

Abstract
Background:
Evidence suggests that single nucleotide polymorphisms (SNPs) in genes involved in serotonergic signaling and stress response pathways moderate associations between PTSD and cortical thickness. This study examined a genetic regulator of these pathways, the PPM1F gene, which has also been implicated in mechanisms of stress responding and is differentially expressed in individuals with comorbid PTSD and depression compared to controls.
Methods:
Drawing from a sample of 240 white non-Hispanic trauma-exposed veterans, we tested 18 SNPs spanning the PPM1F gene for association with PTSD and cortical thickness.
Results:
Analyses revealed six PPM1F SNPs that moderated associations between PTSD symptom severity and cortical thickness of bilateral superior frontal and orbitofrontal regions as well as the right pars triangularis (all corrected p’s<0.05) such that greater PTSD severity was related to reduced cortical thickness as a function of genotype. A whole-cortex vertex-wise analysis using the most associated SNP (rs9610608) revealed this effect to be localized to a cluster in the right superior frontal gyrus (cluster-corrected p<0.02).
Limitations:
Limitations of this study include the small sample size and that the sample was all-white, non-Hispanic predominately male veterans.
Conclusions:
These results extend prior work linking PPM1F to PTSD and suggest that variants in this gene may have bearing on the neural integrity of the prefrontal cortex (PFC).
Keywords: PPM1F, PTSD, cortical thickness, prefrontal cortex, genetics, MRI
Introduction1
Posttraumatic stress disorder (PTSD) is a psychiatric disorder that occurs following exposure to trauma and is defined by reliving of traumatic experiences (through unwanted upsetting memories, nightmares and flashbacks), avoidance of trauma-related stimuli, trauma-related arousal and reactivity (including increased irritability, aggression, startle, hypervigilance, and sleep disturbances) and negative thoughts and feelings (American Psychiatric Association, 2013). Structural neuroimaging studies of PTSD have shown that the disorder is associated with reduced cortical volume and thickness (Bing et al., 2013; Corbo et al., 2014; Geuze et al., 2008; Hayes et al., 2017; Lindemer, Salat, Leritz, McGlinchey, & Milberg, 2013; Liu, Li, Luo, Lu, & Yin, 2012; Woodward et al., 2006; Wrocklage et al., 2017). However, it is unclear to what extent such structural differences in the brain reflect pre-existing vulnerabilities, consequences of PTSD, or some combination of these factors, although previous longitudinal and monozygotic twin studies provide some insight on this question. For example, Lyoo et al. (2011) found that increased dorsolateral prefrontal cortex thickness in trauma-exposed individuals early after trauma was associated with later PTSD symptom reductions and recovery. Utilizing the twin study design, Kasai et al. (2008) found that combat-exposed Vietnam era twins with PTSD exhibited decreased gray matter density compared to their combat-unexposed twins as well as when compared to combat-exposed veterans without PTSD and their unexposed twins, suggesting that some morphological reductions may be consequences of PTSD.
Another potential way to shed light on this question is through psychiatric neuroimaging-genetic studies, which harness both neuroimaging and genetic approaches to identify genetic loci and biological pathways that influence brain morphology directly and/or in association with psychiatric disorders. Using this neuroimaging-genetic approach, our group has shown that the association between PTSD and cortical thickness may be moderated by genetic factors (Miller et al., 2015; Sadeh et al., 2016; Wolf et al., 2017). Decreased cortical thickness may be the result of several mechanisms including alterations in serotonergic signaling and the stress response. For example, prior work has shown positive associations between cortical thickness and 5-HT1A receptor concentration in several cortical regions (Pillai et al., 2018). Further, treatment with selective serotonin reuptake inhibitors (SSRIs) has been associated with enhanced cortical thickness over a two-month period in individuals with major depressive disorder (MDD; Bartlett et al., 2018). Additionally, negative associations between levels of the primary stress hormone cortisol and thickness of frontal regions have been reported (Kremen et al., 2010; Liu et al., 2015). Collectively, these studies suggest that cortical thickness is mediated by serotonergic and stress response pathways which may be further influenced by genetic factors.
The protein phosphatase gene PPM1F is an intriguing genetic candidate that plays a broad role in both the stress response and serotonergic signaling and may be particularly relevant to PTSD-associated disruptions in the prefrontal cortex (PFC). This gene is within the protein phosphatase 2C (PP2C) family of serine/threonine protein phosphatases, which are known to negatively regulate cell stress response pathways. PPM1F is involved in the dephosphorylation and subsequent inactivation of calmodulin-dependent protein kinase II (CAMK-II) (Harvey, Banga, & Ozer, 2004; Ishida, Kameshita, & Fujisawa, 1998), which is critically important for serotonergic signaling (Moyano, Del Río, & Frechilla, 2004). Research investigating PPM1F in the stress response has shown that PPM1F is differentially regulated in the medial prefrontal cortex (mPFC) and amygdala of mice following the immobilization on boards (IMO) stress behavioral protocol (Wingo et al., 2018). Specifically, Wingo et al. (2018) showed that after stress exposure, PPM1F mRNA was decreased in the mPFC and increased in the amygdala of mice. Interrogating the translational relevance of this finding, the authors found that individuals with comorbid PTSD and depression in one cohort and individuals with anxiety in a separate cohort, had significantly lower blood PPM1F expression compared to controls without psychopathology, and that a single nucleotide polymorphism (SNP) in PPM1F (rs17759843) was significantly associated with comorbid PTSD and depression pathology. Furthermore, using the BrainCloud dataset (Colantuoni et al., 2011), Wingo et al. (2018) reported associations between rs17759843 and PPM1F mRNA in human postmortem PFC samples. These results suggest that PPM1F is an important genetic candidate in PTSD and point to the potential relevance of PPM1F in frontal regions via serotonin signaling mechanisms. However, despite this demonstrated role of PPM1F in pathways that have significant effects on cortical thickness, it remains unclear whether PPM1F has effects on the structural integrity of the human PFC in PTSD.
The primary goals of this study were to replicate and extend the work by Wingo et al. (2018) by investigating whether PPM1F genotype influences the association between PTSD (both alone and in comorbidity with depression) and cortical thickness in vivo using a neuroimaging-genetic approach in a large cohort of Iraq and Afghanistan veterans. We hypothesized that PPM1F would: (1) be associated with comorbid PTSD and depression as well as PTSD alone and; (2) moderate the association between comorbid pathology and/or PTSD alone and cortical thickness, with specific influence on the PFC.
Materials and Methods
Participants
Participants were 240 white non-Hispanic military veterans of Operations Enduring Freedom, Iraqi Freedom, and New Dawn (OEF/OIF/OND) or active duty service members not yet deployed to OEF/OIF/OND currently serving in the reserves or national guard enrolled in the Translational Research Center for TBI and Stress Disorders (TRACTS) located on the Jamaica Plain campus of VA Boston Healthcare System. The sample was limited to genotype-confirmed white non-Hispanic veterans (the largest racial and ethnic subgroup in the sample) to avoid genetic ancestry confounds. Exclusion criteria included history of seizures or neurological illness (unrelated to head injuries), serious mental illness such as bipolar disorder or other psychotic disorders (unrelated to PTSD), active suicidal or homicidal ideation, cognitive disorder due to a general medical condition, unstable psychological diagnosis that would interfere with accurate data collection (determined by consensus of at least two doctorate-level psychologists), and incompatibility with MRI due to ferromagnetic objects or pregnancy. Men comprised 95% of the sample and the mean age was 31.3 years (SD=8.0; see Table 1 for participant characteristics).
Table 1.
Demographic and Psychiatric Participant Characteristics (N=240)
| Variable | M (SD) | n (%) |
|---|---|---|
| Demographic | ||
| Sex (male) | 228 (95) | |
| Age | 31.3 (8.0) | |
| SSRI medication | 31 (12.9) | |
| Psychiatric | ||
| Current PTSD severity | 51.3 (28.6) | |
| Current PTSD diagnosis | 155 (64.5) | |
| DASS21: Anxiety total score | 7.3 (7.7) | |
| DASS21: Depression total score | 9.5 (9.6) | |
| Current MDD diagnosis | 59 (24.7) | |
| Current GAD diagnosis | 17 (7.1) | |
| Comorbid current MDD and PTSD diagnoses | 55 (40.4) |
Note: SSRI medication usage was based on self-report. Responses on the DASS21 were unavailable for ten participants (total n=230). Current diagnoses for the SCID-I (MDD and GAD) were unavailable for one participant (total n=239). Individuals with only one diagnosis of either current MDD or PTSD were excluded from the PTSD+MDD analysis (total n=136). DASS21=Depression Anxiety Stress Scales 21; GAD=general anxiety disorder; MDD=major depressive disorder; PTSD=posttraumatic stress disorder; SCID-I=Structured Clinical Interview-I for DSM-IV; SSRI=selective serotonin reuptake inhibitors.
All participants provided written and informed consent. This study was approved by the appropriate institutional review board and was conducted in accordance with the Declaration of Helsinki.
Clinical Assessments
Several clinical assessments were used for the purposes of this study to assess PTSD, comorbid pathology, and potential confounding clinical factors. All clinical diagnoses are determined by consensus of three or more psychologists or psychiatrists.
PTSD was assessed with the Clinician-Administered PTSD Scale (CAPS) for DSM-IV (Blake et al., 1995) by doctoral-level psychologists. The CAPS is a structured interview that assesses the frequency and intensity of DSM-IV PTSD criteria and is the gold standard for PTSD assessment and diagnosis. Frequency and intensity scores within the 30 days prior to assessment were summed to yield a total severity score (current PTSD severity), with higher scores indicative of greater symptom severity. For this study, both current PTSD symptom severity and current PTSD diagnosis were analyzed.
Clinical diagnosis of current MDD was assessed with the Structured Clinical Interview-I for DSM-IV (SCID-I), which was administrated by doctoral-level psychologists. The SCID-I is a semi-structured interview that is considered to be the gold standard for assessment and diagnosis of Axis I disorders (First, Spitzer, Gibbon, & Williams, 1997). Current (past month) single and recurrent episodes of MDD were coded as positive for the MDD diagnosis. Participants who had the condition in the past but did not currently meet DSM-IV criteria for MDD (i.e., lifetime only) at the time of assessment were included in the no diagnosis group. Diagnosis for one individual was unavailable for the SCID-I (n = 239).
To determine current comorbid PTSD and MDD diagnosis (referred to here on as PTSD+MDD), individuals were stratified as either presenting with both current PTSD and MDD diagnoses (n = 55) or without (n = 81). Individuals presenting with only one of the disorders were excluded from these analyses (n = 103).
Clinical diagnosis of current generalized anxiety disorder (GAD) was assessed with the SCID-I. Participants were stratified as either no or yes on current diagnosis for GAD and those who had the condition in the past but did not currently meet DSM-IV criteria for GAD (i.e., lifetime only) at the time of assessment were included in the no diagnosis group. Diagnosis for one individual was unavailable (n = 239).
Anxiety and depression symptomatology was assessed with the self-report Depression Anxiety Stress Scales 21 (DASS21), which is a shorter version of the 42-item self-report instrument that measures depression, anxiety and tension/stress (Lovibond & Lovibond, 1995). The DASS21 includes three scales that each consist of seven questions to measure Depression, Anxiety, and Tension/Stress. Responses are rated on a 0–3-point scale that reflects the extent to which an item applies to the individual (not at all to very much or most of the time). Items were summed for each subscale to create a total score for each domain and then multiplied by a constant (two) to allow for equitable comparison to the DASS 42-item subscale scores (total possible subscale score = 42). Higher scores on these subscales reflect greater severity. Responses on the DASS21 were unavailable for ten participants (n = 230). For the purposes of this study, only depression and anxiety subscale scores were analyzed.
MRI Acquisition and Processing
For the first 222 participants, two Magnetization Prepared Rapid Gradient Echo (MP-RAGE) T1-weighted structural scans were acquired on a 3-Tesla Siemens Trio whole-body TIM Trio MRI scanner with the following parameters: TR=2530ms, TE=3.32ms, flip angle=7°, FOV=256, Matrix=256×256, voxel size=1mm3. Due to an upgrade, the remaining 18 participants had two MP-RAGE T1-weighted structural scans acquired on a Siemens Prisma scanner with Syngo D13D software with the following parameters: TR=2530ms, TE=3.35ms, flip angle=7°, FOV=256, Matrix=256×256, voxel size=1mm3. To account for potential scanner software differences, a scanner flag was included in analyses as a covariate. Scans were averaged to create a single high contrast-to-noise image. A second MP-RAGE was unavailable for four individuals and cortical thickness analyses for those individuals were completed with a single MP-RAGE.
Cortical thickness analysis was performed using the FreeSurfer image analysis suite (version 5.3), which is documented and freely available for download online (http://surfer.nmr.mgh.harvard.edu). Processing of the images included reconstruction of the cortical surface and volumetric segmentation, the technical details of which are reported in prior publications (Dale, Fischl, & Sereno, 1999; Fischl & Dale, 2000; Fischl, Sereno, Tootell, & Dale, 1999). To extract cortical thickness metrics, cortical parcellations were created for each individual via FreeSurfer (Fischl et al., 2002) with the Desikan-Killiany parcellation (34 regions per hemisphere; see Figure 1) (Desikan et al., 2006). Then, parcellations were manually checked and edited for accuracy by members of the research team. Finally, the mean cortical thickness for each parcellation (68 regions in total) was extracted for each individual and inputted into R and SPSS for further analyses.
Figure 1. Anatomical atlas.
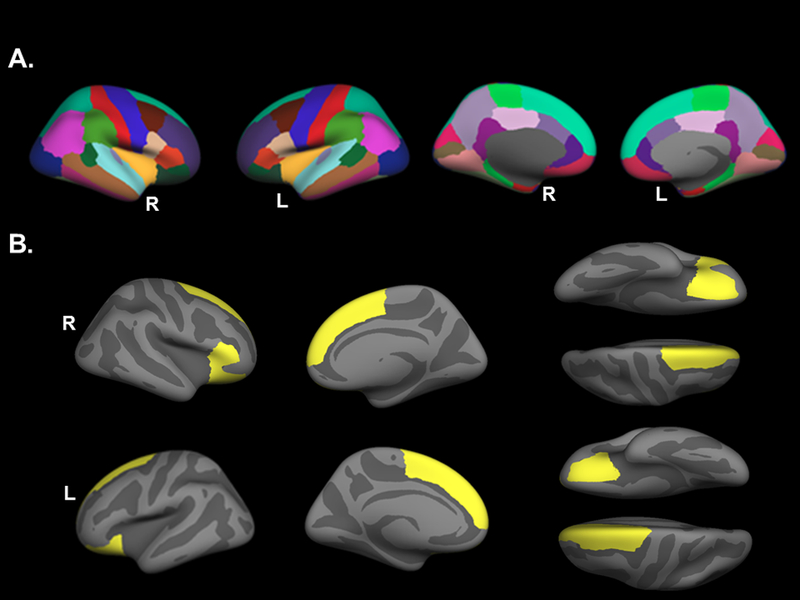
(A) Parcellations of the Desikan-Killiany anatomical atlas used in the PPM1F genotype by current PTSD symptom severity regional cortical thickness analysis. (B) Regions that were significantly associated with the PPM1F genotype by current PTSD symptom severity interaction are highlighted in yellow. These regions include bilateral superior frontal gyri, bilateral orbitrofrontal gyri and the right pas triangularis. L=left; PTSD=posttraumatic stress disorder; R=right.
A follow-up whole brain cortical thickness analysis investigating the PPM1F genotype by PTSD symptom severity interaction was performed with the most significant SNP of the parcellation analysis described above. This whole brain analysis included FreeSurfer version 5.3 command line tools mris_preproc, mri_surf2surf, and mri_glmfit and spatial smoothing of 10mm full-width half-maximum. Analyses were corrected for multiple comparisons with the command line tool mri_glmfit-sim using a vertex-wise/cluster forming threshold of p < 0.0001 and a cluster-wise p-value of p < 0.05.
Genotyping
Genotyping for this sample was performed as described previously in our prior work (Miller et al., 2015). Briefly, DNA was extracted from peripheral blood samples, were whole-genome amplified, fragmented, and hybridized to the Illumina HumanOmni2.5–8 microarray (Illumina, San Diego, CA) per manufacturer’s instructions. Genotypes were generated using the GenomeStudio V2011.1 software (with v1.9.4 Genotyping Module). Samples were examined for concordance between X chromosome homozygosity and reported sex using PLINK v1.07 (Purcell et al., 2007), confirming 228 males and 12 females. Two hundred and forty cases were identified as having white non-Hispanic (European) ancestry based on an analysis of the genome-wide SNP data using SNPweights (Chen et al., 2013) and had both cortical thickness and PTSD data available for investigation. To account for potential substructure within this sample, principal components (PCs) for use as covariates were computed within the white non-Hispanic-subgroup, based on 100,000 randomly chosen common (minor allele frequency >5%) SNPs using PLINKv1.9 (Chang et al., 2015). We investigated all genotyped SNPs with minor allele frequency >5%, that passed quality controls, had <5% missing calls, and were within 5,000 base pairs of the PPM1F gene (GRCh37/hg19 build).
Statistical Analyses
The R (http://www.R-project.org) and SPSS, version 25 (IBM Corp., Armonk, NY) platforms were used for statistical analyses.
We first evaluated the main effects of the 18 PPM1F SNPs as predictors of psychiatric phenotype (i.e., PTSD symptom severity or PTSD+MDD), controlling for age, sex, and the top three ancestry PCs in the first step of the regression. We used Monte Carlo null simulation with 10,000 replicates permuting genotypes across subjects at random, to generate corrected p-value estimates. That is, the observed p-values were compared to the distribution of the minimum p-value across all PPM1F SNPs, and the percentile of the observed p-value in the simulated minimum p-value distribution is taken as the corrected p-value, similar to the Max(T) correction as implemented in PLINK.
Next, the 68 mean cortical thickness measures for each parcellation were extracted for each individual and submitted to omnibus linear regression analyses in which age, sex, scanner flag, PCs 1–3, and either PTSD symptom severity or PTSD+MDD were entered in an initial step, the 18 PPM1F SNPs (coded additively and evaluated individually) were investigated in second steps of the regressions, and the interaction term (PTSD symptom severity or PTSD+MDD by PPM1F SNP) was evaluated in a third step. A Monte-Carlo null simulation was used to correct the significance levels for SNP main effects and interactions. This approach controlled for multiple-testing across both the 18 SNPs and the numerous cortical thickness parameters, taking into account the correlated structure of the morphological parameters as well as within the 18 SNPs. Statistically significant effects were those with corrected p-values <0.05.
We performed secondary PTSD symptom severity by PPM1F interaction analyses in separate regression models for each cortical thickness region that showed a significant association in the primary model. These secondary analyses were performed using the PPM1F SNP that showed the strongest interaction with PTSD symptom severity on cortical thickness in the primary analyses. Secondary analyses investigated additional potential effects of clinical diagnoses and symptoms including (1) current GAD and MDD diagnoses (which are highly comorbid with PTSD) as covariates (along with age, sex, scanner flag, and PCs 1–3) in the model with PTSD symptom severity and PPM1F SNP and (2) depression or anxiety symptomatology as measured by the DASS21 total subscale scores as covariates (along with age, sex, scanner flag, and PCs 1–3) in the model with PTSD symptom severity and PPM1F SNP. Additionally, because PPM1F is believed to be involved with serotonin signaling, we also examined whether SSRI medication usage, as measured by self-report, had any effect on our results. These analyses included SSRI medication (yes/no) as an additional covariate in the first step of each model (along with age, sex, scanner flag, and PCs 1–3) examining PTSD symptom severity, the PPM1F SNP, and cortical thickness. Finally, to determine if results for PTSD symptom severity generalized to PTSD diagnosis, we performed several follow-up regression analyses (i.e., separate models for each of the significant cortical thickness regions) replacing PTSD severity with current PTSD diagnosis, controlling for age, sex, scanner flag, and PCs 1–3.
Results
In separate models, there were no corrected significant main effects of any PPM1F SNP on PTSD symptom severity, PTSD+MDD, or cortical thickness (all corrected p’s>0.1, see Tables S1, S2, and S3 for unstandardized estimates and uncorrected p-values). However, regression models separately examining main effects of each psychiatric phenotype on cortical thickness revealed that there was a significant negative main effect of PTSD symptom severity on thickness in the right caudal (unstandardized β estimate = −0.0006, p = 0.05) and rostral middle frontal region (unstandardized β estimate = −0.00055, p = 0.02) as well as the right precentral (unstandardized β estimate = −0.00066, p = 0.03) and left postcentral gyrus (unstandardized β estimate = −0.00056, p = 0.03) and a significant negative main effect of PTSD+MDD on thickness in the right middle temporal gyrus (unstandardized β estimate = −0.055, p = 0.01). These findings did not survive multiple comparison correction across the 68 cortical thickness regions.
Examination of psychiatric phenotype by SNP interactions on cortical thickness revealed no corrected significant PTSD+MDD by SNP interactions on any region (all corrected p’s>0.2, see Table S4 for unstandardized estimates and p-values for significant uncorrected results). However, there were corrected significant SNP by PTSD symptom severity effects on cortical thickness in bilateral superior frontal and orbitofrontal regions as well as the right pars triangularis (all corrected p’s<0.05; Table 2, Figure 1) for six PPM1F SNPS in linkage disequilibrium (LD; rs9610608, rs62237483, rs9610645, rs62234965, rs199725385, rs9610690; all in high or 100% LD with one another, see Figure S1)2. The SNP that showed the strongest interaction with PTSD symptom severity on cortical thickness (unstandardized β=−0.0034, p=0.003), rs9610608, was examined in all follow-up analyses.
Table 2.
Regression results from models with corrected significant SNPs x PTSD symptom severity interaction terms predicting cortical thickness
| Variable | LH lateral orbitofrontal thick | LH superior frontal thick | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| β | Std Error |
T value |
P
value |
Corrected P |
β | Std Error |
T value |
P
value |
Corrected P |
|
| 1. Covariates | ||||||||||
| Intercept | 2.79 | 0.07 | 40.51 | 1.37 × 10−112 | N/A | 2.94 | 0.06 | 47.02 | 4.79 × 10−127 | N/A |
| Age | −6.54 × 10−3 | <0.001 | −5.70 | 3.39 × 10−8 | N/A | −7.27 × 10−3 | <0.001 | −6.97 | 2.83 × 10−11 | N/A |
| Sex | −2.34 × 10−2 | 0.04 | −0.55 | 0.58 | N/A | −1.87 × 10−2 | 0.04 | −0.48 | 0.63 | N/A |
| PC1 | −0.13 | 0.17 | −0.72 | 0.47 | N/A | −0.18 | 0.16 | −1.14 | 0.25 | N/A |
| PC2 | −0.23 | 0.18 | −1.25 | 0.21 | N/A | −0.12 | 0.16 | −0.73 | 0.47 | N/A |
| PC3 | 0.13 | 0.17 | 0.76 | 0.45 | N/A | 9.17 × 10−2 | 0.16 | 0.58 | 0.56 | N/A |
| Scanner flag | 4.07 × 10−2 | 0.04 | 1.14 | 0.26 | N/A | 3.19 × 10−2 | 0.03 | 0.99 | 0.33 | N/A |
| CAPS | −1.54 × 10−4 | <0.001 | −0.49 | 0.62 | N/A | 1.48 × 104 | <0.001 | −0.52 | 0.60 | N/A |
| 2. SNP | ||||||||||
| rs9610608, G | −1.11 × 10−2 | 0.02 | −0.55 | 0.58 | 1.00 | −1.84 × 10−2 | 0.02 | −1.01 | 0.31 | 1.00 |
| rs62237483, C | −1.06 × 10−2 | 0.02 | −0.54 | 0.59 | 1.00 | −1.83 × 10−2 | 0.02 | −1.03 | 0.30 | 1.00 |
| rs9610645, A | −8.06 × 10−3 | 0.02 | −0.41 | 0.68 | 1.00 | −1.61 × 10−2 | 0.02 | −0.90 | 0.37 | 1.00 |
| rs62234965, G | −1.06 × 10−2 | 0.02 | −0.54 | 0.59 | 1.00 | −1.83 × 10−2 | 0.02 | −1.03 | 0.30 | 1.00 |
| rs199725385, C | −1.06 × 10−2 | 0.02 | −0.54 | 0.59 | 1.00 | −1.83 × 10−2 | 0.02 | −1.03 | 0.30 | 1.00 |
| rs9610690, A | −1.06 × 10−2 | 0.02 | −0.54 | 0.59 | 1.00 | −1.83 × 10−2 | 0.02 | −1.03 | 0.30 | 1.00 |
| 3. Interaction | ||||||||||
| rs9610608, G | −3.41 × 10−3 | <0.001 | −4.55 | 8.33 × 10−6 | 0.007* | −2.77 × 10−3 | <0.001 | −4.04 | 7.21 × 10−5 | 0.048* |
| rs62237483, C | −3.14 × 10−3 | <0.001 | −4.34 | 2.08 × 10−5 | 0.017* | −2.56 × 10−3 | <0.001 | −3.87 | 1.40 × 10−4 | 0.085 |
| rs9610645, A | −3.12 × 10−3 | <0.001 | −4.30 | 2.46 × 10−5 | 0.019* | −2.54 × 10−3 | <0.001 | −3.83 | 1.63 × 10−4 | 0.096 |
| rs62234965, G | −3.14 × 10−3 | <0.001 | −4.34 | 2.08 × 10−5 | 0.017* | −2.56 × 10−3 | <0.001 | −3.87 | 1.40 × 10−4 | 0.085 |
| rs199725385, C | −3.14 × 10−3 | <0.001 | −4.34 | 2.08 × 10−5 | 0.017* | −2.56 × 10−3 | <0.001 | −3.87 | 1.40 × 10−4 | 0.085 |
| rs9610690, A | −3.14 × 10−3 | <0.001 | −4.34 | 2.08 × 10−5 | 0.017* | −2.56 × 10−3 | <0.001 | −3.87 | 1.40 × 10−4 | 0.085 |
| Variable | RH lateral orbitofrontal thick | RH superior frontal thick | ||||||||
| β | Std Error |
T value |
P
value |
Corrected P |
β | Std Error |
T value |
P
value |
Corrected P |
|
| 1. Covariates | ||||||||||
| Intercept | 2.55 | 0.07 | 38.55 | 6.62 × 10−108 | N/A | 2.81 | 0.06 | 47.88 | 7.61 × 10−129 | N/A |
| Age | −6.29 × 10−3 | <0.002 | −5.71 | 3.12 × 10−8 | N/A | −6.51 × 10−3 | <0.001 | −6.66 | 1.69 × 10−10 | N/A |
| Sex | 4.32 × 10−2 | 0.04 | 1.06 | 0.29 | N/A | −6.29 × 10−4 | 0.04 | −0.02 | 0.99 | N/A |
| PC1 | 0.28 | 0.17 | 1.65 | 0.10 | N/A | −3.13 × 10−2 | 0.15 | −0.21 | 0.83 | N/A |
| PC2 | −9.54 × 10−2 | 0.17 | −0.55 | 0.58 | N/A | −8.76 × 10−2 | 0.15 | −0.57 | 0.57 | N/A |
| PC3 | 0.17 | 0.17 | 1.04 | 0.30 | N/A | 0.33 | 0.15 | 2.21 | 0.03 | N/A |
| Scanner flag | 7.06 × 10−2 | 0.03 | 2.06 | 0.04 | N/A | 4.89 × 10−2 | 0.03 | 1.61 | 0.11 | N/A |
| CAPS | −2.76 × 10−4 | <0.001 | −0.92 | 0.36 | N/A | −2.21 × 104 | <0.001 | −0.83 | 0.41 | N/A |
| 2. SNP | ||||||||||
| rs9610608, G | −1.52 × 10−2 | 0.02 | −0.79 | 0.43 | 1.00 | −2.40 × 10−2 | 0.02 | −1.41 | 0.16 | 1.00 |
| rs62237483, C | −1.65 × 10−2 | 0.02 | −0.88 | 0.38 | 1.00 | −2.27 × 10−2 | 0.02 | −1.36 | 0.17 | 1.00 |
| rs9610645, A | −1.77 × 10−2 | 0.02 | −0.93 | 0.35 | 1.00 | −2.37 × 10−2 | 0.02 | −1.41 | 0.16 | 1.00 |
| rs62234965, G | −1.65 × 10−2 | 0.02 | −0.88 | 0.38 | 1.00 | −2.27 × 10−2 | 0.02 | −1.36 | 0.17 | 1.00 |
| rs199725385, C | −1.65 × 10−2 | 0.02 | −0.88 | 0.38 | 1.00 | −2.27 × 10−2 | 0.02 | −1.36 | 0.17 | 1.00 |
| rs9610690, A | −1.65 × 10−2 | 0.02 | −0.88 | 0.38 | 1.00 | −2.27 × 10−2 | 0.02 | −1.36 | 0.17 | 1.00 |
| 3. Interaction | ||||||||||
| rs9610608, G | −3.44 × 10−3 | <0.001 | −4.80 | 2.68 × 10−6 | 0.003* | −2.83 × 10−3 | <0.001 | −4.44 | 1.37 × 10−5 | 0.01* |
| rs62237483, C | −3.25 × 10−3 | <0.001 | −4.70 | 4.27 × 10−6 | 0.004* | −2.56 × 10−3 | <0.001 | −4.15 | 4.50 × 10−5 | 0.03* |
| rs9610645, A | −3.29 × 10−3 | <0.001 | −4.74 | 3.59 × 10−6 | 0.004* | −2.59 × 10−3 | <0.001 | −4.19 | 3.80 × 10−5 | 0.03* |
| rs62234965, G | −3.25 × 10−3 | <0.001 | −4.70 | 4.27 × 10−6 | 0.004* | −2.56 × 10−3 | <0.001 | −4.15 | 4.50 × 10−5 | 0.03* |
| rs199725385, C | −3.25 × 10−3 | <0.001 | −4.70 | 4.27 × 10−6 | 0.004* | −2.56 × 10−3 | <0.001 | −4.15 | 4.50 × 10−5 | 0.03* |
| rs9610690, A | −3.25 × 10−3 | <0.001 | −4.70 | 4.27 × 10−6 | 0.004* | −2.56 × 10−3 | <0.001 | −4.15 | 4.50 × 10−5 | 0.03* |
| Variable | RH pars triangularis | |||||||||
| β | Std Error |
T value |
P
value |
Corrected P |
||||||
| 1. Covariates | ||||||||||
| Intercept | 2.53 | 0.06 | 39.22 | 1.58 × 10−109 | N/A | |||||
| Age | −7.10 × 10−3 | 0.001 | −6.58 | 2.62 × 10−10 | N/A | |||||
| Sex | 3.64 × 10−2 | 0.04 | 0.91 | 0.36 | N/A | |||||
| PC1 | 1.50 × 10−2 | 0.16 | 0.09 | 0.93 | N/A | |||||
| PC2 | −4.61 × 10−3 | 0.17 | −0.03 | 0.98 | N/A | |||||
| PC3 | 0.25 | 0.16 | 1.50 | 0.13 | N/A | |||||
| Scanner flag | 6.15 × 10−2 | 0.03 | 1.84 | 0.07 | N/A | |||||
| CAPS | −2.98 × 10−4 | <0.001 | −1.02 | 0.31 | N/A | |||||
| 2. SNP | ||||||||||
| rs9610608, G | −2.05 × 10−2 | 0.02 | −1.09 | 0.28 | 1.00 | |||||
| rs62237483, C | −1.90 × 10−2 | 0.02 | −1.04 | 0.30 | 1.00 | |||||
| rs9610645, A | −1.87 × 10−2 | 0.02 | −1.02 | 0.31 | 1.00 | |||||
| rs62234965, G | −1.90 × 10−2 | 0.02 | −1.04 | 0.30 | 1.00 | |||||
| rs199725385, C | −1.90 × 10−2 | 0.02 | −1.04 | 0.30 | 1.00 | |||||
| rs9610690, A | −1.90 × 10−2 | 0.02 | −1.04 | 0.30 | 1.00 | |||||
| 3. Interaction | ||||||||||
| rs9610608, G | −2.89 × 10−3 | <0.001 | −4.10 | 5.62 × 10−5 | 0.04* | |||||
| rs62237483, C | −2.62 × 10−3 | <0.001 | −3.83 | 1.60 × 10−4 | 0.095 | |||||
| rs9610645, A | −2.67 × 10−3 | <0.001 | −3.91 | 1.17 × 10−4 | 0.073 | |||||
| rs62234965, G | −2.62 × 10−3 | <0.001 | −3.83 | 1.60 × 10−4 | 0.095 | |||||
| rs199725385, C | −2.62 × 10−3 | <0.001 | −3.83 | 1.60 × 10−4 | 0.095 | |||||
| rs9610690, A | −2.62 × 10−3 | <0.001 | −3.83 | 1.60 × 10−4 | 0.095 | |||||
Note: Corrected P indicates p-value after correction for multiple comparison using Monte-Carlo null simulation with 10,000 replicates. β estimates are unstandardized. Only the results from models in which the interaction term was significantly associated with cortical thickness after correction for multiple testing are shown. The minor allele is shown next to the SNP. CAPS=Clinician-Administered PTSD Scale; LH=left hemisphere; RH=right hemisphere; SNP=single nucleotide polymorphism; std=standard; thick=thickness
Decomposition of the PTSD symptom severity by rs9610608 interaction revealed an effect of genotype on the association between PTSD symptom severity and cortical thickness such that this association was stronger for those with one or more copies of the minor allele (zero copies, AA: n=188, one copy, AG: n=49; two copies, GG: n=3; see Figure 2). Specifically, greater symptom severity was associated with reduced cortical thickness in those with the minor allele. Because so few individuals had two copies of the minor allele, we combined one and two copy minor allele groups (n=52 minor allele carriers vs n=188 non-carriers) and reran the analysis. The pattern of results did not change (corrected p’s<0.05 and unstandardized β estimates ranged between −0.003 to −0.004). The recoded rs9610608 SNP was used in all secondary analyses.
Figure 2. Rs9610608 x PTSD symptom severity interaction on cortical thickness of the right superior frontal gyrus.
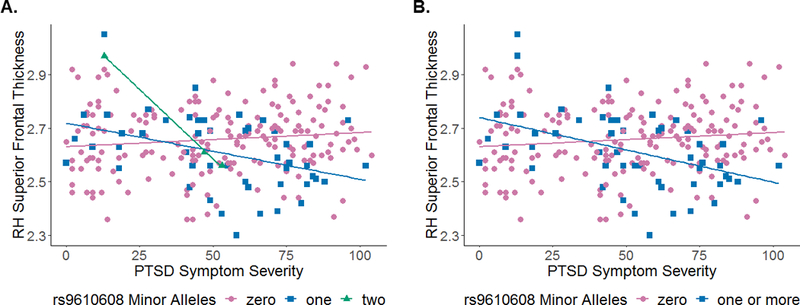
(A) Figure demonstrates carriers of zero (i.e., pink circles), one (i.e., blue squares), or two (i.e., green triangles) minor alleles of the rs9610608 PPM1F SNP. (B) Figure combines one and two allele groups to demonstrate carriers of zero (i.e., pink circles) or one or more (i.e., blue squares) minor alleles of the rs9610608 PPM1F SNP. In both instances, individuals with one or more minor alleles and increasing PTSD symptom severity had reduced cortical thickness in the right superior frontal gyrus. These associations were similar for the left superior frontal, bilateral orbitofrontal, and right pars triangularis regions, which are not pictured here. L=left; PTSD=posttraumatic stress disorder; RH=right hemisphere.
To confirm that the interaction was driven by a negative effect of PTSD symptom severity on cortical thickness within the minor allele carriers and not a positive effect in non-carriers, we performed follow-up regression analyses in which we stratified analyses by the recoded minor allele groups. Regression analyses were performed for each group separately (zero or one or more minor alleles) on cortical thickness of each of the five identified regions (bilateral orbitofrontal, bilateral superior frontal, and right pars triangularis). Results revealed that PTSD symptom severity was negatively associated with cortical thickness of all five identified regions in minor allele carriers (left superior frontal: unstandardized β estimate = −0.002, p=0.005; right superior frontal: unstandardized β estimate = −0., p=0.0; left lateral orbitofrontal: unstandardized β estimate = −0.003, p=0.001; right lateral orbitofrontal: unstandardized β estimate = −0.003, p<0.0001; right pars triangularis: unstandardized β estimate = −0.003, p=0.002). In contrast, there were no significant PTSD symptom severity effects on cortical thickness within any of the five identified regions in the non-carrier group (left superior frontal: unstandardized β estimate = 0.0004, p=0.2; right superior frontal: unstandardized β estimate = 0.0003, p=0.2; left lateral orbitofrontal: unstandardized β estimate = 0.0004., p=0.2; right lateral orbitofrontal: unstandardized β estimate = 0.0003, p=0.4; right pars triangularis: unstandardized β estimate = 0.0002, p=0.5).
Additional analyses were conducted to determine if there was an allele effect in cortical thickness of the five identified regions (bilateral orbitofrontal, bilateral superior frontal, and right pars triangularis) within “no PTSD” individuals (determined via CAPS DSM-IV diagnosis). Results revealed a trending significant effect for thickness in the left lateral orbitofrontal gyrus, such that “no PTSD” individuals in the one or more minor allele group had greater thickness than “no PTSD” individuals in the zero minor allele group (AA, F(1,77)=4.0, p=0.05). However, this finding did not survive multiple testing correction. No other identified regions were significantly different between minor allele groups in “no PTSD” individuals.
We next included current GAD and MDD diagnosis as covariates in the models examining PTSD symptom severity by PPM1F on cortical thickness. Results revealed that the PTSD symptom severity by rs9610608 interaction remained significant in all models (left superior frontal: unstandardized β estimate = −0.003, p<0.0001; right superior frontal: unstandardized β estimate = −0.003, p<0.0001; left lateral orbitofrontal: unstandardized β estimate = −0.004., p<0.0001; right lateral orbitofrontal: unstandardized β estimate = −0.004, p<0.0001; right pars triangularis: unstandardized β estimate = −0.003, p<0.001). GAD was significantly associated with the left lateral orbitofrontal cortex (unstandardized β estimate = −0.07, p<0.05) and left superior frontal gyrus (unstandardized β estimate = −0.07, p<0.04), but not with the right lateral orbitofrontal cortex (unstandardized β estimate = −0.003, p>0.9), pars triangularis (unstandardized β estimate = −0.01, p>0.7), or superior frontal gyrus (unstandardized β estimate = −0.06, p>0.05). MDD was not significantly associated with any of the five cortical thickness regions (left superior frontal: unstandardized β estimate = 0.02, p>0.4; right superior frontal: unstandardized β estimate = 0.007, p>0.7; left lateral orbitofrontal: unstandardized β estimate = −0.001, p<0.001; right lateral orbitofrontal: unstandardized β estimate = −0.03, p>0.2; right pars triangularis: unstandardized β estimate = 0.001, p>0.9).
Additional follow-up analyses were performed with DASS21 Anxiety and Depression total scores in the model as covariates. Once again, the pattern of results did not change, with the PTSD symptom severity by rs9610608 interaction remaining significant in all models with these covariates included in the model (left superior frontal: unstandardized β estimate = −0.003, p<0.001; right superior frontal: unstandardized β estimate = −0.003, p<0.0001; left lateral orbitofrontal: unstandardized β estimate = −0.003, p<0.001; right lateral orbitofrontal: unstandardized β estimate = −0.004, p<0.0001; right pars triangularis: unstandardized β estimate = −0.003, p=0.001). Neither anxiety nor depression symptoms were significantly associated with any of the five cortical thickness regions (DASS21 Anxiety: left superior frontal: unstandardized β estimate = 0.0005, p>0.7; right superior frontal: unstandardized β estimate = −0.00009, p>0.9; left lateral orbitofrontal: unstandardized β estimate = 0.001, p>0.4; right lateral orbitofrontal: unstandardized β estimate = −0.0004, p>0.8; right pars triangularis: unstandardized β estimate = 0.001, p>0.4; DASS21 Depression: left superior frontal: unstandardized β estimate = 0.00008, p>0.9; right superior frontal: unstandardized β estimate = −0.00002, p>0.9; left lateral orbitofrontal: unstandardized β estimate = −0.001, p>0.5; right lateral orbitofrontal: unstandardized β estimate = −0.0002, p>0.8; right pars triangularis: unstandardized β estimate = −0.001, p>0.6).
Next, we reevaluated the model including SSRI medication (yes/no) in the first step of the model. The pattern of results did not change, with the PTSD symptom severity by rs9610608 interaction remaining significant in all models after inclusion of this additional covariate (left superior frontal: unstandardized β estimate = −0.003, p<0.001; right superior frontal: unstandardized β estimate = −0.003, p<0.0001; left lateral orbitofrontal: unstandardized β estimate = −0.004., p<0.0001; right lateral orbitofrontal: unstandardized β estimate = −0.004, p<0.0001; right pars triangularis: unstandardized β estimate = −0.003, p<0.001). Self-reported SSRI medication usage was not significantly associated with any of the five cortical thickness regions (left superior frontal: unstandardized β estimate = 0.01, p>0.5; right superior frontal: unstandardized β estimate = 0.005, p>0.8; left lateral orbitofrontal: unstandardized β estimate = −0.01, p>0.6; right lateral orbitofrontal: unstandardized β estimate = 0.02, p>0.5; right pars triangularis: unstandardized β estimate = −0.001, p>0.9).
To examine if effects observed for PTSD symptom severity generalized to PTSD diagnosis, we evaluated current PTSD diagnosis in place of PTSD symptom severity in separate regression models for each significant cortical thickness region. Analyses revealed a significant PTSD diagnosis by rs9610608 interaction on cortical thickness in all models (left superior frontal: unstandardized β estimate = −0.142, p=0.002; right superior frontal: unstandardized β estimate = −0.121, p=0.004; left lateral orbitofrontal: unstandardized β estimate = −0.155, p=0.001; right lateral orbitofrontal: unstandardized β estimate = −0.151, p=0.001; right pars triangularis: unstandardized β estimate = −0.122, p=0.008). There were no main effects of PTSD diagnosis (left superior frontal: unstandardized β estimate = −0.01, p>0.5; right superior frontal: unstandardized β estimate = −0.01, p>0.4; left lateral orbitofrontal: unstandardized β estimate = −0.005, p>0.7; right lateral orbitofrontal: unstandardized β estimate = −0.03, p=0.1; right pars triangularis: unstandardized β estimate = −0.03, p>0.1) or SNP (left superior frontal: unstandardized β estimate = −0.02, p>0.2; right superior frontal: unstandardized β estimate = −0.03, p>0.09; left lateral orbitofrontal: unstandardized β estimate = −0.009, p>0.6; right lateral orbitofrontal: unstandardized β estimate = −0.02, p>0.2; right pars triangularis: unstandardized β estimate = −0.02, p>0.2) on any of the five cortical thickness regions.
Finally, for a higher-resolution analysis of the specific regions within the cortical parcellations implicated in the omnibus genetic association analysis, we examined whole-brain associations with the PTSD symptom severity by rs9610608 interaction. This analysis yielded a significant cluster in the right superior frontal gyrus that survived multiple comparison correction such that minor allele carriers had reduced cortical thickness in this region with increasing PTSD symptom severity (peak MNI coordinates=9.1 58.6 17.8, peak value=5.2, number of vertices=150, cluster size=94.7mm2, cluster-corrected p<0.02; see Figure 3 for corrected results and Figure S2 for uncorrected results).
Figure 3. Significant whole-brain cortical thickness results.
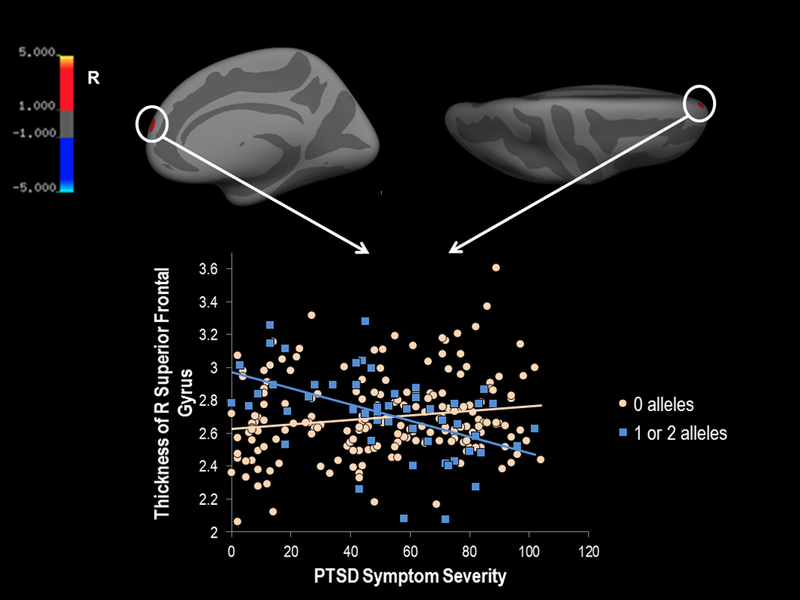
In a whole-brain thickness analysis with vertex-wise threshold of p < 0.0001 and cluster corrected at p < 0.05, results revealed that there was a significant interaction in the right superior frontal gyrus such that individuals with at least one minor allele of the rs9610608 PPM1F SNP had reduced cortical thickness in this region with increased current PTSD symptom severity. The plot of the PPM1F (rs9610608 SNP) by current PTSD symptom severity interaction is plotted below the medial and superior views of the brain (the significant cluster is circled for ease of viewing). Individuals carrying two minor alleles for the PPM1F SNP were combined with individuals with one minor allele for this analysis. Color bar indicates the log 10 value for p-values associated with the cluster corrected results. The red color of the cluster (instead of blue) indicates the association with the interaction of greater PTSD severity and increased minor alleles. PTSD=posttraumatic stress disorder; R=right; SNP=single nucleotide polymorphism.
Discussion
This study investigated the influence of PPM1F, a gene previously implicated in PTSD and depression, on the association between PTSD symptom severity as well as comorbid PTSD with MDD and cortical thickness. We identified six PPM1F SNPs in high LD that moderated the association between PTSD symptom severity and reduced cortical thickness in bilateral superior frontal and orbitofrontal regions and the right pars triangularis. These associations were not substantially altered by self-reported depressive or anxious symptoms, clinician-determined GAD or MDD diagnoses, or self-reported SSRI usage. To confirm the specificity of these results, we performed a whole-cortex vertex-wise analysis with the most significant SNP (rs9610608) and found the reduced cortical thickness effect to be localized to a cluster in the right superior frontal gyrus. While we focused on the gene PPM1F based on its prior association with PTSD and depression (Wingo et al., 2018), we did not replicate the main effects from prior work. These results suggest that PPM1F genotype is particularly important in PTSD-associated reductions of the neural integrity of the PFC.
PPM1F has been shown to dephosphorylate CAMK-II, which plays a role in the neural circuitries underlying depression, anxiety, PTSD, and schizophrenia (Hasegawa et al., 2009; Purkayastha et al., 2012; Robison, 2014; Wen, Li, Han, Wang, & Shi, 2012) and is important in serotonergic regulation of PFC neuronal activity (Cai, Gu, Zhong, Ren, & Yan, 2002; Yuen et al., 2005). The inactivation of CAMK-II via PPM1F suggests that PPM1F may act as an upstream regulator of serotonergic signaling in the PFC. Here, we show that PPM1F in combination with PTSD can have adverse neural consequences within the PFC. This is consistent with previous work that has found an association between PPM1F and postmortem PFC brain tissue (Wingo et al., 2018). One interpretation of these findings is that traumatic stress activates these upstream PPM1F effects on serotonergic signaling, which may in turn have downstream effects on brain health such as the degradation of structural integrity of the PFC. However, longitudinal studies focused on PPM1F, PTSD and neurodegeneration of the PFC are needed to confirm this hypothesis.
We did not find a main effect of PPM1F on our comorbid PTSD and depression phenotype. This is in contrast with prior work by Wingo et al. (2018) who reported that among five genes, PPM1F (rs17759843) was significantly associated with comorbid PTSD with depression and that cases with comorbid pathology had lower PPM1F expression in blood than controls. The discrepancy may be due to the fact that the cohort investigated here was significantly different than that in Wingo et al. (2018). In this study, the sample was a white non-Hispanic predominately male cohort of military veterans, while the cohort investigated in Wingo et al. (2018) was comprised primarily of African American women. Further, the sample size for the current study was small in comparison to the genetic analysis performed in Wingo et al. (2018; n=2361), substantially limiting statistical power. We also did not find evidence for an interaction between comorbid PTSD and depression and PPM1F on cortical thickness, though the effect was evident for the SNP in interaction with both PTSD severity and PTSD diagnosis. This suggests a specific relationship between PPM1F, PTSD and the structural integrity of the PFC and further points to the significance of PPM1F in PTSD. It is important to note that the most significant SNP reported in our analyses of white non-Hispanic participants was not the same as the top SNP reported in African Americans in Wingo et al. (2018).
These findings should be considered with several limitations in mind. First, this was a cross-sectional study and therefore we cannot infer the causal direction of the associations reported. Further longitudinal work is needed to explore the possible directional associations discussed in this study. Second, the sample size was relatively small but comparable with several other neuroimaging-genetics studies investigating psychiatric phenotype (Costafreda et al., 2013; Schultz et al., 2011; Schulz-Heik et al., 2011). Third, this was an all veteran white non-Hispanic predominately male sample. It is unknown whether these findings would extend to civilians, other ethnicities, or to females. Further work is needed to expand this research to other demographic groups. Fourth, this study focused on cortical thickness and did not examine subcortical structures. This decision was based on previous work implicating the cortex in the association between PTSD and PPM1F in human samples (Wingo et al., 2018). Nonetheless, it will be important for future work to examine subcortical structures in PTSD-focused neuroimaging-genetic analyses. Fifth, we did not have a replication cohort to replicate these findings; our results will need to be further evaluated in additional cohorts to ensure their reliability and reproducibility. Sixth, other genes or gene networks were not examined in this study that may be related to PPM1F signaling and are important to investigate in future studies. Finally, this was a candidate-gene study, and we note that genome-wide association studies (GWAS) have not identified PPM1F in studies of PTSD or depression.
In summary, PPM1F genotype was associated with reduced cortical thickness of the PFC in individuals with PTSD. PPM1F is widely expressed in the brain and has important upstream serotonin regulating effects within frontal regions. Future studies should focus on the role of PPM1F in brain alterations and potential neurodegenerative processes induced by traumatic stress. Further insight into this mechanism may be important for identifying novel treatment targets and developing therapeutic interventions for PTSD.
Supplementary Material
Acknowledgements
The contents of this article do not represent the views of the U.S. Department of Veterans Affairs, the National Institutes of Health, or the United States Government. Funding: This work was supported by a VA Clinical Science Research and Development Career Development Award (1IK2CX001772–01) awarded to DRS, National Institutes of Mental Health (NIMH) training grant (T32MH019836–01) awarded to Terence Keane, Ph.D. supporting FGM, a VA Biomedical Laboratory Research and Development grant (I01BX003477) award to MWL, the National Center for PTSD, and the Translational Research Center for TBI and Stress Disorders (TRACTS), a VA Rehabilitation Research and Development Traumatic Brain Injury Center of Excellence (B9254-C). This work was further supported with resources and the use of facilities at the Neuroimaging Research for Veterans Center, VA Boston Healthcare System and the Pharmacogenomics Analysis Laboratory, Research and Development Service, Central Arkansas Veterans Healthcare System, Little Rock, Arkansas.
Footnotes
Declarations of Interest: none
Abbreviations: CAMK-II=calmodulin-dependent protein kinase II; CAPS=Clinician-Administered PTSD Scale; DASS21=Depression Anxiety Stress Scales; GAD=general anxiety disorder; IMO=immobilization on boards; LD=linkage disequilibrium; MDD=major depressive disorder; MP-RAGE= magnetization prepared rapid gradient echo; OEF/OIF/OND=Operations Enduring Freedom/Iraqi Freedom/New Dawn; PFC=prefontal cortex; PP2C=protein phosphatase 2C; PTSD=posttraumatic stress disorder; SCID-I=Structured Clinical Interview-I; SNP=single nucleotide polymorphism; SSRI=serotonin reuptake inhibitors
Given the small number of women in the sample (n=12), we repeated this analysis in the male subset. The pattern of results did not change with significant SNP by PTSD symptom severity effects on bilateral orbitofrontal cortex thickness across the same six SNPs reported when including females (left hemisphere unstandardized β estimates = −0.003 across all six SNPs, corrected p < 0.02; right hemisphere unstandardized β estimates = −0.003 across all six SNPs, corrected p < 0.008) and a PTSD symptom severity by rs9610608 effect on right superior frontal gyrus thickness (unstandardized β estimate = −0.003, corrected p = 0.02). The SNP by PTSD symptom severity effects on bilateral superior frontal cortex and right pars triangularis thickness were trending toward corrected significance when females were removed from the analysis (left superior frontal unstandardized β estimates = −0.003, corrected p = 0.1, uncorrected p < 0.0002; right superior frontal unstandardized β estimates (for SNPs other than rs9610608) = −0.003, corrected p = 0.07, uncorrected p < 0.0001; right pars triangularis unstandardized β estimates ranged between −0.002 to −0.003, corrected p’s ranged between 0.1 to 0.2, uncorrected p’s < 0.0005). Interestingly, a corrected significant effect arose on the right caudal middle frontal thickness such that there was an interaction between PTSD symptom severity and rs9610608 when female veterans were removed from the model (unstandardized β estimate = −0.003, corrected p = 0.03).
References
- American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders (5 ed.). Washington, DC. [Google Scholar]
- Bartlett EA, DeLorenzo C, Sharma P, Yang J, Zhang M, Petkova E, … Ogden RT (2018). Pretreatment and early-treatment cortical thickness is associated with SSRI treatment response in major depressive disorder. Neuropsychopharmacology, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing X, Ming-guo Q, Ye Z, Jing-na Z, Min L, Han C, … Wei C (2013). Alterations in the cortical thickness and the amplitude of low-frequency fluctuation in patients with post-traumatic stress disorder. Brain research, 1490, 225–232. [DOI] [PubMed] [Google Scholar]
- Blake DD, Weathers FW, Nagy LM, Kaloupek DG, Gusman FD, Charney DS, & Keane TM (1995). The development of a Clinician-Administered PTSD Scale. J Trauma Stress, 8(1), 75–90. [DOI] [PubMed] [Google Scholar]
- Cai X, Gu Z, Zhong P, Ren Y, & Yan Z (2002). Serotonin 5-HT1A receptors regulate AMPA receptor channels through inhibiting Ca2+/calmodulin-dependent kinase II in prefrontal cortical pyramidal neurons. Journal of Biological Chemistry, 277(39), 36553–36562. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, & Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4(1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-Y, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, & Price AL (2013). Improved ancestry inference using weights from external reference panels. Bioinformatics, 29(11), 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, … Weinberger DR (2011). Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature, 478(7370), 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo V, Salat DH, Amick MM, Leritz EC, Milberg WP, & McGlinchey RE (2014). Reduced cortical thickness in veterans exposed to early life trauma. Psychiatry Research: Neuroimaging, 223(2), 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costafreda SG, McCann P, Saker P, Cole JH, Cohen-Woods S, Farmer AE, … Fu CH (2013). Modulation of amygdala response and connectivity in depression by serotonin transporter polymorphism and diagnosis. J Affect Disord, 150(1), 96–103. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, & Sereno MI (1999). Cortical surface-based analysis: I. Segmentation and surface reconstruction. Neuroimage, 9(2), 179–194. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, … Killiany RJ (2006). An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage, 31(3), 968–980. [DOI] [PubMed] [Google Scholar]
- First M, Spitzer R, Gibbon M, & Williams JB (1997). Structured clinical interview for DSM-IV clinical version (SCID-I/CV): Washington DC: American Psychiatric Press. [Google Scholar]
- Fischl B, & Dale AM (2000). Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proceedings of the National Academy of Sciences of the United States of America, 97(20), 11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, … Dale AM (2002). Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron, 33(3), 341–355. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Tootell RB, & Dale AM (1999). High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp, 8(4), 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuze E, Westenberg HG, Heinecke A, de Kloet CS, Goebel R, & Vermetten E (2008). Thinner prefrontal cortex in veterans with posttraumatic stress disorder. Neuroimage, 41(3), 675–681. [DOI] [PubMed] [Google Scholar]
- Harvey BP, Banga SS, & Ozer HL (2004). Regulation of the multifunctional Ca2+/calmodulin-dependent protein kinase II by the PP2C phosphatase PPM1F in fibroblasts. Journal of Biological Chemistry [DOI] [PubMed]
- Hasegawa S, Furuichi T, Yoshida T, Endoh K, Kato K, Sado M, … Suzuki R (2009). Transgenic up-regulation of alpha-CaMKII in forebrain leads to increased anxiety-like behaviors and aggression. Molecular brain, 2(1), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JP, Hayes S, Miller DR, Lafleche G, Logue MW, & Verfaellie M (2017). Automated measurement of hippocampal subfields in PTSD: evidence for smaller dentate gyrus volume. Journal of psychiatric research, 95, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida A, Kameshita I, & Fujisawa H (1998). A novel protein phosphatase that dephosphorylates and regulates Ca2+/calmodulin-dependent protein kinase II. Journal of Biological Chemistry, 273(4), 1904–1910. [DOI] [PubMed] [Google Scholar]
- Kasai K, Yamasue H, Gilbertson MW, Shenton ME, Rauch SL, Pitman RK (2008). Evidence for acquired pregenual anterior cingulate gray matter loss from a twin study of combat-related posttraumatic stress disorder. Biological Psychiatry, 63: 550–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremen WS, O’brien RC, Panizzon MS, Prom-Wormley E, Eaves LJ, Eisen SA, … Fischl B (2010). Salivary cortisol and prefrontal cortical thickness in middle-aged men: a twin study. Neuroimage, 53(3), 1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemer ER, Salat DH, Leritz EC, McGlinchey RE, & Milberg WP (2013). Reduced cortical thickness with increased lifetime burden of PTSD in OEF/OIF Veterans and the impact of comorbid TBI. NeuroImage: Clinical, 2, 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kakeda S, Watanabe K, Yoshimura R, Abe O, Ide S, … Watanabe R (2015). Relationship between the cortical thickness and serum cortisol levels in drug‐naive, first‐episode patients with major depressive disorder: a surface‐based morphometric study. Depression and anxiety, 32(9), 702–708. [DOI] [PubMed] [Google Scholar]
- Liu Y, Li Y-J, Luo E-P, Lu H-B, & Yin H (2012). Cortical thinning in patients with recent onset post-traumatic stress disorder after a single prolonged trauma exposure. PloS one, 7(6), e39025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovibond PF, & Lovibond SH (1995). The structure of negative emotional states: comparison of the Depression Anxiety Stress Scales (DASS) with the Beck Depression and Anxiety Inventories. Behaviour research and therapy, 33(3), 335–343. [DOI] [PubMed] [Google Scholar]
- Lyoo IK, Kim JE, Yoon SJ, Hwang J, Bae S, Kim DJ (2011). The neurobiological role of the dorsolateral prefrontal cortex in recovery from trauma. Arch. Gen. Psychiatry, 68(7), 701–713. [DOI] [PubMed] [Google Scholar]
- Miller MW, Wolf EJ, Sadeh N, Logue M, Spielberg JM, Hayes JP, … Carter WC (2015). A novel locus in the oxidative stress-related gene ALOX12 moderates the association between PTSD and thickness of the prefrontal cortex. Psychoneuroendocrinology, 62, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyano S, Del Río J, & Frechilla D (2004). Role of hippocampal CaMKII in serotonin 5-HT 1A receptor-mediated learning deficit in rats. Neuropsychopharmacology, 29(12), 2216. [DOI] [PubMed] [Google Scholar]
- Pillai RL, Zhang M, Yang J, Mann JJ, Oquendo MA, Parsey RV, & DeLorenzo C (2018). Molecular connectivity disruptions in males with major depressive disorder. Journal of Cerebral Blood Flow & Metabolism, 0271678X18764053. [DOI] [PMC free article] [PubMed]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, … Daly MJ (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics, 81(3), 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkayastha S, Ford J, Kanjilal B, Diallo S, Del Rosario Inigo J, Neuwirth L, … Azmitia EC (2012). Clozapine functions through the prefrontal cortex serotonin 1A receptor to heighten neuronal activity via calmodulin kinase II–NMDA receptor interactions. Journal of neurochemistry, 120(3), 396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison A (2014). Emerging role of CaMKII in neuropsychiatric disease. Trends in neurosciences, 37(11), 653–662. [DOI] [PubMed] [Google Scholar]
- Sadeh N, Spielberg JM, Logue MW, Wolf EJ, Smith AK, Lusk J, … McGlinchey RE (2016). SKA2 methylation is associated with decreased prefrontal cortical thickness and greater PTSD severity among trauma-exposed veterans. Molecular psychiatry, 21(3), 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz CC, Nenadic I, Koch K, Wagner G, Roebel M, Schachtzabel C, … Deufel T (2011). Reduced cortical thickness is associated with the glutamatergic regulatory gene risk variant DAOA Arg30Lys in schizophrenia. Neuropsychopharmacology, 36(8), 1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Heik RJ, Schaer M, Eliez S, Hallmayer JF, Lin X, Kaloupek DG, & Woodward SH (2011). Catechol-O-methyltransferase Val158Met polymorphism moderates anterior cingulate volume in posttraumatic stress disorder. Biol Psychiatry, 70(11), 1091–1096. [DOI] [PubMed] [Google Scholar]
- Wen Y, Li B, Han F, Wang E, & Shi Y (2012). Dysfunction of calcium/calmodulin/CaM kinase IIα cascades in the medial prefrontal cortex in post-traumatic stress disorder. Molecular medicine reports, 6(5), 1140–1144. [DOI] [PubMed] [Google Scholar]
- Wingo AP, Velasco ER, Florido A, Lori A, Choi DC, Jovanovic T, … Andero R (2018). Expression of the PPM1F gene is regulated by stress and associated with anxiety and depression. Biological psychiatry, 83(3), 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf EJ, Miller DR, Logue MW, Sumner J, Stoop TB, Leritz EC, … McGlinchey RE (2017). Contributions of polygenic risk for obesity to PTSD-related metabolic syndrome and cortical thickness. Brain, behavior, and immunity, 65, 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward SH, Kaloupek DG, Streeter CC, Martinez C, Schaer M, & Eliez S (2006). Decreased anterior cingulate volume in combat-related PTSD. Biological psychiatry, 59(7), 582–587. [DOI] [PubMed] [Google Scholar]
- Wrocklage KM, Averill LA, Scott JC, Averill CL, Schweinsburg B, Trejo M, … Martini B (2017). Cortical thickness reduction in combat exposed US veterans with and without PTSD. European Neuropsychopharmacology, 27(5), 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Jiang Q, Chen P, Gu Z, Feng J, & Yan Z (2005). Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. Journal of Neuroscience, 25(23), 5488–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.