 5-mC into 5-hydroxymethylcytosine (5-hmC) [12]. These enzymes can also further oxidize 5-hmC into 5-formylcytosine (5-fC) and 5-carboxycytosine (5-caC), a process thought to be involved in active DNA demethylation [13,14]. Like DNMTs, TETs are also involved in the differentiation and regulation of placental trophoblasts [15,16].
Epigenetic marks play a key role in the regulation of genomic imprinting [10]. This epigenetic phenomenon is essential in the development and function of the placenta, and growth of the fetus. Imprinting errors may be caused by epigenetic disruptions that may lead to different phenotypes, such as fetal growth restriction (FGR) [17,18]. The particular characteristic of the imprinted genes is their monoallelic expression, depending on the parent-of-origin. Differentially methylated regions (DMRs) are responsible for this differential expression and are associated with imprinting control regions (ICRs). These ICRs, acording to its specific methylation status, control the expression of neighborhood imprinted genes that usually occur in clusters [10,19]. Imprinted DMRs can also influence expression by acting as methylation-sensitive insulators, such as the H19 DMR found on chromosome 11p15.5. Normally, H19 DMR is methylated on the paternal allele preventing the binding of the zinc finger protein CTCF, which in turn allows the access of the IGF2 promoter to the enhancers located downstream of H19, and consequently, paternal IGF2 is expressed. On the other hand, in the maternal allele this CTCF-binding site is unmethylated, which allows the binding of CTCF and the interaction of the H19 promoter to the enhancers. In this case, the IGF2 is silenced and the H19 is expressed [20–22]. Alternatively, the imprinting DMRs can influence gene expression through non-coding RNA (ncRNA) promoters, such as the KvDMR1, also on 11p15.5 chromosome. This DMR is located in the KCNQ1 gene, which contains a promoter for a ncRNA, the KCNQ10T1. Typically, this DMR is unmethylated at the paternal allele, which allows the transcription of the ncRNA that silences the expression of the cis-linked imprinted genes, such as CDKN1C and PHLDA2 genes. In contrast, the methylation present in the maternal allele leads to ncRNA silencing, and consequently, allows expression of the imprinted genes adjacent to the ICR [21,23,24]. In addition, DMRs may be present in the promoter of a gene controlling its expression, according to the methylation status. This happens with MEST/PEG1 and PEG10 genes that are paternally expressed and located on the long arm of the chromosome 7 [25–27].
Regarding the association of imprinted genes with fetal deaths, some studies have shown the involvement of PEG10 and MEST in embryonic lethality. PEG10 knockout animal models showed early embryonic lethality owing to defects in the placenta [28,29]. Also, according to the literature, a decrease in MEST protein expression was observed in human placentas of missed abortions and this suggested a possible pathological mechanism for missed abortion [30]. Futhermore, we previously showed changes in the expression of imprinted genes, namely CDKN1C, IGF2, KCNQ1 and PHLDA2, in placental samples of SA [31,32]. In addition, deletion of the active copies of several other imprinted genes, including the genes here studied, was shown to result in a noticeable placental phenotype, with most deletions affecting the size of the placenta [33].
Taking this into account and considering the importance of epigenetics and genomic imprinting in fetal and placental development, it is plausible that epigenetic changes may underlie the failure of a gestation. The main goal of this study was to evaluate if alterations in the expression of the epigenetic machinery and imprinted genes were present in samples from SA with an idiopathic cause in the second trimester of pregnancy. Therefore, we analysed the expression of DNMT and TET genes, and six imprinted genes, three paternally expressed – IGF2, MEST, PEG10 – and three maternally expressed – CDKN1C, KCNQ1, PHLDA2. Furthermore, methylation patterns of MEST, KvDMR1, H19 DMR and IGF2 promoters were evaluated in order to investigate a possible correlation between the DNA methylation status and gene expression. In addition, global levels of 5-hmC were determined in SA samples. This work was performed in both placental and fetal tissues and was developed in order to contribute for understanding the etiology of human pregnancy losses in the second trimester of gestation.
Results
Altered expression of epigenetic regulators in pregnancy losses
We analysed the expression of 6 epigenetic modifiers, TET1, TET2, TET3, DNMT1, DNMT3A and DNMT3B in a total of 35 placentas and 35 matched fetal tissues from SA during the second trimester of gestation, namely 16 controls (SA due to infections) and 19 idiopathic SA cases. In SA cases, we observed an upregulation of TET2 and TET3 in placental samples (Figure 1(a)) whilst in fetal tissue a significant downregulation of TET3 was observed (Figure 1(b)). Notably, the changes observed in the placenta showed an opposite trend to the fetal tissue. Of interest, we observed that the upregulation of TET2 and TET3 in placental samples was mainly due to SA cases not showing FGR (Supplementary Fig. S1A). On the other hand, in fetal tissues, both SA cases with and without FGR showed a tendency for TET3 decreased expression (Supplementary Fig. S1B).
Figure 1.
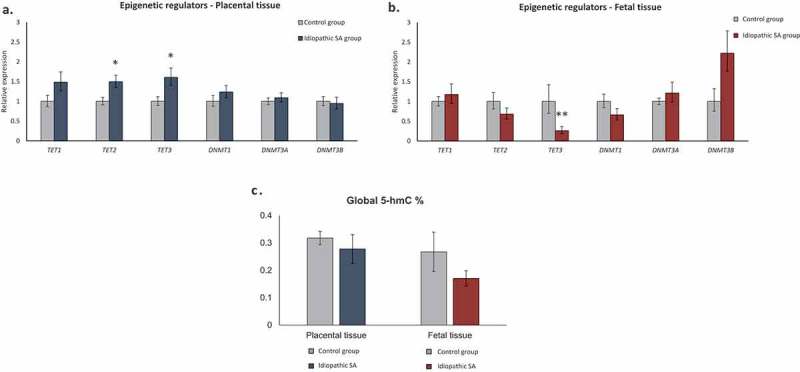
Relative expression of epigenetic regulators (TET1, TET2, TET3, DNMT1, DNMT3A and DNMT3B) in placental and fetal tissues. Bars represent 2ΔΔCt ± SEM; * represents p < 0.05, ** represents p < 0.01, *** represents p < 0.001. (a) Relative expression of epigenetic regulators in placental tissue from idiopathic SA (n = 19) comparing to controls (n = 16). (b) Relative expression of epigenetic regulators in fetal tissue samples from idiopathic SA (n = 19) comparing to controls (n = 16). (c) Global 5-hmC levels in placental and fetal tissues (controls, n = 8 and SA, n = 13). Bars represent 5-hmC (%) ± SEM.
Global hydroxymethylation levels
Since changes in TET genes expression were observed, we analysed global 5-hmC levels of 21 pairs of placenta/fetal tissue from SA by an ELISA assay. Both in placental and fetal samples, no significant changes were observed between controls and idiopathic SA cases (Figure 1(c)). Nevertheless, 5-hmC was present at detectable levels in the placenta and fetal tissues, ranging from 0,12% to 0,85% in placentas and from 0,04% to 0,73% in fetal tissue (Supplementary table S1).
Deregulation of imprinted genes in pregnancy losses
We also analysed the expression of 6 imprinted genes, 3 paternally expressed (IGF2, MEST and PEG10) and 3 maternally expressed (CDKN1C, KCNQ1 and PHLDA2) (Figure 2(a,b)). In placental samples, a significant upregulation of two imprinted genes – IGF2 and CDKN1C – was observed in SA cases, together with a downregulation of MEST gene (Figure 2(a)). In fetal tissue, only IGF2 presented altered expression, with an upregulation in SA cases (Figure 2(b)). Notably, IGF2 was upregulated in both placental and fetal tissues in idiopathic SA samples.
Figure 2.
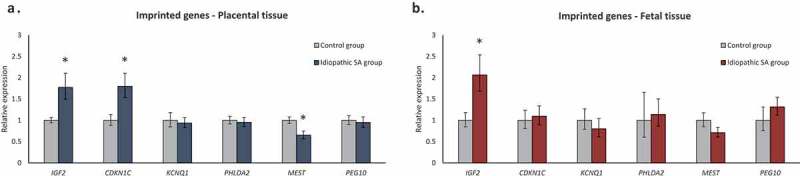
Relative expression of imprinted genes (IGF2, CDKN1C, KCNQ1, PHLDA2, MEST and PEG10) in placental and fetal tissues. (a) Relative expression of imprinted genes in placentas from idiopathic SA (n = 19) comparing to controls (n = 16). (b) Relative expression of imprinted genes in fetal tissue from idiopathic SA (n = 19) comparing to controls (n = 16). Bars represent 2ΔΔCt ± SEM; * represents p < 0.05, ** represents p < 0.01, *** represents p < 0.001.
Of interest, we observed that the upregulation of IGF2 and CDKN1C and downregulation of MEST in placental samples was mainly due to SA cases not showing FGR (Supplementary Fig. S1C). On the other hand, in fetal tissues, both SA cases with and without FGR showed a tendency for IGF2 increased expression (Supplementary Fig. S1D).
Methylation status of imprinted genes
Amongst the six imprinted genes studied, IGF2, CDKN1C and MEST showed an altered expression relatively to control group (Figure 2). Therefore, methylation patterns of ICRs of these imprinted genes were studied in samples showing differences in transcript levels.
For standard bisulfite sequencing, 22 CpGs at MEST promoter DMR, 24 CpGs at KvDMR1 and 18 CpGs at H19 DMR were analysed. Although not significant, we observed an increase in methylated CpGs in the placental samples of SA cases comparing to controls, for MEST (mean methylation percentage of 52,3% vs 36,7%, respectively; p > 0,05), KvDMR1 (mean methylation percentage 78,0% vs 65,2%, respectively; p > 0,05) and H19 DMR (mean methylation percentage 58,5% vs 45,8%, respectively; p > 0,05) (Figure 3(a–c)), which is in accordance with the altered expression patterns (decreased expression of MEST and increased expression of CDKN1C and IGF2) observed in SA samples (Figure 2(a)). Concerning IGF2 gene, we also studied two promoter regions, P0 and P3, by MS-MLPA and bisulfite sequencing, respectively, and did not observe significant changes, although an hypomethylated state was noted in both promoters (P3 mean methylation percentage 3,9% in idiopathic SA vs 7,6% in controls; p > 0,05) (Figure 3(d)). This is in accordance with the high levels of transcription observed for this gene, in both placental and fetal tissues.
Figure 3.
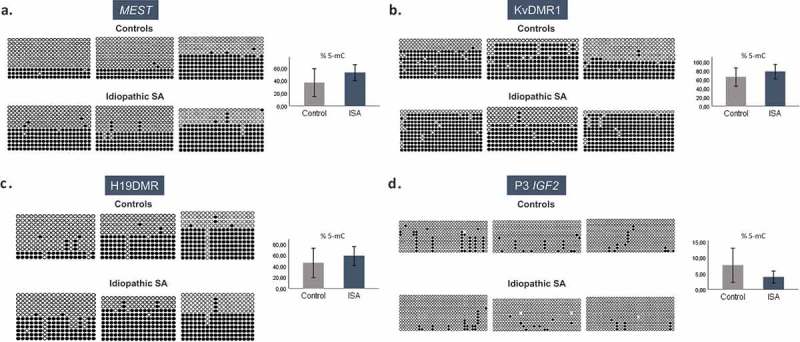
Methylation patterns of MEST, KvDMR1, H19 DMR and P3 promoter IGF2 in placenta samples by bisulfite genomic sequencing. The columns represent the successive CpGs and the rows represent the different clones. Black circles represent methylated CpGs and white circles represent non-methylated CpGs. The graph to the right represents 5-mC (%) ± SEM of each group; ISA – Idiopathic spontaneous abortion. (a) Methylation status of 22 CpGs at MEST in three placenta samples of controls and three placenta samples of idiopathic SA. (b) Methylation status of 24 CpGs at KvDMR1 in three placenta samples of controls and three placenta samples of idiopathic SA. (c) Methylation status of 18 CpGs at H19 DMR in three placenta samples of controls and three placenta samples of idiopathic SA. (d) Methylation status of 33 CpGs at P3 IGF2 in three placenta samples of controls and three placenta samples of idiopathic SA.
Given that we also observed an upregulation of IGF2 in fetal tissues of SA cases, we also studied methylation patterns in these samples; however no differences were observed for H19 DMR (mean methylation percentage 84,4% in idiopathic SA vs 74,8% in controls; p > 0,05) but an increase in methylation was observed at P3 region (mean methylation percentage 6,1% in idiopathic SA vs 2,3% in controls; p > 0,05) (Figure 4(a,b)), albeit not statistically significant.
Figure 4.
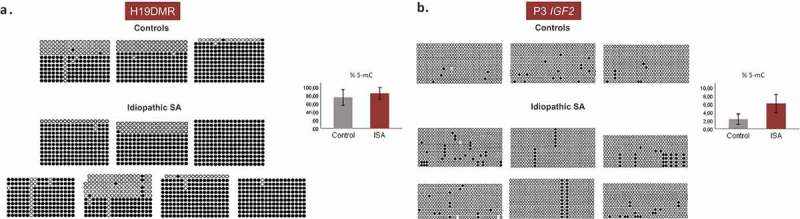
Methylation patterns of H19 DMR and P3 promoter IGF2 in fetal tissue samples by bisulfite genomic sequencing. The columns represent the successive CpGs and the rows represent the different clones. Black circles represent methylated CpGs and white circles represent non-methylated CpGs. The graph to the right represents the 5-mC (%) ± SEM of each group; ISA – Idiopathic spontaneous abortion. (a) Methylation status of 18 CpGs at H19 DMR in three fetal tissue samples of controls and seven fetal tissue samples of idiopathic SA. (b) Methylation status of 33 CpGs at P3 IGF2 in three fetal tissue samples of controls and six fetal tissue samples of idiopathic SA.
To confirm the bisulfite sequencing results described above by another technique, we analysed the samples by MS-MLPA (Methylation-specific Multiplex Ligation-Dependent Probe Amplification). With this method, 4 CpGs at KvDMR1, 4 CpGs at H19 DMR, 1 CpG at IGF2 DMR0 and 1 CpG at exon 1 of CDKN1C were analysed, in both biological tissues of 4 controls and 6 idiopathic SA cases. In accordance with the bisulfite sequencing results, MS-MLPA showed no significant differences between the controls and the idiopathic SA cases, either in fetal or placental tissue (Figure 5(a,b)). The mean methylation percentage of each biological group is depicted in Figure 5(a,b). In addition, the CpGs analysed for the additional regulatory regions of IGF2 (IGF2 DMR0) and CDKN1C (exon 1 of CDKN1C) did not explain the overexpression of these genes in the idiopathic SA cases (Figure 5(a,b)).
Figure 5.
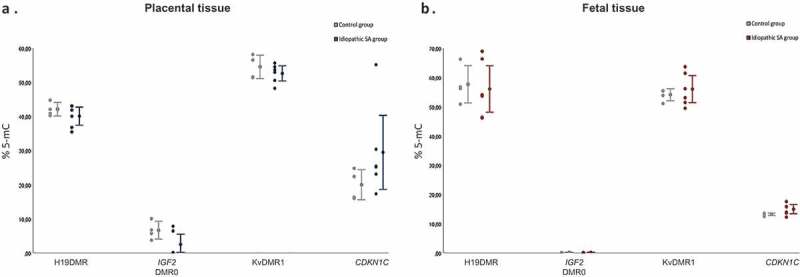
Percentage of methylation at imprinted genes DMRs by MS-MLPA. Methylation results of H19 DMR (4 CpGs), IGF2 DMR0 (1 CpG), KvDMR1 (4 CpGs) and CDKN1C (1 CpG) in controls (n = 4) and idiopathic SA (n = 6). (a) Placental samples. (b) Fetal tissues. Left – the dots represent the percentage value for each sample; Right – mean % 5-mC ± SEM.
Discussion
Spontaneous pregnancy loss is a severe obstetric complication that has clinical and social impact. Pregnant women whom undergo recurrent SA frequently do not have an indentifiable inherent clinical cause. Despite advances in research, there is still limited knowledge about the underlying molecular mechanisms behind this condition. Since epigenetic mechanisms and genomic imprinting play a key role in many processes during placenta and fetal development, we hypothesized that epigenetic deregulation might underlie idiopathic SA. Additionally, it is known that FGR is associated with deregulation of imprinted genes, namely PHLDA2 and CDKN1C upregulation, in term placentas [22,34,35].
To our knowledge, this is the first study that evaluates the expression of epigenetic regulators and global 5-hmC levels in fetal tissue from second trimester pregnancy losses. Our results showed a significant upregulation of TET2 and TET3 in placentas from idiopathic SA. The observed upregulation is in disagreement with previous results by Wu and collaborators [36] in which they observed downregulation of all TET enzymes in villous tissue from early pregnancy loss cases, however, this study was carried out between 6–8 weeks of gestation which might explain the discrepancies observed. Indeed, Rakoczy and collaborators showed a specific cellular localization for the different TET enzymes in human placentas, suggesting a dynamic role in the regulation of transcriptional activity of trophoblast progenitors and differentiated cell subtypes [15].
In our study, despite TET2 and TET3 being upregulated in idiopathic SA placentas, we did not observe significant changes in DNA hydroxymethylation global levels. In this regard, it was described that TET1 is particularly recruited for 5-mC oxidation and, consequently, to originate 5-hmC, while TET2 and TET3 are responsible for the remaining steps of the oxidation cascade, stimulating the removal of 5-hmC. Interestingly, it was shown that TET1 deletion has a greater impact on the loss of 5-hmC, whereas the reduction of TET2 and TET3 levels lead to accumulation of 5-hmC [37]. Taking this into account, it can be suggested that the observed overexpression of TET2 and TET3 in our samples could lead to an increase of the other derivatives of the oxidation cascade. Nevertheless, we confirmed the presence of 5-hmC in placentas, as others have previously shown [38,39]. Mora and collaborators have reported that TET2 and TET3 are highly abundant in the placenta and 5-hmC is particularly located at numerous imprinted genes, with an enrichment on the transcribed allele [39]. Thus, it would be interesting to analyse if 5-hmC levels at these particular regions might be altered in samples from idiopathic spontaneous abortion, correlating with changes in expression of imprinted genes.
One interesting observation was the marked downregulation of TET3 in fetal tissue, but upregulation in the placentas, from SA cases which could indicate that fetal deaths might be associated with changes in the epigenetic machinery in both the fetus and the placenta. Although not significant, we also observed an upregulation of TET1 in these cases, which could indicate a compensatory mechanism [40].
Regarding imprinted genes expression, we found upregulation of IGF2 and CDKN1C and downregulation of MEST, namely in placental tissue. These results corroborate our previous findings, in which we observed in both placental and fetal samples an upregulation of IGF2 and CDKN1C genes in the second trimester of SA [32]. Considering the parental conflict teory, these two genes behave in opposite ways, the paternally expressed IGF2 gene is proposed to promote growth, while the maternally expressed CDKN1C gene may act as growth suppressor limiting resources to fetus; this could suggest that the ‘parental conflict’ theory may not be applied to all imprinted human genes, namely in extreme conditions like a spontaneous loss of pregnancy. It should also be noted that the demand of maternal resources is less strenuous in humans in comparison with mice, since human pregnancies are normally singletons [32,35,41].
It is important to stress that these changes in imprinted genes expression observed in idiopathic SA samples may also be a consequence of aberrant development, being compensatory or adverse mechanisms that could underlie pathology and should therefore be a matter of further investigation.
The observed changes in methylation at the DMRs of the imprinted genes studied showed a tendency for hypermethylation, which is in line with observed increased transcript levels; nevertheless, another layer of epigenetic regulation, such as histone modifications [42], might be controlling the expression of these genes.
A previous report showed that the specific siRNA knockdown of MEST drastically reduced the invasion and migration of extravillous trophoblasts [30]. In the same study, the authors observed decreased MEST expression concomitant with MEST promoter hypermethylation in placentas of SA, corroborating our observations. Therefore, the authors suggested that changes in MEST DNA methylation and MEST expression might have an impact on extravillous trophoblasts invasion that may explain the underlying mechanism of the SA [30]. Zheng and collaborators also associated increased MEST methylation to pregnancy loss [43]. Both studies were performed with chorionic villi samples of early pregnancy losses (6 to 10 weeks), but are in accordance with our results in the second trimester pregnancy. Hence, we postulate that hypermethylation of the MEST promoter affects its expression and may contribute to SA.
One of the limitations of our study is that cell-type specific epigenetic states exist within the human placenta [44]. Also, fetal tissues analysed were limited to skin. Furthermore, we can not ignore that the controls used, although corresponding to pregnancy losses with a non-genetic cause (infections), do not represent normal pregnancies (without any kind of complications). We may also point out that only a few imprinted genes have been studied and that we may be faced with a complex and coordinated network of imprinted genes responsible for intrauterine development [45]. To acquire a better knowledge about the etiology of the loss of human pregnancy more studies on the molecular mechanism underlying the expression of imprinted genes are needed.
Despite the limitations, this study bridged the gap of the lack of studies with SA samples of second trimester. Most of the studies are performed with samples of early or term pregnancy losses and, since pregnancy is a highly dynamic process, as well as all the epigenetic mechanisms involved, it is essential to conduct studies throughout the whole pregnancy period. In addition, there are few studies on SA that use fetal tissue matched to placental samples, which values this study in order to extend the knowledge of the etiology of spontaneous abortion.
Material and methods
Sample collection
A total of 35 placental samples and 35 matched fetal tissues was collected from pregnancy losses with gestational ages between 12 and 24 weeks, with no significant diffrerences on maternal age and gestational age between controls and idiopathic SA cases (supplementary table S2).This collection followed the protocol established with the Gynecology and Obstetric Department of Centro Hospitalar de São João (CHSJ)-Porto; therefore, the placental and fetal samples were always collected by the same team of obstetricians and under the same conditions. All placental samples were taken from the closest region to the umbilical cord, in order to uniformize the composition of the samples. Fetal tissue used for this study corresponded to calcaneus or skin samples, which was available for research purposes and not compromising the anatomo-pathological analysis of the fetus.
Detailed information about the samples is in supplementary table S3. Samples were divided in controls (n = 16; placental and fetal infections as the known cause of abortion) and idiopathic SA (n = 19). The clinical evaluation of the cases was made by an obstetrician (clinical and analytic evaluation) considering also the anatomopathological evaluation. All samples here analysed presented a normal karyotype and the exclusion criteria included twin pregnancies, endocrine diseases, chromosomal abnormalities, immunological diseases, inherited thrombophilia, preeclampsia, cardiopathies, maternal exposure to toxins and anatomical abnormalities of the genital tract.
After collection, placental and fetal samples were fragmented and stored immediately at −80ºC in RNA later (Ambion) until RNA and DNA extraction. This study was approved by the local Ethics Committee – Health Ethical Committee of Faculty of Medicine University of Porto, CHSJ, Porto, Portugal.
RNA and DNA extraction
RNA and DNA extraction was performed using Trizol (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to manufacturers’ protocol with minor modifications. Briefly, 1mL of TRIzol was added to the samples and transferred to a Triple-Pure™ zirconium beads tube (Bertin Technologies) and homogenized by a Minilys homogenizer (Bertin Technologies). DNA was precipitated from the interphase at −20ºC for 2 hours after the addition of absolute ethanol. DNA and RNA quantification and purity were analysed by NanoDrop 2000 UV-vis Spectrophotometer (Nanodrop Technologies, Wilmington, USA).
Quantitative real time PCR (qRT-PCR)
Total RNA was subjected to treatment with DNaseI (Thermo Scientific™) and cDNA was synthesized using 1 µg of the DNase-treated total RNA and 4 µL of qScriptTM cDNA SuperMix (Quanta Biosciences, Inc., Gaithersburg, USA).
Transcript levels of DNMT family (DNMT1, DNMT3A and DNMT3B), TET family (TET1, TET2 and TET3), six imprinted genes (IGF2, CDKN1C, KCNQ1, PHLDA2, MEST and PEG10) and three housekeeping genes (ACTB, TBP and RPLP0) were analysed by Real-Time PCR on a 96-well StepOnePlus™ Real-Time PCR System (Life Technologies Corporation, California, USA) (Primer sequences are described in supplementary table S4 [46,47]). For each gene, cases and control samples were run in the same plate to minimize intra-plate variations and each reaction was performed in duplicate. In addition, a negative control for each pair of primers was included in the reaction plate. Primers were designed to be exon-spanning, in order to avoid amplification of contaminating genomic DNA.
The mastermix used was the 2× PowerUp SYBR Green Master Mix (Thermo Ficher Scientific), following the manufacturer’s instructions. This mastermix has the particularity of preventing carry-over contaminations by previous PCR products. PCR reaction was performed in a 10 μL volume per well and the PCR parameters were as follows: 50°C for 2 min, 95°C for 2 min, followed by 40 cycles at 95°C for 3 s and 60°C for 30 s.
Global DNA hydroxymethylation levels
Global 5-hmC content of the DNA samples (100 ng) was measured using the QUEST 5-hmCTM DNA ELISA Kit (Zymo Research, USA), according to the manufacturer’s instructions. For this procedure, 42 samples (21 fetal samples and 21 respective placentas) were used. This included, for each tissue, 8 controls and 13 idiopathic SA cases. A control DNA set, with known 5-hmC quantities, was used to build a standard curve and calculate 5-hmC levels in DNA samples. All samples were run in duplicates.
The intensity of the signal was measured after 60 min of incubation and the reading was performed at an absorbance of 405nm on an ELISA plate reader (Sunrise, Tecan).
Bisulfite genomic sequencing
Methylation at regions of interest was analysed in samples with significantly altered imprinted gene expression by standard bisulfite sequencing. We analysed 18 CpGs of the H19 DMR that includes one CTCF-binding site (GenBank Accession Number AF087017; nucleotides 6006–6328), 33 CpGs of P3 promoter of IGF2 (GenBank Accession Number NT_009237; nucleotides 2079781-2080144), 24 CpGs of KvDMR1 (Genbank Accession Number U90095; nucleotides 66536-66800) and 22 CpGs of MEST promoter (GenBank Accession Number Y10 620; nucleotides 609–898) (Primer sequences are described in supplementary table S4 [48–50]). Briefly, 1 µg of each DNA sample was treated with sodium bisulfite, according to the standard protocol of EpiTect Bisulfite Kit (Qiagen, Hiden, Germany). Afterwards, modified DNA was subjected to PCR amplification of the regions of interest using Type-it Microsatellite PCR Kit (Qiagen, Hiden, Germany). The PCR conditions were as follows: initial denaturation during 5 min at 95°C, followed by 35 cycles of denaturation for 1 min at 95°C, annealing for 90 s at 60ºC and extension for 1 min at 72°C, ending with a final extension step for 30 min at 60ºC. Bisulfite PCR products were run in a QIAxcel Eletrophoresis Unit (QIAGEN) and cloned with the TOPO® TA Cloning Kit (Invitrogen), using pCR™II-TOPO® (Invitrogen) and DH5α Chemically Competent E. coli cells (Invitrogen). Colony PCR was performed with Taq DNA Polymerase (Thermo Fisher Scientific) and the conditions were as follows: 94°C for 10 min, followed by 35 cycle at 94°C for 45 s, 50°C for 45 s and 72°C for 1 min, ending with a final extension step for 10 min at 72ºC. Around 12 clones for each sample were purified using Agencourt® AMPure® XP (Beckman Coulter Krefeld, Germany), and sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), in a 3500 Genetic Analyzer (Applied Biosystems).
Methylation specific-multiplex ligation-dependent probe amplification (MS-MLPA)
For MS-MLPA, 10 samples were selected, 4 controls and 6 cases. This was performed as a complementary technique and the samples used in the cloning were also included in this analysis. For this, 50–100 ng of DNA samples, a negative control, as well as two reference samples as endogenous control, were included. Furthermore, all samples of the same tissue type were simultaneously subjected to MS-MLPA (SALSA MS-MLPA probemix ME030-C3 BWS/RSS (MRC Holland, Amsterdam, The Nederland’s)). This mix includes 4 specific methylation probes for each H19 DMR and KVDMR1 as well as 1 specific methylation probe for each 5ʹDMR0 IGF2 and for the exon 1 of CDKN1C, which contain a HhaI recognition site.
The hybridization step was performed at 60ºC for 16 hours and, subsequently, samples were equally split into two aliquots, the first underwent the ligation step, whereas the second one underwent the ligation step and an enzymatic digestion step with HhaI. Subsequently, the probes were amplified using universal primers. Lastly, 1 µl of MS-MLPA PCR product was mixed with 0,3 µl of internal size standard (GeneScan™ 600 LIZ; Applied Biosystems) and 13,7 µl of highly deionized formamide and analysed in a 3500 Genetic Analyzer (Applied Biosystems).
Statistical analysis
Raw data obtained from qRT-PCR was imported to qbasePlus software (BioGazelle) which calculated the relative expression values. The far outliers in each group were identified in SPSS Statistics 25 (IBM) and were excluded from the calculations. We assumed that the value of the last cycle of amplification (Ct = 40 cycles) corresponded to the absence of relative expression.
For the hydroxymethylation analysis, the 5-hmC percentages for each sample were calculated from the absorbance values and the values of the linear regression equation, following the manufacturer’s instructions. Firstly, the arithmetic mean of the duplicates from each sample was calculated and then the arithmetic mean of all samples from each biological group was calculated.
The bisulfite sequencing data was analysed in BiQ Analyzer software. The methylation percentage for each sample was calculated by dividing the total number of methylated CpGs by the total number of CpGs analysed. Then, the arithmetic mean was calculated between the samples of each group. Only clones with more than 95% of non-CpG cytosines converted were included in the analysis. Regarding the results of the MS-MLPA, the data was exported from the GeneMapper software (Applied Biosystems). The calculation of the percentages of methylation was based on the difference between the values obtained from the enzymatic non-digestion and the values obtained from the enzymatic digestion.
The expression values and the global hydroxymethylation data was evaluated by the Mann-Whitney U test and the methylation data was evaluated by the t-test, using SPSS Statistics 25 software (IBM). The results of all tests to assess the significance of the observed differences between groups were considered significant when p < 0,05.
Funding Statement
This work was partly supported by (Foundation for Science and Technology (FCT)) through Investigador FCT programme, with a researcher contract to C.J.M. [IF/00047/2012 and CEECIND/00371/2017].
Acknowledgments
We would like to thank Liliana Capela for help with MS-MLPA and Joel Pinto for help with processing the samples (Department of Genetics, FMUP).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Regan L, Rai R.. Epidemiology and the medical causes of miscarriage. Baillieres Clin Obstet Gynaecol. 2000;14:839–854. [DOI] [PubMed] [Google Scholar]
- [2].Tur-Torres MH, Garrido-Gimenez C, Alijotas-Reig J.. Genetics of recurrent miscarriage and fetal loss. Best Pract Res Clin Obstet Gynaecol. 2017;42:11–25. [DOI] [PubMed] [Google Scholar]
- [3].van Den Berg MMJ, van Maarle MC, van Wely M, et al. Genetics of early miscarriage. Biochim Biophys Acta. 2012;1822:1951–1959. [DOI] [PubMed] [Google Scholar]
- [4].Nikitina TV, Sazhenova EA, Tolmacheva EN, et al. Comparative cytogenetic analysis of spontaneous abortions in recurrent and sporadic pregnancy losses. Biomed Hub. 2016;1:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Christiansen OB, Steffensen R, Nielsen HS, et al. Multifactorial etiology of recurrent miscarriage and its scientific and clinical implications. Gynecol Obstet Invest. 2008;66:257–267. [DOI] [PubMed] [Google Scholar]
- [6].Warren JE, Silver RM. Genetics of pregnancy loss. Clin Obstet Gynecol. 2008;51:84–95. [DOI] [PubMed] [Google Scholar]
- [7].Jaenisch R, Bird A. Epigenetic regulation of gene expression : how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. [DOI] [PubMed] [Google Scholar]
- [8].Adalsteinsson BT, Ferguson-Smith AC. Epigenetic control of the genome — lessons from genomic imprinting. Genes (Basel). 2014;5:635–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tanaka S, Nakanishi MO, Shiota K. DNA methylation and its role in the trophoblast cell lineage. Int J Dev Biol. 2014;58:231–238. [DOI] [PubMed] [Google Scholar]
- [10].Monk D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: the selected survival of imprints. Int J Biochem Cell Biol. 2015;67:128–138. [DOI] [PubMed] [Google Scholar]
- [11].Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. [DOI] [PubMed] [Google Scholar]
- [12].Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rakoczy J, Padmanabhan N, Krzak AM, et al. Dynamic expression of TET1, TET2, and TET3 dioxygenases in mouse and human placentas throughout gestation. Placenta. 2017;59:46–56. [DOI] [PubMed] [Google Scholar]
- [16].Logan PC, Mitchell MD, Lobie PE. DNA methyltransferases and TETs in the regulation of differentiation and invasiveness of extra-villous trophoblasts. Front Genet. 2013;4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Piedrahita JA. The role of imprinted genes in fetal growth abnormalities. Birth Defects Res A Clin Mol Teratol. 2011;91:682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ishida M, Moore GE. The role of imprinted genes in humans. Mol Aspects Med. 2013;34:826–840. [DOI] [PubMed] [Google Scholar]
- [19].Mackay DJG. Human imprinting disorders: principles, practice, problems and progress. Eur J Med Genet. 2017;60:618–626. [DOI] [PubMed] [Google Scholar]
- [20].Hark AT, Schoenherr CJ, Katz DJ, et al. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. [DOI] [PubMed] [Google Scholar]
- [21].Kalish JM, Jiang C, Bartolomei MS. Epigenetics and imprinting in human disease. Int J Dev Biol. 2014;58:291–298. [DOI] [PubMed] [Google Scholar]
- [22].Koukoura O, Sifakis S, Spandidos DA. DNA methylation in the human placenta and fetal growth (review). Mol Med Rep. 2012;5:883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Smilinich NJ, Day CD, Fitzpatrick GV, et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith – Wiedemann syndrome. Genetics. 1999;96:8064–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Co Biol. 2014;141:1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ono R, Kobayashi S, Wagatsuma H, et al. A retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics. 2001;73:232–237. [DOI] [PubMed] [Google Scholar]
- [26].Rahat B, Mahajan A, Bagga R, et al. Epigenetic modifications at DMRs of placental genes are subjected to variations in normal gestation, pathological conditions and folate supplementation. Sci Rep. 2017;7:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kobayashi S, Kohda T, Miyoshi N, et al. Human PEG1/MEST, an imprinted gene on chromosome 7. Hum Mol Genet. 1997;6:781–786. [DOI] [PubMed] [Google Scholar]
- [28].Chen H, Sun M, Liu J, et al. Silencing of paternally expressed gene 10 inhibits trophoblast proliferation and invasion. PLoS One. 2015;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ono R, Nakamura K, Inoue K, et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat Genet. 2006;38:101–106. [DOI] [PubMed] [Google Scholar]
- [30].Peng W, Chen Y, Luo X, et al. DNA methylation-associated repression of MEST/PEG1 expression contributes to the invasion of extravillous trophoblast cells. Placenta. 2016;46:92–101. [DOI] [PubMed] [Google Scholar]
- [31].Cordeiro A, Neto AP, Carvalho F, et al. Relevance of genomic imprinting in intrauterine human growth expression of CDKN1C, H19, IGF2, KCNQ1 and PHLDA2 imprinted genes. J Assist Reprod Genet. 2014;31:1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dória S, Sifakis S, Soufla G, et al. Gene expression pattern of IGF2, PHLDA2, PEG10 and CDKN1C imprinted genes in spontaneous miscarriages or fetal deaths. Epigenetics. 2010;5:444–450. [DOI] [PubMed] [Google Scholar]
- [33].Cleaton MAM, Edwards CA, Ferguson-Smith AC. Phenotypic outcomes of imprinted gene models in mice: elucidation of pre- and postnatal functions of imprinted genes. Annu Rev Genomics Hum Genet. 2014;15:93–126. [DOI] [PubMed] [Google Scholar]
- [34].Koukoura O, Hwang H-J, Kim I-H, et al. Loss of imprinting and aberrant methylation of IGF2 in placentas from pregnancies complicated with fetal growth restriction. Int J Mol Med. 2011;28:481–487. [DOI] [PubMed] [Google Scholar]
- [35].McMinn J, Wei M, Schupf N, et al. Unbalanced placental expression of imprinted genes in human intrauterine growth restriction. Placenta. 2006;27:540–549. [DOI] [PubMed] [Google Scholar]
- [36].Wu A, Yang D-Y, Liu Y-D, et al. Expression of TET and 5-HmC in trophoblast villi of women with normal pregnancy and with early pregnancy loss. Curr Med Sci. 2018;38:505–512. [DOI] [PubMed] [Google Scholar]
- [37].Putiri EL, Tiedemann RL, Thompson JJ, et al. Distinct and overlapping control of by the TET proteins in human cancer cells. Genome Biol. 2014;15:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nestor CE, Ottaviano R, Reddington J, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hernandez Mora JR, Sanchez-Delgado M, Petazzi P, et al. Profiling of oxBS-450K 5-hydroxymethylcytosine in human placenta and brain reveals enrichment at imprinted loci. Epigenetics. 2018;13:182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development. 2012;139:1895–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Monk D, Arnaud P, Apostolidou S, et al. Limited evolutionary conservation of imprinting in the human placenta. Proc Natl Acad Sci. 2006;103:6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lewis A, Mitsuya K, Umlauf D, et al. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet. 2004;36:1291–1295. [DOI] [PubMed] [Google Scholar]
- [43].Zheng HY, Shi X-Y, Wu F-R, et al. Assisted reproductive technologies do not increase risk of abnormal methylation of PEG1/MEST in human early pregnancy loss. Fertil Steril. 2011;96:84–89.e2. [DOI] [PubMed] [Google Scholar]
- [44].Fogarty NME, Burton GJ, Ferguson-Smith AC. Different epigenetic states define syncytiotrophoblast and cytotrophoblast nuclei in the trophoblast of the human placenta. Placenta. 2015;36:796–802. [DOI] [PubMed] [Google Scholar]
- [45].Sazhenova EA, TV Nikitina, Skryabin NA, et al. Epigenetic status of imprinted genes in placenta during recurrent pregnancy loss. Hum Genet. 2017;53:376–387. [Google Scholar]
- [46].Joana Marques C, Pinho MJ, Carvalho F, et al. DNA methylation imprinting marks and DNA methyltransferase expression in human spermatogenic cell stages. Epigenetics. 2011;6:1354–1361. [DOI] [PubMed] [Google Scholar]
- [47].Klug M, Schmidhofer S, Gebhard C, et al. 5-Hydroxymethylcytosine is an essential intermediate of active DNA demethylation processes in primary human monocytes. Genome Biol. 2013;14:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Marques CJ, Costa P, Vaz B, et al. Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. MHR-Basic Sci Reprod Med. 2008;14:67–73. [DOI] [PubMed] [Google Scholar]
- [49].Khoueiry R, Ibala-Romdhane S, Al-Khtib M, et al. Abnormal methylation of KCNQ1OT1 and differential methylation of H19 imprinting control regions in human ICSI embryos. Zygote. 2012;21:129–138. [DOI] [PubMed] [Google Scholar]
- [50].Li Y, Meng G, Ba LH, et al. Hypomethylation of the P3 promoter is associated with up-regulation of IGF2 expression in human osteosarcoma. Hum Pathol. 2009;40:1441–1447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.