

ABSTRACT
Oral squamous cell carcinoma (OSCC) ranks as the sixth most common carcinoma worldwide, and the third most common carcinoma in developing countries as well. Recently, the aberrant expression of lncRNA CCAT1 has been revealed to play an important role in the development of several cancers. However, its role in OSCC remains unknown. The expression levels of CCAT1 and miR-181a were determined in 15 paired primary OSCC tissues and their adjacent noncancerous tissues and cell lines with qPCR. shRNA against CCAT1 was employed to investigate the impact of CCAT1 on proliferation and metastasis. Then dual luciferase reporter and RIP assays were utilized to study the interaction between CCAT1 and miR-181a. Cells transfected with sh-CCAT1 or treated with miR-181a inhibitor were subjected to western blot to investigate the role of Wnt/β-catenin signaling in CCAT1-mediated proliferation and metastasis. Finally, the role of CCAT1 in OSCC was confirmed with tumor xenografts mice model. CCAT1 was upregulated in OSCC tissues and cell lines. Knockdown of CCAT1 inhibited the proliferation, migration and invasion of OSCC cells, while the cell apoptosis was enhanced. Luciferase and RIP assays revealed that miR-181a was a direct target of CCAT1. Inhibition of miR-181a partially reversed the efficacy of sh-CCAT1. Moreover, sh-CCAT1 inhibited OSCC tissues growth through inhibiting Wnt signaling in a miR-181a-dependent manner in vivo. lncRNA CCAT1 activated Wnt/β-catenin signaling via inhibiting miR-181a, resulting in the cell proliferation, migration and invasion of OSCC, suggesting that CCAT1 might serve as a potential target of OSCC treatment.
Abbreviation: LncRNA: long non-coding RNA; OSCC: oral squamous cell carcinoma; 3’ UTR: 3’ untranslated region; ANOVA: one-way analysis of variance; CDK: cyclin-dependent kinase; ceRNA: competing endogenous RNA; FBS: fetal bovine serum; HGF: human gingival fibroblasts; MAPK: mitogen-activated protein kinase; miRNA: micro RNA; ncRNA: noncoding RNAs; PBS: phosphate-buffered saline; PI3K: phosphatidylinositol 3-kinase
KEYWORDS: Oral squamous cell carcinoma, lncRNA CCAT1, miR-181a, Wnt/β-catenin signaling
Introduction
Approximately 300,000 patients worldwide are diagnosed as Oral squamous cell carcinoma (OSCC) each year, thus OSCC is recognized as one of the top ten malignant tumor globally [1,2]. It demonstrated aggressive syndromes with a 5-year survival of 40–50% [3]. Because of poor early diagnosis and awareness, usually OSCC is not recognized until the advanced stages [3]. The potential risk factors that contribute to OSCC include smoking, alcohol consumption and betel quid chewing [4]. The current treatment of OSCC, such as surgery, chemoradiotherapy, photodynamic therapy, drugs like EGFR inhibitors and COX-2 inhibitors, could cause noticeable esthetic and functional side effects because of non-specific cell death [5,6]. Thus, improving the treatment outcomes of OSCC is urgent and requires a better understanding of the disease progression.
Noncoding RNAs can be divided into small ncRNAs (<200nt) and long ncRNAs (>200nt, lncRNA) [7,8]. Small ncRNAs, including microRNA (miRNAs) have been reported to regulate gene expression and inhibit targeting protein expression post-transcriptionally through targeting complementary sequences in the 3’ untranslated region (3’ UTR) of mRNA [9]. Increasing evidences suggested that dysregulated lncRNAs expression in cancer may be involved in the pathogenesis of cancer and could serve as predictors for outcomes [10–16]. LncRNAs function through regulating the translation of mRNA by competitively combining with miRNA response element. For example, lncRNA CRNDE was reported to promote colorectal cancer cell proliferation and chemoresistance through Wnt/β-catenin signaling, which was regulated by miR-181a [17]. Chang et al. demonstrated that lncRNA MALAT1 functioned as a competing endogenous RNA (ceRNA) to increase the expression of STAT3 through interacting with miR-125b, leading to an promotion of OSCC development [2]. LncRNA CCAT1 is reported to be upregulated in numerous cancers, including colon cancer, gastric cancer and ovarian cancer [18–22]. And CCAT1 is known to promote proliferation, migration and invasion of cancer cells. However, its role in OSCC remains elusive.
In the past decades, emerging evidences demonstrated that miRNAs also contributed to the development of tumors. Lyu et al. reported that miR-181a promoted the cell proliferation in acute lymphoblastic leukemia through activating Wnt signaling [23]. However, miR-9 was reported to deliver inhibitory effect on the proliferation of OSCC through suppressing CXCR4 expression via Wnt/β-catenin signaling [24]. Despite that Shin et al. demonstrated that miR-181a showed tumor suppressive effects in OSCC through downregulating K-ras [25], the function and regulation mechanism of miR-181a is not fully understood.
In the present study, we showed that CCAT1 is aberrantly expressed in OSCC and increased the malignancy of OSCC cells. Notably, the upregulation of CCAT1 was negatively related to the miR-181a level. Further investigations revealed that CCAT1 functioned through targeting on miR-181a, which inhibited Wnt/β-catenin signaling. Our study depicted a lncRNA CCAT1/miR-181a/Wnt signaling pathway regulatory network, providing a novel target for OSCC treatment.
Materials and methods
Clinical sample
Fifteen paired primary OSCC tissues and their adjacent noncancerous tissues were collected from The First Affiliated Hospital of Zhengzhou University according to standard operation procedures. All subjects did not have received chemotherapy or radiotherapy. The research protocol was designed and approved by the ethical committee of the Medical Research Ethics Committee of The First Affiliated Hospital of Zhengzhou University, and informed consent was obtained from all patients.
Cell culture
The normal human gingival fibroblasts (HGF-1) and human OSCC cell lines SCC-9, KB, Cal-27, SCC-25, HN6 and SCC-090 were cultured with appropriate medium supplemented with 10% fetal bovine serum (FBS, GIBCO, Rockville, MD, USA), 100 U/ml penicillin and 100 μg/ml streptomycin in humidified incubator with 5% CO2 at 37°C.
Cell transfection
The short hairpin RNA against CCAT1 (sh-CCAT1) and negative control (sh-NC) were purchased by Genepharma (Shanghai, China). MiRNA mimics/inhibitor (for miR-181a) and negative control oligonucleotides (miR-NC/inhibitor NC) were purchased from RiboBio (Guangzhou, China). Transfection assay was performed using Lipofectamine 3000 (Invitrogen, CA, USA) according to the manufacturer’s protocol.
Cell proliferation detection
Cell proliferation was measured with CCK8 assay (Dojindo, Japan). Briefly, cells transfected with sh-CCAT1 or sh-NC were seeded in 96-well plates at a density of 1 × 104 cells/well. At 24 h, 48 h and 72 h, 10 μl of CCK8 reagent (100 μl medium/well) was added and incubated with cells for 1 h at 37°C, 5% CO2. The absorbance of optical density at 450 nm was detected using a microplate reader Model 680 (Bio-Rad, California, USA).
RNA extraction, reverse transcription and quantitative PCR (qPCR)
Total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA USA) according to the manufacturer’s instructions and transcribed to complementary DNA (cDNA) using the PrimeScript® RT reagent Kit with gDNA Eraser (Takara, Dalian, China). qPCR was employed to determine the relative transcription level of target genes using the SYBR® Premix Ex TaqII kit (Takara). And GAPDH or U6 was used as the internal control. The reactions were performed in triplicate for each cDNA on the Applied Biosystems Step One Plus Real-Time PCR System (Applied Biosystems, Foster city, CA, USA). The relative expressions of CCAT1 and miR-181a were calculated by 2−ΔΔct, according to the widely accepted method.
Crystal violet staining assay
The colony formation capability of OSCC cells was analyzed using a crystal violet staining method. Briefly, cells were rinsed with PBS and fixed with PBS containing 1% glutaraldehyde overnight in 4°C. Then cells were stained with 0.02% crystal violet for 30 min at room temperature. After 3 times water rinses, 70% ethanol was added to extract crystal violet that bound to cells at 4°C. Measure the absorbance of each well at 570 nm using a microplate reader Model 680 (Bio-Rad, California, USA).
Flow cytometry for cell apoptosis analysis
Flow cytometry was employed to detect the cell apoptosis. After treatment, cells were collected with trypsin digestion solution, washed twice with phosphate-buffered saline (PBS) and resuspended in 200 μl PBS. For apoptosis analysis, Annexin V/Dead Cell Apoptosis Kit (Invitrogen, CA, USA) was utilized according to the manufacturer’s instructions. Briefly, 5 µl Annexin V-FITC and 5 µl PI was added to each well and cells were incubated in the dark for 15 min. Then the cell apoptosis was measured using the FACScan flow cytometer (Becton, CA) equipped with CellQuest Software (Becton Dickinson).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde, then 0.1% Triton X-100 was added and permeabilization for 5 min. Samples were blocked for 1 h with 5% bovine serum albumin in PBS. Then cells were incubated with a rabbit anti-β-catenin (Abcam, Cambridge, MA, USA) primary antibody for 1 h at room temperature. Specific secondary antibodies conjugated with RFP fluorochrome (Invitrogen, Carlsbad, CA) were incubated for 1 h at room temperature. After PBS washes, cells were co-stained with DAPI dye and mounted. Images were captured using an optical microscope (DP73, Olympus Corporation, Tokyo, Japan).
Luciferase reporter assay
The dual-luciferase reporter assay was utilized to determine the interaction of miR-181a and CCAT1 and was conducted according to the method described by Han et al. [17]. Briefly, the mutant CCAT1 3’UTR (CCAT1-mut) was prepared with one-step overlap extension PCR. The fragments including the 3’UTR-wild-type (CCAT1-wt) or CCAT1-mut were cloned in XhoI/NotI-digested psiCHECK-2 vector (Promega). Then cells were co-transfected with CCAT1-wt or CCAT1-mut and miR-NC or miR-181a. Twenty-four hours later, cells were harvested, and the firefly and renilla luciferase activities were measured with a dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions.
Rna-binding protein immunoprecipitation (RIP) assay
To elucidate the interaction between CCAT1 and miR-181a, RIP Assay Kit (Millipore, USA) was used for evaluation. Firstly, cells were collected followed by resuspension with RIP lysis buffer (Solarbio). Then, the cell extracts were incubated overnight at 4°C with RIP buffer containing magnetic beads bound to anti-Ago2 antibody or immunoglobulin G (IgG). After 3 times of washing, the magnetic beads were incubated with proteinase K and subsequently total RNA isolation. Lastly, qPCR was performed to analyze the relative enrichment of CCAT1 and miR-181a.
Cell migration and invasion assays
Transwell plates were pre-coated with Matrigel Matrix (BD Biosciences) and cells were seeded onto matched transwell membrane inserts. After 24 to 48 h incubation in humidified incubator with 5% CO2 at 37°C, cells on the lower membrane surface were fixed with 4% paraformaldehyde for 15 min and stained with crystal violet. The experimental procedures of migration assay was similar with invasion without coating with Matrigel Matrix. Five randomly selected fields was counted and cell number was analyzed with microscope software (Olympus, Japan).
Western blotting
Whole cell extracts were extracted using RIPA and protein concentrations were determined by BCA assay. Proteins were separated by SDS-PAGE and transferred to a PVDF membrane. Membranes were blocked with 5% bovine serum albumin in TBST and then incubated with primary antibodies overnight at 4°C. Next day, the membranes were incubated with secondary antibodies for 1 h at room temperature. Primary antibodies against Cyclin D1, CDK4, Bcl-2, Bax, cleaved Caspase3, cleaved Caspase9, phosphorylated-GSK-3β (p-GSK-3β), GSK-3β and c-myc were purchased from Cell signaling technology (Boston, MA, USA) and used with the dilution of 1: 1000. β-actin (1: 5000) antibody was purchased from Sigma Aldrich (St Louis, MO, USA). Finally, signal was detected with a chemiluminescent detection system (Pierce ECL Substrate Western blot detection system, Thermo, Rockford, IL, USA).
Animals
Twelve-week-old female nude BALB/c mice were purchased and housed at the animal research facility of The First Affiliated Hospital of Zhengzhou University. All experiments were conducted in accordance with the protocols approved by the Animal Care and Use Committee of The First Affiliated Hospital of Zhengzhou University.
Tumor xenografts in athymic nude mice
The Cal-27 cells were transfected with negative control sh-NC or sh-CCAT1 and were uspended in PBS at a final concentration of 1 × 108 cells/ml. Then a volume of 0.1 ml of suspended cells were injected subcutaneously into left side of the posterior flank of mice (n = 6/group). Tumor growth was examined every week for at least 5 weeks. At the end of treatment, mice were sacrificed and tumors were isolated and weighed. Tumor volumes were calculated as previously described [26].
Histology
Isolated tumor tissues were embedded in paraffin, and cut into 4 μM sections using a rotary microtome (Leica, Mannheim, Germany). Hematoxylin and eosin (H&E) staining was used to visualize the tissue structure. The dried slices soaked in xylene, dewaxing for 10 min, rehydrated, hematoxylin rinse for 5 min and washed, followed by differentiation with 1% hydrochloric acid alcohol. Then slides were stained with eosin for 30 seconds, dehydrated with gradient alcohol, soaked in xylene 3 times. Finally, mounted slides with neutral gum. For immunohistchmistry (IHC) staining, slides were stained with antibodies against Ki-67 (Cell Signaling Technologies, Boston, MA, USA) before hematoxylin co-staining. The histopathological changes of the tissues were analyzed under an optical microscope (DP73; Olympus Corporation, Tokyo, Japan).
Statistical analysis
All experiments were performed in triplicates and data were presented as mean ± SD. The student’s t-test and one-way analysis of variance (ANOVA) followed by Tukey post hoc test were employed for comparison between two groups and multiple comparison, respectively. The correlation between CCAT1 and miR-181a was determined by Pearson’s correlation analysis. A two-side value of p < 0.05 was considered statistically significant. Graphpad Prim 5 (GraphPad Software, La Jolla, CA, USA) was utilized for statistical analysis.
Results
CCAT1 was highly expressed in OSCC
Firstly, we collected 15 pairs of cancer tissues and corresponding adjacent non-cancer tissues from patients diagnosed as oral squamous cell carcinoma. We found that lncRNA CCAT1 was significantly upregulated in cancer sites compared with the normal (Figure 1(a)). Then we confirmed the expression level of CCAT1 with 7 oral-originated cell lines. In normal human gingival fibroblasts HGF-1, CCAT1 expression was kept in a relatively low base level. However, in cells from oral carcinomas, CCAT1 expression was upregulated, especially in KB and Cal-27 (Figure 1(b)). Thus, we concluded that CCAT1 might be an oncogene in OSCC.
Figure 1.

CCAT1 expression in OSCC tissues and cell lines. (a) The level of CCAT1 was assayed using qPCR in OSCC tissues and the paired normal tissues. (b) Quantified expression level of CCAT1 in normal or tumor oral cell lines. Data are means ± SD. *, p < 0.05, **, p < 0.01.
Knockdown of CCAT1 inhibited the development of OSCC
To understand the role of CCAT1 in OSCC, we constructed plasmids to knockdown the cellular level of CCAT1. The knockdown efficacy was confirmed with qPCR as sh-CCAT1 strongly repressed the CCAT1 expression level compared to the shame knockdown (sh-NC) group (Figure 2(a)). Knockdown of CCAT1 significantly reduced the cell proliferation rate and the colony formation ability in both KB and Cal-27 (Figure 2(b–d)). And flow cytometry revealed that sh-CCAT1 dramatically increased the cell apoptosis in the two cell lines (Figure 2(e–f)). Moreover, an weakened migration and invasion capability of both cells were confirmed with transwell assays in cells transfected with sh-CCAT1 compared to the sh-NC (Figure 2(g–j)). Taken together, these data indicated that knockdown of CCAT1 could inhibite the development of OSCC and reduces its malignancies.
Figure 2.
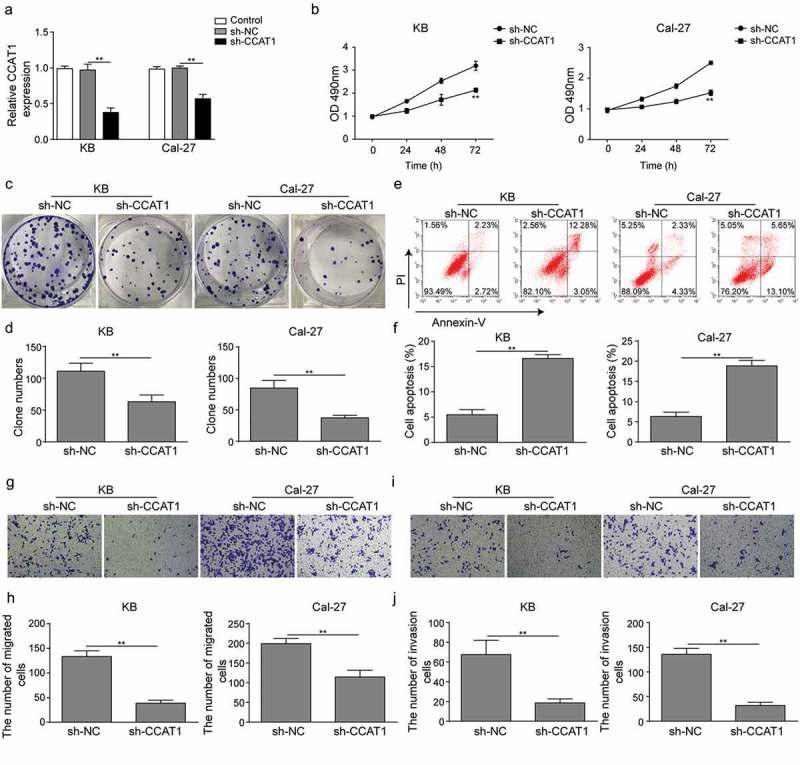
Effects of sh-CCAT1 on OSCC cells. (a) The expression level of CCAT1. Cells were transfected with empty vectors (sh-NC) or sh-RNA of CCAT1 (sh-CCAT1) and the level was assayed using qPCR. (b) Cell proliferation measured with CCK8 assay. (c–d) Colony formation of cells. (e–f) Cell apoptosis detected with flow cytometry. (g–j) Cell migration and invasion measured with transwell assay. Data are means ± SD. *, p < 0.05, **, p < 0.01.
miR-181a is a direct target of CCAT1 in OSCC
CCAT1 was reported to regulate numerous miRNAs in different cancers. To understand whether CCAT1 regulated OSCC through miRNAs, online miRNA target prediction databases were employed. These databases showed that miR-181a could be the potential target of CCAT1. Thus, we measured the expression level of miR-181a in OSCC tissues and cells. qPCR showed a relatively low expression level of miR-181a in OSCC tissues compared to their adjacent noncancerous tissues (Figure 3(a)). Pearson’s correlation analysis indicated a negative correlation between miR-181a and CCAT1 (Figure 3(b)). Similarly, the miR-181a was significantly down-regulated in oral carcinomas cell lines, especially in Cal-27 and KB (Figure 3(c)). Knockdown of CCAT1 in Cal-27 and KB increased miR-181a level, indicating that CCAT1 might regulate the miR-181a negatively (Figure 3(d)).
Figure 3.
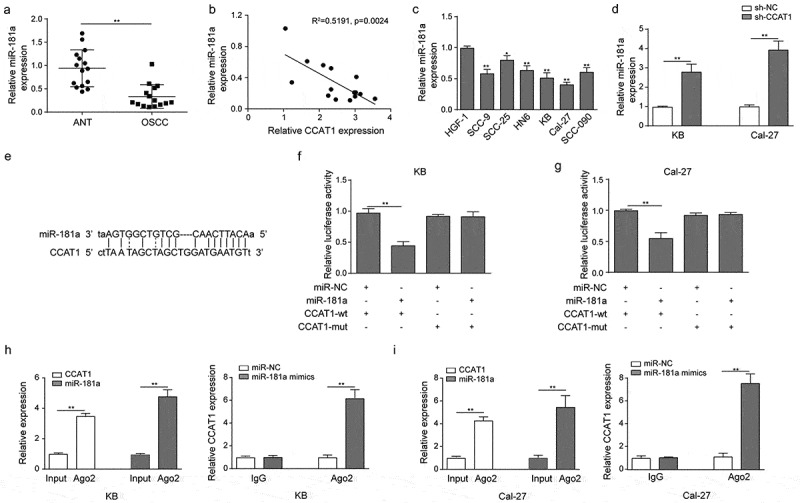
The interaction between CCAT1 and miR-181a. (a) The level of miR-181a in OSCC tissues and the paired normal tissues. Relative expression was assayed using qPCR. (b) The Pearson’s correlation analysis of miR-181a and CCAT1. (c) Quantified expression level of miR-181a in normal or tumor oral cell lines. (d) Quantified level of miR-181a in KB and Cal-27. Cells were transfected with sh-NC or sh-CCAT1 before qPCR. (e–g) Illustration of plasmids sequences for luciferase reporter assay and quantified luciferase read. (h–i) RNA immunoprecipitation was performed to analyze the potentially endogenous interaction between CCAT1 and miR-181a. Data are means ± SD. *, p < 0.05, **, p < 0.01.
Next, we investigated whether miR-181a is the direct target of CCAT1 using the dual-luciferase reporter assay and RIP assay. We mutated the 3’-UTR fragment of CCAT1, by which recognized miR-181a. Then cells were transfected with miR-181a and wild-type CCAT1 (CCAT1-wt) or mutant CCAT1 (CCAT1-mut). Luciferase assay demonstrated that miR-181a significantly reduced the luciferase activity in wild-type KB, while CCAT1 mutation abrogated the suppression effects on luciferase activity. Similar data were observed in Cal-27 (Figure 3(e–g)). Furthermore, RIP results showed that CCAT1 and miR-181a were preferentially enriched in the Ago2 pellets and endogenous CCAT1 was specifically enriched in miR-181a-transfected cells (Figure 3(h–i)). In general, these results made clear that miR-181a is one direct target of CCAT1.
CCAT1 regulated the malignancies of OSCC via miR-181a
Since we found CCAT1 directly binds to miR-181a, the regulation of CCAT1 in combination with miR-181a was investigated. In both Cal-27 and KB, sh-CCAT1 significantly suppressed the colony formation ability, which was conversely enhanced by miR-181a inhibitor. As a rescue experiment, inhibition of miR-181a in CCAT1-knockdown cells partly reversed the decrease of cell colony formation ability (Figure 4(a–b)). Flow cytometry showed that sh-CCAT1 increased the apoptosis in OSCC cells, and simultaneous transfection of shCCAT1 and miR-181a inhibitor partially removed the pro-apoptotic efficacy of sh-CCAT1 alone (Figure 4(c–d)). Meanwhile, western blot revealed a reduction of proteins that regulated cell cycle, including Cyclin D1, CDK4 in sh-CCAT1 group. And pro-apoptotic proteins Bax, cleaved Caspase3 and cleaved Caspase9 were up-regulated, while anti-apoptotic protein Bcl-2 reduced, indicating that apoptosis was enhanced by knockdown of CCAT1. Consistently, the above proteins expression in sh-CCAT1 transfected cells could be partially restored by miR-181a inhibitor (Figure 4(e)). Transwell assay was employed and the data showed that the migration and invasion ability of KB and Cal-27 were impaired by sh-CCAT1 and enhanced by miR-181a inhibitor. And the co-treatment of both demonstrated the inhibitory effects of sh-CCAT1 on migration and invasion could be partially restored by miR-181a inhibitor (Figure 4(f–i)). Overall, these results provided evidences that CCAT1 regulates OSCC cell proliferation, migration and invasion via supressing miR-181a.
Figure 4.
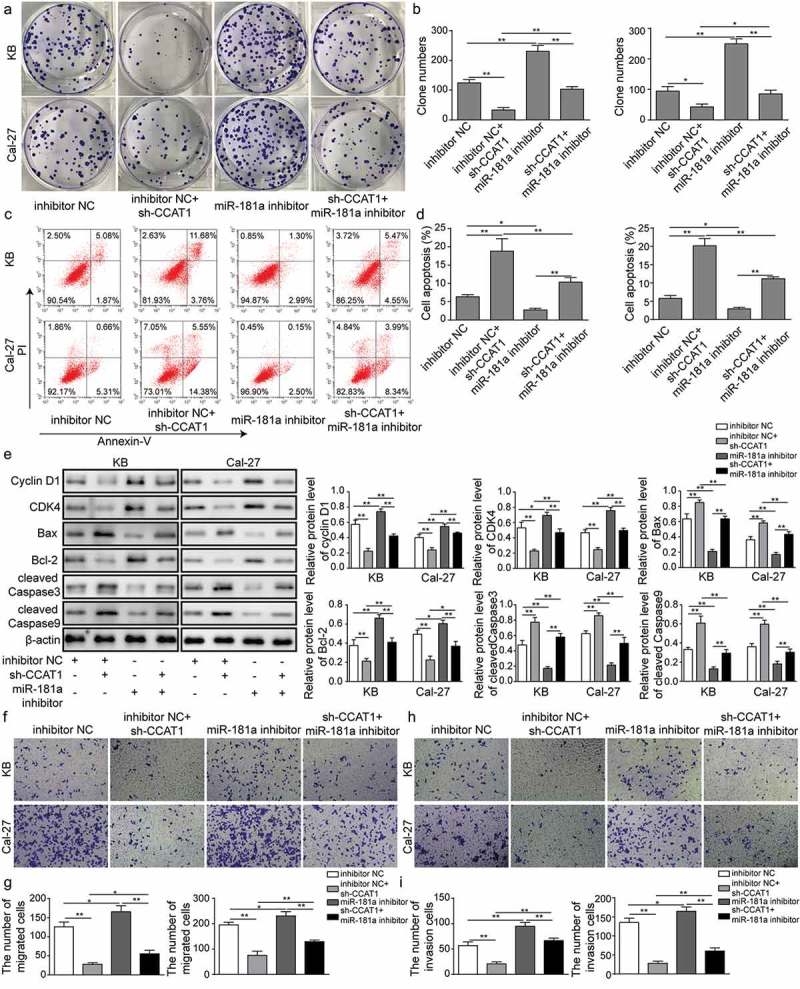
Effects of sh-CCAT1 and miR-181a inhibitor on OSCC cells. (a–b) Colony formation of cells. Cells were transfected with sh-CCAT1 and/or treated with miR-181a inhibitor. (c–d) Cell apoptosis detected with flow cytometry. (e) Expression of proteins that regulated cell cycle and apoptosis determined with western blot. (f–i) Cell migration and invasion measured with transwell assay. Data are means ± SD. *, p < 0.05, **, p < 0.01.
CCAT1 mediated Wnt/β-catenin signaling through miR-181a
miR-181a is known to induce Wnt signaling through downregulating Wnt inhibitory factor-1 (WIF1) expression and facilitate cell proliferation in acute lymphoblastic leukemia [23]. We hypothesized that CCAT1 might regulate the Wnt/β-catenin signaling via miR-181a. To test the hypothesis, we investigated the proteins in Wnt signaling. The expression levels of phosphorylated GSK-3β, β-catenin and c-myc were reduced by sh-CCAT1. miR-181a inhibitor increased their expression, while sh-CCAT1 and miR-181a inhibitor together partially abrogated the changes caused by either alone (Figure 5(a)). To validate the activation of Wnt signaling, cells were stained with β-catenin antibody. In negative control, β-catenin was localized both in cytoplasm and nuclei, but sh-CCAT1 significantly reduced the cytoplasm level of β-catenin. Meanwhile, miR-181a inhibitor activated Wnt as β-catenin was observed to translocated from nuclei to cytoplasm (Figure 5(b)). However, co-treating cells with miR-181a inhibitor and sh-CCAT1 partially recovered the inhibitory effects of sh-CCAT1. In summary, the impact of CCAT1 on OSCC progress may depend on miR-181a/Wnt/β-catenin axis.
Figure 5.

Effects of sh-CCAT1 and miR-181a inhibitor on Wnt signaling. (a) Expression of the proteins in Wnt signaling determined with western blot. (b) β-catenin expression visualized with immunocytochemistry. *, p < 0.05, **, p < 0.01.
CCAT1 promoted the tumor growth via miR-181a in vivo
Finally, we validated the role of CCAT1 with mice received tumor xenografts. Tumor size was measured weekly, cells transfected with sh-CCAT1 demonstrated impaired proliferation capability as tumor size was significantly smaller than the control group from 5-week post transplantation (Figure 6(a–b)). At the end of culture, the tumor weight was determined as well, the tumor weight was dramatically lighter than sh-NC (Figure 6(c)). H&E and IHC staining demonstrated that proliferation marker Ki-67 was significantly reduced in sh-CCAT1 group (Figure 6(d)). Next, we detected the relative expression of CCAT1 and miR-181a in tumor tissues, downregulated CCAT1 was accompanied with miR-181a upregulation, validating that miR-181a was negatively regulated by CCAT1 (Figure 6(e)). Moreover, the phosphorylated GSK-3β, β-catenin, c-myc and cyclin D1 were reduced in sh-CCAT1, indicating that tumor growth was inhibited by sh-CCAT1 through suppressing Wnt signaling (Figure 6(f)). Thus, CCAT1 may be a novel indicator for diagnosis and prognosis of OSCC.
Figure 6.
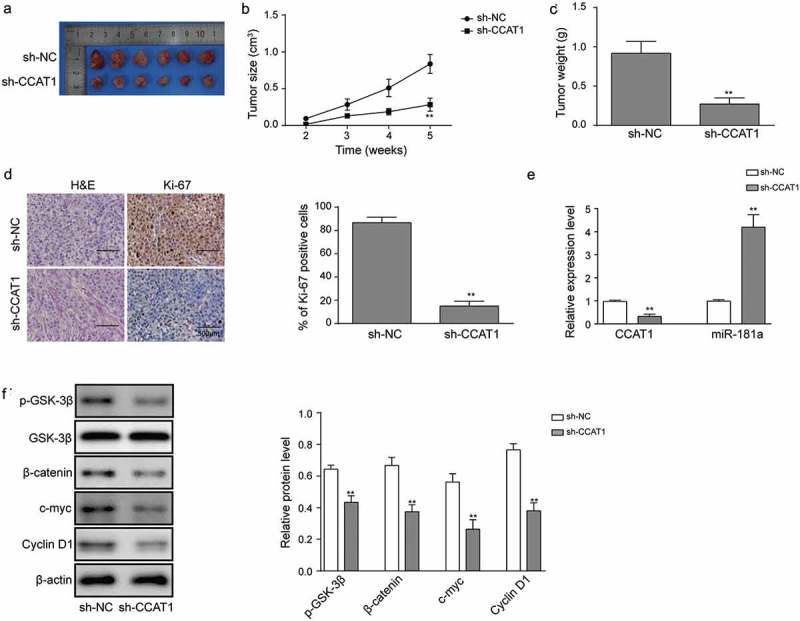
Effects of sh-CCAT1 on tumor growth in vivo. (a) Representative images of tumor tissues after 5-week. (b) Tumor size measured at each week. (c) Tumor weight measured after 5-week. (d) Ki-67 expression visualized with H&E/IHC staining. (e) The level of CCAT1 and miR-181a in OSCC tissues. Relative expression was assayed using qRT-PCR. (f) Expression of the proteins in Wnt signaling determined with western blot. Data are means ± SD. *, p < 0.05, **, p < 0.01.
Discussion
lncRNA CCAT1 is a non-coding RNA with the length of 2628 nt and first reported to be upregulated in colon cancer [27]. Later, aberrant overexpression of CCAT1 was identified in numerous cancers, including gastric cancer, hepatocellular cancer, breast cancer, ovarian cancer and lung cancer [20,21,28–30]. But its role in oral squamous cell carcinoma is not clear. Here we reported that CCAT1 was significantly upregulated in OSCC tissues, as well as OSCC cells. And the overexpression of CCAT1 was closely linked to the proliferation, migration and invasion of cancer cells. Knockdown of CCAT1 reduced the malignancies of OSCC, suggesting that CCAT1 contributes to the progression of OSCC.
In the past decades, increasing studies investigated the mechanism by which CCAT1 exerts its oncogenic efficacy. Li et al. demonstrated that CCAT1 promoted cell proliferation through regulating cell cycle as knockdown of CCAT1 led to the inhibition of gastric cancer cell proliferation and increase the G1 phase cell population [19]. Recently, emerging evidences suggested that CCAT1 could serve as ceRNA, which function abolish the suppressive effects of common miRNA on key targets [31–36]. Deng et al. reported that in hepatocellular carcinoma, CCAT1 interacted with let-7 and removed its suppression on HMGA2 and c-myc, resulting in facilitated cell proliferation [37]. miR-143 was reported to be one target of CCAT1 in thyroid cancer, where CCAT1 activated PI3K/AKT and MAPK signaling through miR-143 and promoted the development of cancer [31]. In the present study, we reported miR-181a as a novel target of CCAT1 in human cancer. miR-181a inhibitor treatment mimicked the efficacy of CCAT1 overexpression as enhanced cell proliferation and invasion were observed compared to the normal cells. And co-treating OSCC cells with sh-CCAT1 and miR-181a inhibitor abrogated the efficacy of sh-CCAT1 alone, indicating that miR-181a is essential to CCAT1-mediated proliferation and invasion.
Previously, miR-181a was reported to show tumor suppressive effect against OSCC by downregulating K-ras oncogene, which encodes small guanosine triphosphatase that regulates cell proliferation, differentiation, survival [25]. Han et al. demonstrated that miR-181a was regulated by another lncRNA CRNDE and modulated the colorectal cancer cell proliferation and chemoresistance through Wnt signaling [17]. Wnt signaling is a well-known signaling that modulate cell proliferation, differentiation and survival. Proteins in Wnt signaling are recognized as prognosis biomarkers or therapy targets [38–40]. Our further studies demonstrated that in OSCC, miR-181a functions through Wnt signaling, consistent with previous report [23]. Inhibiting miR-181a promoted the activation of Wnt signaling, resulting in enhanced tumor growth.
To conclude, we reported an upregulation of CCAT1, together with reduced miR-181a in OSCC tissues and cell lines. Further studies unraveled that CCAT1 promoted tumor cell proliferation and invasion through miR-181a-mediated Wnt signaling.
Funding Statement
This work was supported by National Natural Science Foundation of China (No. 31400839).
Acknowledgments
We would like to give our sincere gratitude to the reviewers for their constructive comments.
Data availability statement
All data generated or analyzed during this study are included in this published article.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Yu T, Li C, Wang Z, et al. Non-coding RNAs deregulation in oral squamous cell carcinoma: advances and challenges. Clin Transl Oncol. 2016;18(5):427–436. [DOI] [PubMed] [Google Scholar]
- [2].Chang SM, Hu WW.. Long non-coding RNA MALAT1 promotes oral squamous cell carcinoma development via microRNA-125b/STAT3 axis. J Cell Physiol. 2018;233(4):3384–3396. [DOI] [PubMed] [Google Scholar]
- [3].Markopoulos AK. Current aspects on oral squamous cell carcinoma. Open Dent J. 2012;6:126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gharat SA, Momin M, Bhavsar C. Oral squamous cell carcinoma: current treatment strategies and nanotechnology-based approaches for prevention and therapy. Crit Rev Ther Drug Carrier Syst. 2016;33(4):363–400. [DOI] [PubMed] [Google Scholar]
- [5].Villaret AB, Cappiello J, Piazza C, et al. Quality of life in patients treated for cancer of the oral cavity requiring reconstruction: a prospective study. Acta Otorhinolaryngol Ital. 2008;28(3):120–125. [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang B, Huang H-Z, Pan C-B, et al. Aesthetic and functional radical surgery in young patients with stage one or two tongue cancer: a preliminary report. J Craniomaxillofac Surg. 2011;39(3):209–214. [DOI] [PubMed] [Google Scholar]
- [7].Jiang L, Yu X, Ma X, et al. Identification of transcription factor-miRNA-lncRNA feed-forward loops in breast cancer subtypes. Comput Biol Chem. 2018;78:1–7. [DOI] [PubMed] [Google Scholar]
- [8].Hao NB, He Y-F, Li X-Q, et al. The role of miRNA and lncRNA in gastric cancer. Oncotarget. 2017;8(46):81572–81582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4(3):143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1(5):391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ji N, Wang Y, Bao G, et al. LncRNA SNHG14 promotes the progression of cervical cancer by regulating miR-206/YWHAZ. Pathol Res Pract. 2019;215(4):668–675. [DOI] [PubMed] [Google Scholar]
- [12].Zhang S, Zhang X, Sun Q, et al. LncRNA NR2F2-AS1 promotes tumourigenesis through modulating BMI1 expression by targeting miR-320b in non-small cell lung cancer. J Cell Mol Med. 2019;23(3):2001–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bo H, Fan L, Li J, et al. High expression of lncRNA AFAP1-AS1 promotes the progression of colon cancer and predicts poor prognosis. J Cancer. 2018;9(24):4677–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao J, Liu L, Li G, et al. LncRNA GAS5 confers the radio sensitivity of cervical cancer cells via regulating miR-106b/IER3 axis. Int J Biol Macromol. 2018;126:994–1001. [DOI] [PubMed] [Google Scholar]
- [15].Huang P, Zhang L, Li T, et al. lncRNA profile study reveals the mRNAs and lncRNAs associated with docetaxel resistance in breast cancer cells. Sci Rep. 2018;8(1):17970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yao N, Yu L, Zhu B, et al. LncRNA GIHCG promotes development of ovarian cancer by regulating microRNA-429. Eur Rev Med Pharmacol Sci. 2018;22(23):8127–8134. [DOI] [PubMed] [Google Scholar]
- [17].Han P, Li J-W, Zhang B-M, et al. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR-181a-5p-mediated regulation of Wnt/beta-catenin signaling. Mol Cancer. 2017;16(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu J, Ding D, Jiang Z, et al. Long non-coding RNA CCAT1/miR-148a/PKCzeta prevents cell migration of prostate cancer by altering macrophage polarization. Prostate. 2019;79(1):105–112. [DOI] [PubMed] [Google Scholar]
- [19].Li N, Jiang K, Fang LP, et al. Knockdown of long noncoding RNA CCAT1 inhibits cell growth, invasion and peritoneal metastasis via downregulation of Bmi-1 in gastric cancer. Neoplasma. 2018;65(5):736–744. [DOI] [PubMed] [Google Scholar]
- [20].Coni P, Madeddu A, Kuqi L, et al. LncRNA colon cancer-associated transcript 1 (CCAT1) in ovarian cancer. Eur Rev Med Pharmacol Sci. 2018;22(6):1525–1527. [DOI] [PubMed] [Google Scholar]
- [21].McCleland ML, Mesh K, Lorenzana E, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J Clin Invest. 2016;126(2):639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].He X, Tan X, Wang X, et al. C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biol. 2014;35(12):12181–12188. [DOI] [PubMed] [Google Scholar]
- [23].Lyu X, Li J, Yun X, et al. miR-181a-5p, an inducer of Wnt-signaling, facilitates cell proliferation in acute lymphoblastic leukemia. Oncol Rep. 2017;37(3):1469–1476. [DOI] [PubMed] [Google Scholar]
- [24].Yu T, Liu K, Wu Y, et al. MicroRNA-9 inhibits the proliferation of oral squamous cell carcinoma cells by suppressing expression of CXCR4 via the Wnt/beta-catenin signaling pathway. Oncogene. 2014;33(42):5017–5027. [DOI] [PubMed] [Google Scholar]
- [25].Shin KH, Bae SD, Hong HS, et al. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem Biophys Res Commun. 2011;404(4):896–902. [DOI] [PubMed] [Google Scholar]
- [26].Yang YT, Wang Y-F, Lai J-Y, et al. Long non-coding RNA UCA1 contributes to the progression of oral squamous cell carcinoma by regulating the WNT/beta-catenin signaling pathway. Cancer Sci. 2016;107(11):1581–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li DY, Chen W-J, Luo L, et al. Prospective lncRNA-miRNA-mRNA regulatory network of long non-coding RNA LINC00968 in non-small cell lung cancer A549 cells: a miRNA microarray and bioinformatics investigation. Int J Mol Med. 2017;40(6):1895–1906. [DOI] [PubMed] [Google Scholar]
- [28].Yao YX, Xu BH, Zhang Y. CX-3543 promotes cell apoptosis through downregulation of CCAT1 in colon cancer cells. Biomed Res Int. 2018;2018:9701957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li Y, Jing F, Ding Y, et al. Long noncoding RNA CCAT1 polymorphisms are associated with the risk of colorectal cancer. Cancer Genet. 2018;222–223:13–19. [DOI] [PubMed] [Google Scholar]
- [30].Lai Y, Chen Y, Lin Y, et al. Down-regulation of LncRNA CCAT1 enhances radiosensitivity via regulating miR-148b in breast cancer. Cell Biol Int. 2018;42(2):227–236. [DOI] [PubMed] [Google Scholar]
- [31].Yang T, Zhai H, Yan R, et al. lncRNA CCAT1 promotes cell proliferation, migration, and invasion by down-regulation of miR-143 in FTC-133 thyroid carcinoma cell line. Braz J Med Biol Res. 2018;51(6):e7046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [32].Cui B, Li B, Liu Q, et al. lncRNA CCAT1 promotes glioma tumorigenesis by sponging miR-181b. J Cell Biochem. 2017;118(12):4548–4557. [DOI] [PubMed] [Google Scholar]
- [33].Arunkumar G, Murugan AK, Prasanna Srinivasa Rao H, et al. Long non-coding RNA CCAT1 is overexpressed in oral squamous cell carcinomas and predicts poor prognosis. Biomed Rep. 2017;6(4):455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lv L, Jia JQ, Chen J. The lncRNA CCAT1 upregulates proliferation and invasion in melanoma cells via suppressing miR-33a. Oncol Res. 2018;26(2):201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dou C, Sun L, Jin X, et al. Long non-coding RNA colon cancer-associated transcript 1 functions as a competing endogenous RNA to regulate cyclin-dependent kinase 1 expression by sponging miR-490-3p in hepatocellular carcinoma progression. Tumour Biol. 2017;39(4):1010428317697572. [DOI] [PubMed] [Google Scholar]
- [36].Chen L, Wang W, Cao L, et al. Long non-coding RNA CCAT1 acts as a competing endogenous RNA to regulate cell growth and differentiation in acute myeloid leukemia. Mol Cells. 2016;39(4):330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Deng L, Yang S-B, Xu -F-F, et al. Long noncoding RNA CCAT1 promotes hepatocellular carcinoma progression by functioning as let-7 sponge. J Exp Clin Cancer Res. 2015;34:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Santiago L, Daniels G, Wang D, et al. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am J Cancer Res. 2017;7(6):1389–1406. [PMC free article] [PubMed] [Google Scholar]
- [39].Li L, Wang Z, Wang R, et al. Overexpression of beta-catenin induces cisplatin resistance in oral squamous cell carcinoma. Biomed Res Int. 2016;2016:5378567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ogoshi K, Kasamatsu A, Iyoda M, et al. Dickkopf-1 in human oral cancer. Int J Oncol. 2011;39(2):329–336. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.