

ABSTRACT
Growing evidence suggests that aberrant epigenetic regulation of gene function is strongly related to the genesis of cancer. Unlike genetic mutations, the ability to reprogram the epigenetic landscape in the cancer epigenome is one of the most promising target therapies in both treatment and reversibility of drug resistance. Epigenetic alterations in cancer development and progression may be the basis for the individual variation in drug response. Thus, this review focuses on the emerging area of pharmaco(epi)genomics, specifically highlighting epigenetic reprogramming during tumorigenesis and how epigenetic markers are targeted as a therapy (epidrugs) and the clinical implications of this for cancer treatment.
KEYWORDS: Epigenetics, reprogramming, pharmacology, epidrugs, cancer, treatment
Epigenetic mechanism and cancer
Epigenetic alterations are the main mechanisms underlying many human diseases, especially growth and developmental disorders, including Beckwith-Wiedemann (BWS) [1], Silver-Russell [2], Prader-Willi and Angelman [3] syndromes. Due to their important role in growth-related pathways, epigenetic mutations (epimutations) participate in the earliest stages of neoplasia and have been increasingly recognized as a hallmark of cancer [4,5]. Cancer is a group of diseases characterized by the dysregulation of important pathways that control cellular processes involved in DNA repair, cell survival, proliferation and mortality [6]. Cell transformation, tumor progression and metastasis are orchestrated by a complex and intriguing network of interactions where genomic and epigenomic mutations, especially in oncogenes and tumor suppressor genes, and environmental factors induce malignancy and tumorigenesis [7,8].
The complexity, and multifactorial nature of cancer present a great challenge for diagnosis and treatment. The epigenetic basis of cancer development revolutionized the field of cancer genetics in the post-genomic era, providing new targets for cancer therapy [8,9]. First described by Waddington (1942), the epigenetic landscape represents the developmental pathways which a cell can take from an undifferentiated to a differentiated state. Epigenetics determine how the genotype of an organism responds to the environment in a coordinated way, and this response can be fixed independent of mutational effects. Epigenetic mechanisms modify chromatin structure and confer a differential gene expression program without changing the DNA sequence, which is stably maintained during cell growth [10–12].
Epimutations in cancer cells change the structure and stability of the genome and have been proposed as driver mutation in tumor initiation and together with genetic lesions, they propagate carcinogenesis [13]. It is important to highlight the dynamic and reversible nature of epigenetic modifications, which can establish new epigenetic programs according to each cell type. Epigenetic reprogramming is strongly affected by environmental factors, which play an important role in the acquisition and maintenance of epigenetic marks, especially DNA methylation [14,15]. Environmentally induced epigenetic changes may explain the sporadic origin of most cancers, since evidences shows nearly 10% of some cancers has genetically inherited [16]. Therefore, epigenetics changes in cancer are a growing area of interest, and the possibility of reprogramming the cancer epigenome is emerging as a promising therapy for cancer treatment as well as in regenerative medicine. Thereby, the main mechanisms and elements involved in epigenetic alterations in cancer are discussed below.
DNA modification
DNA methylation is the main epigenetic modification, and a well-known epigenetic marker, in which the cytosine (C) in a dinucleotide CpG (cytosine-phosphate-guanine) is covalently modified by a methyl group (–CH3) resulting in a ‘fifth’ base in the DNA sequence, the 5-methylcytosine (5mC) [17,18]. This modification is catalyzed by DNA methyltransferase (DNMT) enzymes [19]. DNMT1 is responsible for maintaining existing DNA methylation, and the de novo methyltransferases DNMT3A and DNMT3B act on hemimethylated and/or unmethylated CpG sites, establishing new methylation patterns [20]. DNA methylation regulates important biological processes in the mammalian genome, including transcription and post-transcriptional processing, post-translational modifications, chromatin remodeling, genomic imprinting, X-chromosome inactivation and repression of repetitive DNA elements [21,22].
The CpG-rich domains in the vertebrate genome, known as CpG islands (CGI), are predominantly nonmethylated, such as in repetitive sequences, transcription start sites (TSS) and promoter regions. However, the global genome is CG-deficient and consequently highly methylated (hypermethylated), which is important for chromosomal stability [23]. Thus, hypomethylation and hypermethylation can occur at the same time depending on the genomic region and can have different effects on the disease phenotype. Global loss of genomic DNA methylation (hypomethylation) is often found in several types of tumor and is related to genomic instability, DNA damage, and reactivation of transposons and retroviruses [21,24,25]. Whereas aberrant CpG methylation in promoter regions can inactivate tumor suppressor genes or activate proto-oncogenes [26].
DNA hypomethylation of proto-oncogenes and the activation of transposable elements are related to carcinogenesis and metastasis [27–29]. Also, global hypomethylation contributes to loss of imprinting (LOI), such as in the IGF2/H19 imprinting control region (ICR) that promotes overexpression of the IGF2 gene, which is suggested to be an epigenetic progenitor mutation that leads to tumorigenesis [30]. Mutations in DNMTs are often related to overall DNA hypomethylation and malignant transformation [31]. However, hypermethylation and silencing of tumor suppressor genes, e.g., RAS, BRCA1, BCL2, occurs in many neoplastic cells and also promotes malignancy [9].
Recently, hydroxymethylation has been implicated in cancer development. 5-mC is oxidized to 5-hydroxymethylcytosine (5-hmC) in a reaction catalyzed by a methylcytosine oxygenase and involving members of the TET protein family, in an active DNA demethylation process. Reduced TET gene expression and consequent lower hydroxymethylation levels were reported in human cancers [32]. In addition to 5-mC and 5-hmC methylation, the N6-methyladenine (N6-mA) DNA modification is a repressive epigenetic marker which silences long interspersed nuclear element (LINE) transposons [33]. Higher levels of N6-mA have been recently reported in glioblastoma, however the role of this epigenetic marker in human cancer is poorly understood [34].
Histone modification
Other important elements of epigenetic regulation are histone proteins, a central component of the nucleosome that are responsible for the stable maintenance of repressive chromatin. The nucleosome is composed of strands of DNA wrapped around two copies each of the histones H2A, H2B, H3 and H4 in an octameric core with a linker histone, H1. Chromatin is composed of repeating subunits of nucleosomes, and has the potential to define the state in which genetic information is structured within a cell. Conformational changes in the chromatin structure confer a particular arrangement of the genome, in a condensed or non-condensed state, that alters and controls gene expression [35].
Epigenetic information in the histone core modifies the chromatin structure for transcription activation or repression [36]. Histone post-translational modification refers to the addition of chemical groups to the tails of these nucleosome-forming proteins. The most common chemical modifications are methylation and acetylation, which generally occur next to promoter and enhancer regions [37]. These modifications are catalyzed by multiple enzymes, such as histone acetyltransferases (HATs) and deacetylases (HDACs), as well as histone methyltransferases (HMTs) and demethylases (HDMs) that can modify amino acid residues in the histone tail. In the mammalian genome, the ‘histone code’, plays an essential role in regulating the accessibility of genomic DNA, thereby controlling gene expression [38]. Histones can also be modified by phosphorylation, ubiquitination, and other atypical modifications such as citrullination, ADP-ribosylation, deamination, formylation, O-GlcNAcylation, propionylation, butyrylation, crotonylation and proline isomerization [39]. Errors in histone post-transcriptional modification may alter gene expression patterns and cause human disease due to changes (epimutations) at the chromatin level [40].
Aberrant epigenetic histone modifications have been implicated in cancer pathogenesis and may impact clinical outcomes, especially in the prognosis and invasiveness of cancer [41,42]. Reduced levels of lysine acetylation (H3K9ac, H3K18ac, H4K12ac) and methylation (H3K4me2, H4K20me3) and arginine methylation (H4R3me2) are associated with a poor prognosis in breast cancer [41]. A decreased H3K4me2 level is also a marker of shorter survival times, while a reduced level of H3K4me3 is associated with a better prognosis [43,44].
Post-translational histone modifications are important regulators of cellular growth and development, including cell cycle control, replication, DNA damage response, cell signaling and metabolic pathways and gene expression, and are potentially useful as biomarkers and a therapeutic target for cancer treatment [42,45].
Non-coding RNA and RNA modification
As mentioned, epigenetic elements are versatile and have an important role in cancer development. Among these elements, non-coding RNAs (ncRNA) have recently received special attention, due to the discovery that they participate in several epigenetic mechanisms controlling gene expression. This control is exerted at the level of chromatin structure modulation, transcriptional regulation and post-transcriptional modification [46]. In eukaryotic cells, a greater proportion of the genome is transcribed than translated, since approximately 2–3% of the genome encodes functional proteins whereas 80% is transcribed into non-coding RNA [47,48]. Non-coding RNAs are classified according to length, basically divided into micro RNAs (miRNA, 19–31nt) and long ncRNAs (lncRNA, >200nt) [49].
In addition to DNA methylation and histone modification, ncRNAs have also been implicated in carcinogenesis as oncogenic drivers and/or tumor suppressors in a complex regulatory network. These regulatory ncRNAs elements interact through physical ligation of a target molecule [50]: RNA-DNA interactions, such as the lncRNA XIST that modulates chromatin structure in the X-chromosome inactivation process [51]; RNA-RNA interactions that can regulate stability or protein translation; and RNA-protein interactions, (ribonucleoproteins) such as the telomerase complex that is responsible for telomere elongation and maintenance [52]. Several miRNAs have been reported to have aberrant expression in many different types of cancer, including leukemia [52] and colorectal cancer [53], and therefore represent a new molecular marker and therapeutic strategy to regulate the epigenetic machinery for chromatin remodeling and gene expression [52]. Regarding lncRNA, the imprinted H19 gene has a controversial role in cancer initiation, progression and metastasis, and is a key gene in tumorigenesis [54].
A newly discovered role of methylation is in the modulation of messenger RNA (mRNA). The most important and known epitranscriptome modification is N6-methyladenosine (m6A) [55], which represents 0.4% of all adenine residues in mammalian mRNA [56]. It is mainly located in 3’ untranslated regions (3’UTRs), stop codons, translation start sites [57] and also in long exons [58] of protein-coding genes. These positions suggest that m6A has a biological function related to splicing control, translation and stability [59]. Therefore, the proper function of RNA methyltransferases (METTL3 and METTL4) and demethylases (FTO, ALKBH5 and YTHDF2) properly keeps the RNA methylation pattern in the transcripts. Disruption in these processes with the increased activity or the inactivation of this enzymes contribute to the initiation of tumorigenesis and cancer development [60].
Epigenetic reprogramming and drug therapy in cancer
The epigenetic landscape in cancer gives to the tumor cell a peculiar phenotype that may trigger the carcinogenesis process, followed by genetic alterations, that propagate tumorigenesis [26]. These alterations make it difficult to understand the malignancy, make a prognosis, and prescribe the most effective course of treatment and surveillance, because the (epi)genetic variation of each cancer type or even individual patient may be essential for the development of new therapeutic strategies in a precision medicine era [13,61]. Due to its role in the modulation of gene expression and chromosomal stability, epigenetic disruption has considerable consequences for many physiological and pathological processes, resulting in epigenetic disorders such as cancer and mental retardation [62]. However, unlike genetic mutations, epigenetic alterations are potentially reversible and have great plasticity, as the epigenome can be reprogrammed [26]. The possibility of reprogramming the epigenome and changing the cell landscape, represents a new and promising therapeutic strategy.
Epigenetic drugs (epidrugs) are chemical compounds that alter DNA and chromatin structure, promoting the disruption of transcriptional and post-transcriptional modifications, mainly by regulating the enzymes necessary for their establishment and maintenance, reactivating epigenetically silenced tumor-suppressor and DNA repair genes [63,64]. The design of therapeutic strategies involving epidrugs is a growing field of drug discovery, which focuses on the cancer epigenome to develop pharmacological compounds which could restore a ‘normal’ epigenetic landscape [63]. Epidrugs act on the enzymes necessary for the maintenance and establishment of epigenetic modifications, with the main strategy being the inhibition of DNMTs and HDACs [63] (Figure 1). These drugs have implications for the regulation and dysregulation of physiological and pathological processes, and the epigenetic modifications induced can control the chronological and spatial expression of genes [65]. Targeting epigenetic marks has great potential to provide molecular biomarkers for diagnosis [62] and treatment options for cancer therapy, since they are strictly linked to the type of tumor and stage of the disease, as well as to individual genetic variation, as in personalized medicine [66,67].
Figure 1.
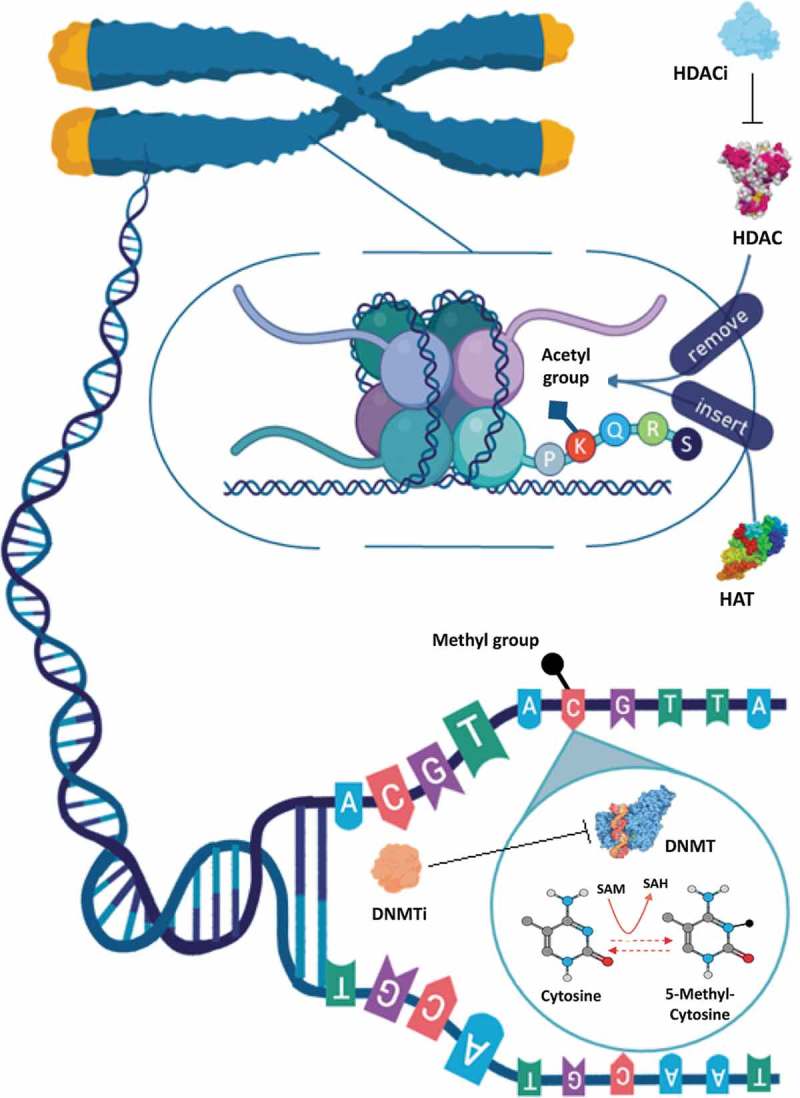
Schematic representation of epigenetic modifications and epidrugs action. The modification of cytosine to 5-methylcytosine (5mC) DNA methylation is catalyzed by DNA methyltransferases (DNMTs). DNMTs require S-Adenosylmethionine (SAM) as a cofactor for methyl group donation that is converted to S-adenosylhomocysteine (SAH). DNA methylation at a promoter region is related to transcriptional silencing. The DNMT inhibitors (DNMTi) block DNA methylation and restore the function of aberrantly silenced genes. At the chromatin level, histone post-translational acetylation is catalyzed by histone acetyltransferases (HATs) and deacetylation by histone deacetylases (HDACs). Such modifications occur into amino acid residues (such as lysine) at histone tails. HDAC inhibitors (HDACi) induce the acetylation, an epigenetic modification associated with transcriptional activation and gene expression
Studies in vitro have demonstrated that there are many implications under the regulation of tumor suppressor genes and DNA repair enzymes [68]. More recently, the great potential of using a combination of epigenetic drugs in in vitro assays [69,70], and also in clinical trials for chemotherapy treatment [71,72] has been shown. Epigenetic therapy has also been associated with cell differentiation, cell cycle arrest and cell death, energy metabolism and other cellular issues involving a large number of genes and proteins [69,70,73,74]. These features play an important role in cancer development and help in understanding the evolution of some cancer hallmarks including progression, survival and regulation [75].
Several epigenetic therapies have been approved by the U.S. Food and Drug Administration (FDA) and used for cancer treatment. However, new epidrugs compounds are constantly being evaluated for cytotoxicity, pharmacological parameters and to better understand their mechanism of action in pre-clinical studies (in vitro and in vivo), as well as in clinical trials for the development and release of new therapies. A Web of Science database search for the terms ‘epigenetics and drug discovery processes’ (accessed in June 2019) currently lists 2772 publications, which has increased over the years (Supplementary Table S1). These studies include epigenetic compounds and their mechanisms of regulation, and the different pharmacological phases such as; in vitro (1217), in vivo (952), pre-clinical (465) and clinical trials (phases I, II, III and IV) (137). According to clinical trial database (Figure 2) most investigations involving epigenetic drugs are related to cancer detection, treatment and prognosis. In addition to the clinical studies in progress, six new epidrugs and multi-drug therapies have been approved by the FDA (Table 1).
Figure 2.
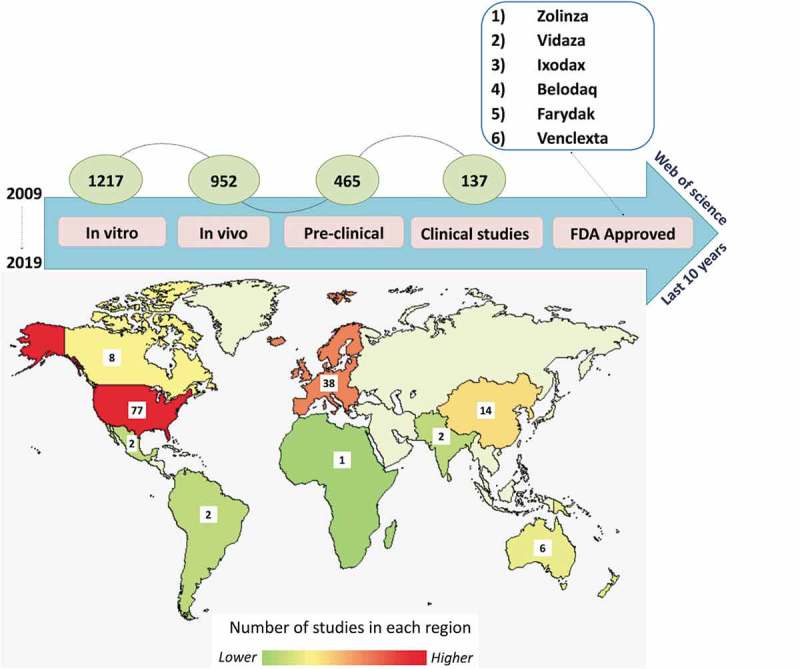
Progress of epigenetic drugs in different stages of pharmacological studies. The number of epigenetic studies from 2009 to 2019 according to ‘web of science’ database is showed according to each study phase: in vitro (1217) in vivo (952), preclinical (465) and clinical (137). The map represents the global distribution of clinical epigenetic drug studies according to clinical trial database (www.clinicaltrial.org, june, 2019), and the box show the exact number of studies.
Table 1.
Epigenetic compounds approved by Food and Drug Administration (FDA), treatment indication and epigenetic mechanism of action.
| FDA approved Drug (commercial name) |
Year | Treatment | Mechanism of action | Structural formula |
|---|---|---|---|---|
| Azacitidine+ decitabine or low-dose cytarabine (Venclexta) |
2018 | Acute myeloid leukemia | DNTMi | 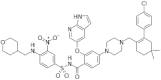 |
| Panobinostat + Bortezomib + Dexamethasone (Farydak) |
2015 | Multiple myeloma | HDACi | 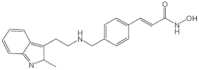 |
| Belinostat (Belodaq) |
2014 | Peripheral T Cells Lymphoma | HDACi | 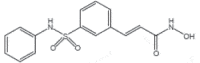 |
| Valproic Acid | 2010 | Antidepressive Neurologic disorders |
HDACi | 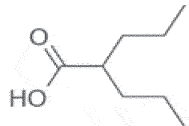 |
| Romidepsin (Ixodax) |
2009 | Cutaneous T-cell lymphoma | HDACi | 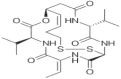 |
| 5 Azacitidine (Vidaza) |
2009 | Myelodysplastic syndrome | DNTMi | 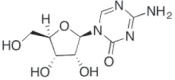 |
| Vorinostat (Zolinza) |
2006 | Cutaneous T-cell lymphoma | HDACi | 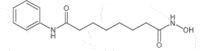 |
DNMTi, DNA methyltransferase inhibitor; HDACi, Histone desacetilase inhibitor
The first DNMT inhibitors (DNMTi) or hypomethylating agents approved by the FDA were cytidine analogs, i.e., Azacitidine (Vidaza) and Decitabine (5-aza- and 5-aza-2’-deoxycytidine) respectively, and were primarily used to treat myelodysplastic syndrome (MDS). The mechanism of action of these compounds is to inhibit DNA methylation, by acting especially on DNMT1 enzymes (Figure 1), leading to a hypomethylated profile and restoring the function of aberrantly silenced genes [76]. As well as their clinical use in MDS treatment, these cytidine analogs have also been used to treat other hematologic malignancies and solid tumors [9].
The majority of compounds approved by the FDA, including the first epidrug approved for cancer treatment, Vorinostat® (2006), are HDAC inhibitors (HDACi), which show promising results in selectivity index and toxicity. The hydroxamic acids are the predominant class of HDACi, which includes: Vorinostat, used for the treatment of cutaneous T-cell lymphoma; Panobinostat and Belinostat, used for treatment of multiple myeloma; and Romidepsin, also used to treat lymphoma [77–79]. The general feature of an HDACi, such as Vorinostat, is to enable histone acetylation to increase by blocking the catalytic sites of HDACs (Figure 1). This dramatically alters cellular acetylation patterns, and causes growth arrest and death in a wide range of transformed cells [80].
Recently, new therapies have emerged, including the use of microRNA [81], multi-drug combinations [82] and immunotherapy [83], helping to improve cancer treatment and reduce drug resistance. Many studies have reported a great potential of combining epidrugs with chemotherapy or immunotherapy, both in vitro [69,70] and in clinical trials [71,72] (Table 2), with increased apoptosis induction as compared to each compound alone [73,74] and tumor growth inhibition in xenograft nude mice [69].
Table 2.
In vitro effect of epigenetic drugs association.
| Drug/Compound | Cell Lines | Cell Cycle Arrested | Target | Findings | Reference |
|---|---|---|---|---|---|
| Vorinostat/Etoposide/Cisplatin | H209 H143 |
G0/G1 and S | H3 histone Aceltil alfa-tubulin Thymidylate synthase PARP | In vivo study showed significant tumor growth inhibition in xenograft nude mice | [69] |
| Trichostatin A/ Lomustine (CCNU) |
U87MG | G0/G1 S and G2/M |
pChk1 Chk1 Chk2 Caspase-3 PARP H3 histone | The combined treatment showed a greater efficiency compared to single-drug treatment. | [74] |
| Panobinostat + topoisomerase inhibitors | Hela SiHa |
G0/G1 | H3 histone Bcl-xL p21 Caspase-9 |
Panobinostat has a synergistic effect in association with Topotecan and Etoposide | [73] |
ncRNA targets and ncRNA based therapy
Non-coding RNA may be a target for inhibitory drugs or, the opposite, since its supplementation can restore endogenous function in cases of downregulation or gene disruption [84]. The latter case has been observed when ncRNAs act as tumor suppressors and their lack contributes to the disruption of important pathways related to carcinogenesis [85,86]. Despite RNA diversity, microRNAs are the main targets for research and clinical applications [87]. As an example, the microRNA family MiR-34 is silenced in a wide variety of cancers and seems to regulate important genes related to cell cycle control and proliferation, such as MYC [88,89]. Synlogic Therapeutics, a biopharmaceutical company, synthesized the first microRNA similar to mir-34, MRX34 [89]. During the preclinical phase, MRX34 was efficiently delivered (using nanocarriers) into bone metastasis and colon cancer in xenograft models, resulting in a significant decrease of tumor size in vivo [90,91].
The future of anticancer miRNA replacement therapy comes with several potential advantages. Due its small size, miRNAs are well suited for cell delivery. In addition, they are associated with the RNA-induced silencing complex (RISC) to modulate translation in the cytoplasm [92]. In addition to miRNAs, long non-coding RNAs (lncRNAs) are emerging as new targets for cancer development arrest. Several lncRNAs work as natural antisense transcripts (NATs) to genes of therapeutic interest; both cis- and trans-acting lncRNAs have been reported as important modulators that induce transcriptional silencing [93]. A second strategy deals with the use of Antisense Oligonucleotides (ASO), which are short DNA sequences complementary to the RNA of interest. Although ASOs still need to be approved as an anticancer therapy, MALAT-1 ASOs have already shown efficacy against breast cancer in a preclinical mouse MMTV-PyMT model [94].
Given the importance of miRNA in the process of tumorigenesis, miRNAs associated with cancer (oncomirs) are therapeutic targets [95]. Anti-miR oligonucleotides are capable of binding with the guide strand of miRNAs. They have been shown to be a powerful tool for understanding microRNA action and have potential therapeutic use [96]. Multiple miRNA inhibition therapy has also been used, for example, a combination of a low dose of the chemotherapeutic drug Sunitinib and anti-miRNA oligonucleotides (targeting overexpressed miR-21, miR-221/222 and miR-10) achieved a notable synergistic antitumor effect in pancreatic ductal adenocarcinoma cells, indicating that a combinatory approach might be of great importance for therapeutic applications in cancer treatment [97,98].
Epigenetic drugs in multi-drug treatment
The use of epigenetic drugs seems to have the ability to interfere in many biological processes [99] and may play an important role in multiple drug therapy [100–102]. A big challenge to the success of traditional chemotherapy treatment is chemoresistance, possibly due to genetic mutations that affect cell cycle regulation, apoptosis and cellular adhesion in cancer cells [103]. The use of epidrugs with other compounds in a multi-drug therapy, have shown an improvement in cancer treatment, including tumor remission, reduction of chemoresistance, increased life expectancy and a reduction in adverse events [101,102,104].
The epidrugs Panobinostat and Decitabine have been combined with a traditional chemotherapy, Temozolomide, to treat resistant melanoma, and showed great improvements in disease stabilization and remission [71]. A study performed by Pili et al (2017) using Vorinostat and the antibody drug Bevacizumab, showed higher tolerance effects, increasing the efficacy of renal carcinoma treatment when compared to the single drug therapy [102]. In contrast, treatments using the compounds Azacitidine, Valproic Acid and Carboplatin separately decrease tumor size, however a combined therapy of these three drugs showed some adverse effects such as fatigue, neutropenia and altered mental status [104]. Epigenetic combinatory drugs are also being tested with promising results in some models of leukemia [105], including in clinical trials [106,107]. Despite the seeming promise of these therapies, the synergistic action of the drugs, the pathways involved, the mechanism of action and the clinical effects are poorly understood and require more investigation [108].
In addition to traditional chemotherapy and new therapies using epidrugs, immunotherapy has emerged as a potent adjunct to cancer treatment, especially monoclonal antibodies, which in association with chemotherapy are first-line treatment for some types of tumors, such as non–small cell lung cancer, breast cancer, colorectal carcinoma and non-Hodgkin lymphoma [109,110]. The 2018 Nobel Prize in Physiology or Medicine, awarded to James P. Allison and Tasuku Honjo for their discovery of CTLA4 and PD1 immune checkpoints (two important targets for therapeutic monoclonal antibodies), recognized the significant advance in cancer treatment obtained through immunotherapy. Currently, dozens of monoclonal antibodies targeting diverse tumor markers are used in the medical clinic, especially for the treatment of cancer and autoimmune diseases [111].
Interestingly, recent studies have shown a close relationship between epidrugs and the enhancement of immune checkpoint therapy. As mentioned, the actions of DNMTs and HDACs are generally associated with transcriptional repression, thereby the use of epidrugs such as DNMTi and HDACi can change the expression of genes involved in immune checkpoint pathways, potentiating the immunotherapeutic effects [112]. Further, drugs that promote epigenetic changes can improve antigen presentation by tumor cells, boosting CD8 T-cell killing [113]. In this context, Woods et. al., (2015) showed that HDACi upregulated PD-1 in melanoma cells in vitro and in a mouse model, and when combined with a PD-1–blocking antibody, the treatment decreased tumor progression and improved survival in mice [114]. Thereafter, several clinical trials have been performed using anti-PD1/PD-L1 or anti-CTL4 antibodies in combination with DNMTi or HDACi drugs like Entinostat, Vorinostat, and Azacytidine [115,116].
Other antibody-based therapies have emerged as important breakthroughs in cancer treatment, such as antibody-drug conjugates, bispecific antibodies, and recently the CAR-T cell technology, approved by the FDA for the treatment of non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL). Although promising, these therapies are expensive, which impairs the universalization of treatment. Thus, a better understanding of the epigenetic changes related to the onset and progression of cancer is essential for the development of more cost-effective and efficient anti-tumor drugs and treatments.
Conclusions and perspectives
The complex and multifactorial nature of carcinogenesis, in which different pathways can be activated or silenced, both for tumor formation and in response to treatment, direct the research to systemic studies which take into account the complex networks of interactions and regulatory mechanisms existing in the cellular context, to explain the biological phenomena present in living organisms [117]. Since cancer is a complex multifactorial disease, understanding the genomic and epigenomic alterations, the cell microenvironment and how it can be reprogrammed, combined with individual information is the most promising therapeutic strategy for cancer treatment in the personalized medicine and precision oncology era [118].
There is a close relationship between epigenetic mechanisms and cancer progression. The original discovery of DNA methylation, and the more recent studies of RNA methylation and the uses of different RNA molecules for epigenetic reprogramming, have been essential for the development of more cost-effective and efficient anti-tumor drugs and treatments. The use of epidrugs in single- or multi-drug therapy, or immunotherapy are promising avenues for clinical studies. While clinical studies have been carried out using diverse combinations of traditional and new chemotherapeutics for cancer treatment, some results expected from these trials include increased life expectancy and fewer side effects from chemotherapy. On the other hand, the scientific community is still attempting to understand the mechanism of action of these epidrugs when administered in single- or multi-drug therapies. The in vitro assays with translational feedback from clinical reports are great tools for illustrating and understanding some of the proposed mechanisms, associating epigenetic modification with the modulation of phenotypes.
Funding Statement
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Programa de Excelência Acadêmica (CAPES-PROEX) and Programa de Apoio à Pós-Graduação (CAPES-PROAP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Grant number: 437,037/2018-5, CLMF; 440755/2018-2, CP; 439019/2018-4, GPF) and Fundação Cearense de Apoio ao Desenvolvimento Cientí;fico e Tecnológico (FUNCAP, PPSUS PP3-0118-00070.01.00).
Acknowledgments
The authors are grateful to the members of the Experimental Oncology Laboratory (LOE) at Drug Research and Development Center (NPDM), Federal University of Ceara (UFC). We also thank Kalil Andrade Mubarac Romcy for helping in the design of the Figure 1. We would like to thank Glen McGugan, Dr Daniel Pascoalino Pinheiro and Dr Catherine J. E. for the English review of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Horike S, Mitsuya K, Meguro M, et al. Targeted disruption of the human LIT1 locus defines a putative imprinting control element playing an essential role in Beckwith-Wiedemann syndrome. Hum Mol Genet. 2000;9:2075–2083. [DOI] [PubMed] [Google Scholar]
- [2].Eggermann T, Schönherr N, Meyer E, et al. Epigenetic mutations in 11p15 in Silver-Russell syndrome are restricted to the telomeric imprinting domain. J Med Genet. 2006;43:615–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Buiting K. Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:365–376. [DOI] [PubMed] [Google Scholar]
- [4].Murphy SK, Jirtle RL. Imprinting evolution and the price of silence. BioEssays. 2003;25:577–588. [DOI] [PubMed] [Google Scholar]
- [5].Das R, Hampton DD, Jirtle RL. Imprinting evolution and human health. Mamm Genome. 2009;20:563–572. [DOI] [PubMed] [Google Scholar]
- [6].Burstein HJ, Krilov L, Aragon-ching JB, et al. Clinical cancer advances 2017 : annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2017;35:1342–1368. [DOI] [PubMed] [Google Scholar]
- [7].Werner HMJ, Mills GB, Ram PT. Cancer systems biology: A peek into the future of patient care? Nat Rev Clin Oncol. 2014;11:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Park JW, Han PJ. Targeting epigenetics for cancer therapy. Arch Pharm Res. 2019;42:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bennett RL, Licht JD. Targeting epigenetics in cancer. Annu Rev Pharmacol Toxicol. 2018;58:187–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Waddington CH. Canalization of development and the inheritance of acquired characters. Nature. 1942. [DOI] [PubMed] [Google Scholar]
- [11].Waddington CH. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183:1654–1655. [DOI] [PubMed] [Google Scholar]
- [12].Haig D. The (dual) origin of epigenetics Cold spring harbor symposia on quantitative biology. 2004;69:67–70. [DOI] [PubMed] [Google Scholar]
- [13].Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. [DOI] [PubMed] [Google Scholar]
- [14].Yamada Y, Yamada Y. The causal relationship between epigenetic abnormality and cancer development: in vivo reprogramming and its future application. Proc Jpn Acad Ser B. 2018;94:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sogabe Y, Seno H, Yamamoto T, et al. Unveiling epigenetic regulation in cancer, aging, and rejuvenation with in vivo reprogramming technology. Cancer Sci. 2018;109:2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zervos EE, Tanner SM, Osborne DA, et al. de la Chapelle A. Differential gene expression in patients genetically predisposed to pancreatic cancer. J Surg Res. 2006;135:317–322. [DOI] [PubMed] [Google Scholar]
- [17].Widschwendter M. 5-Methylcytosine - The fifth base of DNA: the fifth wheel on a car or a highly promising diagnostic and therapeutic target in cancer? Dis Markers. 2007;1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Geiman TM, Muegge K. DNA methylation in early development. Mol Reprod Dev. 2010;77:105–113. [DOI] [PubMed] [Google Scholar]
- [19].Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000. [DOI] [PubMed] [Google Scholar]
- [20].Lyko F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92. [DOI] [PubMed] [Google Scholar]
- [21].Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139–3155. [DOI] [PubMed] [Google Scholar]
- [22].Kurihara Y, Kawamura Y, Uchijima Y, et al. Maintenance of genomic methylation patterns during preimplantation development requires the somatic form of DNA methyltransferase 1. Dev Biol. 2008;313:335–346. [DOI] [PubMed] [Google Scholar]
- [23].Deaton A, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Brothman AR, Swanson G, Maxwell TM, et al. Global hypomethylation is common in prostate cancer cells: A quantitative predictor for clinical outcome? Cancer Genet Cytogenet. 2005;156:31–36. [DOI] [PubMed] [Google Scholar]
- [25].Hon GC, Hawkins RD, Caballero OL, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zelic R, Fiano V, Grasso C, et al. Global DNA hypomethylation in prostate cancer development and progression: a systematic review. Prostate Cancer Prostatic Dis. 2014;18:1–12. [DOI] [PubMed] [Google Scholar]
- [28].Agrawal A, Murphy RF, Agrawal DK. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20:711–721. [DOI] [PubMed] [Google Scholar]
- [29].Hur K, Cejas P, Feliu J, et al. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer. Gut. 2014;63:635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Leick MB, Shoff CJ, Wang EC, et al. Loss of imprinting of IGF2 and the epigenetic progenitor model of cancer. Am J Stem Cells. 2012;1:59–74. [PMC free article] [PubMed] [Google Scholar]
- [31].Zhang W, Xu J. DNA methyltransferases and their roles in tumorigenesis. Biomark Res. 2017;5:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Haffner MC, Chaux A, Meeker AL, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget. 2011;2:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wu TP, Wang T, Seetin MG, et al. DNA methylation on N 6 -adenine in mammalian embryonic stem cells. Nature. 2016;532:329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xie Q, Wu TP, Gimple RC, et al. N 6 -methyladenine DNA modification in glioblastoma. Cell Press. 2018;175:1228–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Müller O, Kepper N, Schöpflin R, et al. Changing chromatin fiber conformation by nucleosome repositioning. Biophys J. 2014;107:2141–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Barth TK, Imhof A. Fast signals and slow marks: the dynamics of histone modifications. Cell Press. 2010;35:618–626. [DOI] [PubMed] [Google Scholar]
- [37].Wang Z, Schones DE, Zhao K. Characterization of human epigenomes. Curr Opin Genet Dev. 2009;19:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications Sarah. Biochim Biophys Acta Biophys Incl Photsynth. 2014;1839:728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tweedie-Cullen RY, Brunner AM, Grossmann J, et al. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS One. 2012;38:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dieker J, Muller S. Epigenetic histone code and autoimmunity. Clin Rev Allergy Immunol. 2010;39:78–84. [DOI] [PubMed] [Google Scholar]
- [41].Elsheikh SE, Green AR, Rakha EA, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009;69:3802–3809. [DOI] [PubMed] [Google Scholar]
- [42].Shanmugam MK, Arfuso F, Arumugam S, et al. Role of novel histone modifications in cancer. Oncotarget. 2018;9:11414–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu BL, Cheng JX, Zhang X, et al. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol Biomarkers Prev. 2010;19:2888–2896. [DOI] [PubMed] [Google Scholar]
- [44].Li S, Shen L, Chen K-N. Association between H3K4 methylation and cancer prognosis: A meta-analysis. Thorac Cancer. 2018;9:794–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kaypee S, Sudarshan D, Shanmugam MK, et al. Aberrant lysine acetylation in tumorigenesis: implications in the development of therapeutics. Pharmacol Ther. 2016;162:98–119. [DOI] [PubMed] [Google Scholar]
- [46].Penna I, Gigoni A, Costa D, et al. The inhibition of 45A ncRNA expression reduces tumor formation, affecting tumor nodules compactness and metastatic potential in neuroblastoma cells. Oncotarget. 2017;8:8189-8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen J, Miao Z, Xue B, et al. Long non-coding RNAs in urologic malignancies: functional roles and clinical translation. J Cancer. 2016;7:1842–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Barata P, Sood AK, Hong DS. RNA-targeted therapeutics in cancer clinical trials: current status and future directions. Cancer Treat Rev. 2016;50:35–47. [DOI] [PubMed] [Google Scholar]
- [49].Qi P, Zhou X, Du X. Circulating long non-coding RNAs in cancer: current status and future perspectives. Mol Cancer. 2016;15:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2017;18:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mohammad F, Mondal T, Kanduri C. Epigenetics of imprinted long noncoding RNAs. Epigenetics. 2009;4:277–286. [PubMed] [Google Scholar]
- [52].Memari F, Joneidi Z, Taheri B, et al. Epigenetics and Epi-miRNAs: potential markers/therapeutics in leukemia. Biomed Pharmacother. 2018;106:1668–1677. [DOI] [PubMed] [Google Scholar]
- [53].Hernández R, Sánchez-Jiménez E, Melguizo C, et al. Downregulated microRNAs in the colorectal cancer: diagnostic and therapeutic perspectives. BMB Rep. 2018;51:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Raveh E, Matouk IJ, Gilon M, et al. The H19 Long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol Cancer. 2015;14:184–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Visvanathan A, Somasundaram K. mRNA traffic control reviewed: N6-Methyladenosine (m6A) takes the driver’s seat. BioEssays. 2017;40:1–12. [DOI] [PubMed] [Google Scholar]
- [56].Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975;4:379–386. [DOI] [PubMed] [Google Scholar]
- [57].Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. [DOI] [PubMed] [Google Scholar]
- [59].Zhao BS, Wang X, Beadell AV, et al. M6 A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542:475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ke S, Alemu EA, Mertens C, et al. A majority of m6 A residues are in the last exons, allowing the potential for 3’ URT regulation. Genes Dev. 2015;29:2037–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Dugger SA, Platt A, Goldstein DB. Drug development in the era of precision medicine. Nat Rev Drug Discov. 2018;17:183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Egger G, Liang G, Aparicio A, et al. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. [DOI] [PubMed] [Google Scholar]
- [63].Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. [DOI] [PubMed] [Google Scholar]
- [64].Salarinia R, Sahebkar A, Peyvandi M, et al. Epi-drugs and Epi-miRs: moving beyond current cancer therapies. Curr Cancer Drug Targets. 2016;16:773–788. [DOI] [PubMed] [Google Scholar]
- [65].Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. [DOI] [PubMed] [Google Scholar]
- [66].Mund C, Lyko F. Epigenetic cancer therapy: proof of concept and remaining challenges. BioEssays. 2010;32:949–957. [DOI] [PubMed] [Google Scholar]
- [67].Jones PA, Issa JPJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17:630–641. [DOI] [PubMed] [Google Scholar]
- [68].Alcazar O, Achberger S, Aldrich W, et al. Epigenetic regulation by decitabine of melanoma differentiation in vitro and in vivo. Int J Cancer. 2012;131:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pan C-H, Chang Y-F, Lee M-S, et al. Vorinostat enhances the cisplatin-mediated anticancer effects in small cell lung cancer cells. BMC Cancer. 2016;16:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hsu CC, Chang WC, Hsu TI, et al. Suberoylanilide hydroxamic acid represses glioma stem-like cells. J Biomed Sci. 2016;23:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xia C, Leon-Ferre R, Laux D, et al. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother Pharmacol. 2014;74:691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Voso MT, Santini V, Finelli C, et al. Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin Cancer Res. 2009;15:5002–5007. [DOI] [PubMed] [Google Scholar]
- [73].Wasim L, Chopra M. Panobinostat induces apoptosis via production of reactive oxygen species and synergizes with topoisomerase inhibitors in cervical cancer cells. Biomed Pharmacother. 2016;84:1393–1405. [DOI] [PubMed] [Google Scholar]
- [74].Staberg M, Michaelsen SR, Rasmussen RD, et al. Inhibition of histone deacetylases sensitizes glioblastoma cells to lomustine. Cell Oncol. 2017;40:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hanahan D, Weinberg RA. Hallmarks of Cancer: the Next Generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- [76].Kaminskas E, Farrell AT, Wang Y-C, et al. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10:176–182. [DOI] [PubMed] [Google Scholar]
- [77].Yazbeck VY, Grant S. Romidepsin for the treatment of non-Hodgkin’s lymphoma. Expert Opin Investig Drugs. 2015;24:965–979. [DOI] [PubMed] [Google Scholar]
- [78].Iyer SP, Foss FF. Romidepsin for the treatment of peripheral T-cell lymphoma. Oncologist. 2015;20:1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Coiffier B, Pro B, Prince HM, et al. Romidepsin for the treatment of relapsed/refractory peripheral T-cell lymphoma: pivotal study update demonstrates durable responses. J Hematol Oncol. 2014;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280:201–210. [DOI] [PubMed] [Google Scholar]
- [81].Garofalo M, Croce CM. MicroRNAs as therapeutic targets in chemoresistance. Drug Resist Updat. 2013;16:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Glasgow MDK, Chougule MB. Recent developments in active tumor targeted multifunctional nanoparticles for combination chemotherapy in cancer treatment and imaging. J Biomed Nanotechnol. 2015;11:1859–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Crea F, Nobili S, Paolicchi E, et al. Epigenetics and chemoresistance in colorectal cancer: an opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist Updat. 2011;14:280-96. [DOI] [PubMed] [Google Scholar]
- [84].Vitiello M, Tuccoli A, Poliseno L. Long non-coding RNAs in cancer: implications for personalized therapy. Cell Oncol (Dordr). 2015;38:17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Inamura K. Major tumor suppressor and oncogenic non-coding RNAs: clinical relevance in lung cancer. Cells. 2017;6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shao Q, Xu J, Deng R, et al. Long non-coding RNA-422 acts as a tumor suppressor in colorectal cancer. Biochem Biophys Res Commun. 2018;495:539–545. [DOI] [PubMed] [Google Scholar]
- [87].Ji W, Sun B, Su C. Targeting microRNAs in cancer gene therapy. Genes (Basel). 2017;8:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hosseinahli N, Aghapour M, Duijf PHG, et al. Treating cancer with microRNA replacement therapy: A literature review. J Cell Physiol. 2018;233:5574–5588. [DOI] [PubMed] [Google Scholar]
- [89].Farooqi AA, Tabassum S, Ahmad A. MicroRNA-34a: a versatile regulator of myriads of targets in different cancers. Int J Mol Sci. 2017;18:2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Gaur S, Wen Y, Song JH, et al. Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget. 2015;6:29161–29177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tazawa H, Tsuchiya N, Izumiya M, et al. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci. 2007;104:15472–15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Shah MY, Ferrajoli A, Sood AK, et al. microRNA therapeutics in cancer — an emerging concept. EBioMedicine. 2016;12:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Renganathan A, Felley-Bosco E. Long noncoding RNAs in cancer and therapeutic potential Adv Exp Med Biol. 2017;1008:199–222. [DOI] [PubMed] [Google Scholar]
- [94].Jadaliha M, Zong X, Malakar P, et al. Functional and prognostic significance of long non-coding RNA MALAT1 as a metastasis driver in ER negative lymph node negative breast cancer. Oncotarget. 2016;7:40418-40436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Svoronos AA, Engelman DM, Slack FJ. OncomiR or tumor suppressor? The duplicity of microRNAs in cancer. Cancer Res. 2016;76:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jahanafrooz Z, Motamed N, Bakhshandeh B. Effects of miR-21 downregulation and silibinin treatment in breast cancer cell lines. Cytotechnology. 2017;69:667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Song J, Ouyang Y, Che J, et al. Potential value of miR-221/222 as diagnostic, prognostic, and therapeutic biomarkers for diseases. Front Immunol. 2017;8:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Fan M, Li X, Jiang W, et al. A long non-coding RNA, PTCSC3, as a tumor suppressor and a target of miRNAs in thyroid cancer cells. Exp Ther Med. 2013;5:1143–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Prachayasittikul V, Prathipati P, Pratiwi R, et al. Exploring the epigenetic drug discovery landscape. Expert Opin Drug Discov. 2017;12:345–362. [DOI] [PubMed] [Google Scholar]
- [100].Momparler RL, Idaghdour Y, Marquez VE, et al. Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk Res. 2012;36:1049–1054. [DOI] [PubMed] [Google Scholar]
- [101].Shah K, Mirza S, Desai U, et al. Synergism of curcumin and cytarabine in the down regulation of multi drug resistance genes in acute mieloyd leukemia. Anticancer Agents Med Chem. 2016;16:128–135. [DOI] [PubMed] [Google Scholar]
- [102].Pili R, Liu G, Chintala S, et al. Combination of the histone deacetylase inhibitor vorinostat with bevacizumab in patients with clear-cell renal cell carcinoma: A multicentre, single-arm phase I/II clinical trial. Br J Cancer. 2017;116:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Alimbetov D, Askarova S, Umbayev B, et al. Pharmacological targeting of cell cycle, apoptotic and cell adhesion signaling pathways implicated in chemoresistance of cancer cells. Int J Mol Sci. 2018;19:1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Falchook GS, Fu S, Naing A, et al. Methylation and histone deacetylase inhibition in combination with platinum treatment in patients with advanced malignancies. Invest New Drugs. 2013;31:1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Liu Z, Ding K, Li L, et al. A novel histone deacetylase inhibitor chidamide induces G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomed Pharmacother. 2016;83:1032–1037. [DOI] [PubMed] [Google Scholar]
- [106].Grishina O, Schmoor C, Döhner K, et al. DECIDER: prospective randomized multicenter phase II trial of low-dose decitabine (DAC) administered alone or in combination with the histone deacetylase inhibitor valproic acid (VPA) and all-trans retinoic acid (ATRA) in patients >60 years with acute. BMC Cancer. 2015;15:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kim E, Ilagan JO, Liang Y, et al. SRSF2 mutations contribute to myelodysplasia through mutant-specific effects on exon recognition. Cancer Cell. 2015;27:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yang AS, Yang BJ. The failure of epigenetic combination therapy for cancer and what it might be telling us about DNA methylation inhibitors. Epigenomics. 2016;8:9–12. [DOI] [PubMed] [Google Scholar]
- [109].Karachaliou N, Fernandez-Bruno M, Rosell R. Strategies for first-line immunotherapy in squamous cell lung cancer: are combinations a game changer? Transl Lung Cancer Res. 2018;7:S198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Jakovljevic M, Gutzwiller F, Schwenkglenks M, et al. Costs differences among monoclonal antibodies-based first-line oncology cancer protocols for breast cancer, colorectal carcinoma and non-Hodgkin’s lymphoma. J Buon. 2014;19:1111–1120. [PubMed] [Google Scholar]
- [111].Kimiz-Gebologlu I, Gulce-Iz S, Biray-Avci C. Monoclonal antibodies in cancer immunotherapy. Mol Biol Rep. 2018;45:2935–2940. [DOI] [PubMed] [Google Scholar]
- [112].Chiappinelli KB, Zahnow CA, Ahuja N, et al. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016;76:1683–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gallagher SJ, Shklovskaya E, Hersey P. Epigenetic modulation in cancer immunotherapy. Curr Opin Pharmacol. 2017;35:48–56. [DOI] [PubMed] [Google Scholar]
- [114].Woods DM, Sodre AL, Villagra A, et al. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res. 2015;3:1375–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Mazzone R, Zwergel C, Mai A, et al. Epi-drugs in combination with immunotherapy: a new avenue to improve anticancer efficacy. Clin Epigenetics. 2017;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol. 2017;87:227–239. [DOI] [PubMed] [Google Scholar]
- [117].Welch GR, Clegg JS. From protoplasmic theory to cellular systems biology: a 150-year reflection. Am J Physiol Physiol. 2010;298:C1280–90. [DOI] [PubMed] [Google Scholar]
- [118].Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.