

Abstract
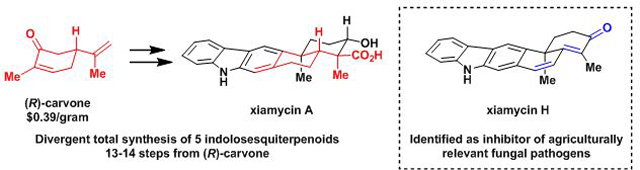
Divergent and enantiospecific total syntheses of the indolosesquiterpenoids xiamycins A, C, F, H and oridamycin A have been accomplished. The syntheses, which commence from (R)-carvone, employ a key photoinduced benzannulation sequence to forge the carbazole moiety characteristic of these natural products. Late-stage diversification from a common intermediate enabled the first syntheses of xiamycins C and F, and an unexpected one-pot oxidative decarboxylation, which may prove general, led to xiamycin H. All synthetic intermediates and the natural products were tested for anti-fungal activity. Xiamycin H emerged as an inhibitor of three agriculturally relevant fungal pathogens.
Keywords: total synthesis, benzannulation, chiral pool, divergent synthesis, fungitoxicity
Graphical Abstract
A benzannulation of carvone strategy has enabled the enantiospecific and divergent syntheses of indolosesquiterpenoids xiamycin A, C, F, H, and oridamycin A in 13–14 steps from (R)-carvone. Evaluation of their anti-fungal properties revealed xiamycin H to be an inhibitor of three agriculturally relevant fungal pathogens.
The ‘xiamycin-type’ secondary metabolites (Figure 1) were first isolated from a range of Streptomyces species in 2010. These molecules represent the first examples of indolosesquiterpenoids from bacterial sources, and new members continue to be discovered to this day.1 The emerging biological activity of these indolosesquiterpenoids has sparked interest in employing them as a starting point for the development of pharmaceuticals and agrochemicals. For example, xiamycin A (1) displays antibiotic and anti-HIV activity,2a whereas its C-16 epimer, oridamycin A (2), exhibits modest activity against the water mold Saprolegnia parasitica.1b On the basis of this latter bioactivity, we have become especially interested in profiling the agrochemical potential of the xiamycins as fungicides against a broad range of fungal pathogens that significantly impact crop yields. Notably, we sought to identify new chemotypes active against wheat leaf blotch (Zymoseptoria tritici) which has been known to cause up to 50% crop yield loss in the European Union (EU).3a While there are known fungicides to combat this fungus, newer compounds that possess novel modes of action are critical to addressing the resistance shifts that continue to emerge for many damage-causing fungi.3b
Figure 1.
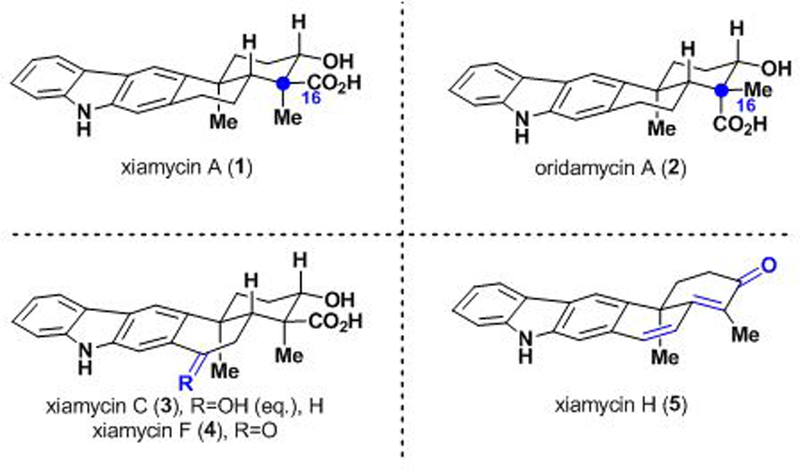
Selected members of the xiamycin and oridamycin families.
Xiamycin A and oridamycin A have drawn significant attention from synthetic chemists as reflected in the number of total syntheses of these molecules completed over the last five years (Scheme 1a).2 Structurally, they are composed of a challenging pentacyclic framework, including a carbazole nucleus fused to a trans-decalin ring system that bears four contiguous stereocenters.
Scheme 1a.
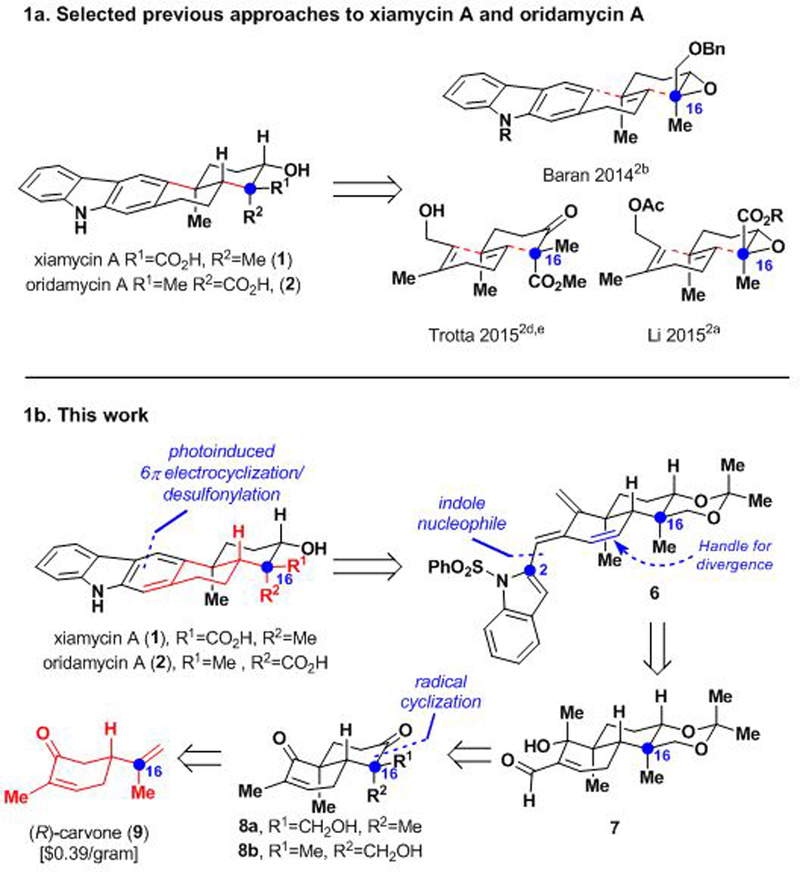
Summary of selected previous approaches. 1b. Xiamycin family retrosynthesis from carvone.
Two elegant previous syntheses of these natural products by Li and Trotta employ a benzannulation strategy to forge the carbazole moiety. Notably, Baran, Li, and Trotta all utilize polycyclizations to form the trans-decalin system.2 Our group has had a standing interest in divergent4 syntheses of terpenoid natural products utilizing carvone as a starting material.5 Carvone is of particular interest to us because of its ready availability in either enantiomeric form,6 and its highly modifiable cyclic structure, which allows for rapid elaboration.7 The syntheses that we report herein are complementary to previous approaches to the xiamycin-type indolosesquiterpenoids. Specifically, we begin from a cyclic ‘chiral pool’ compound (carvone), which obviates the need for a polycyclization to build the framework of these molecules, thus presenting a unique opportunity for divergence that has culminated in the synthesis of additional congeners of the xiamycin-type indolo-terpenoids than had been previously achieved. In our initial synthesis considerations, we envisioned an expansion of our reported5d ‘benzannulation of carvone’ strategy whereby the α−methyl group of carvone would be exploited in a C–C bond and concomitant six-membered ring formation. Here, we report the realization of this strategy in the total syntheses of xiamycins A, C, F, H (1, 3, 4, 5) and oridamycin A (2). As part of an ongoing program to identify novel natural-product derived small molecules relevant to crop protection, we also describe the first systematic investigation of their anti-fungal activity.
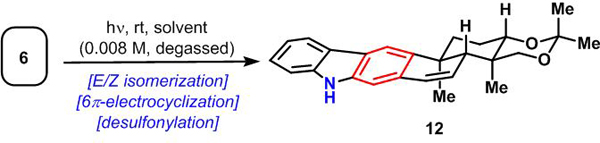
Retrosynthetically (Scheme 1b), we envisioned 1 and 2 arising from a late-stage carbazole-forming benzannulation sequence. However, unlike in some of the previous syntheses of these indolosesquiterpenoids, which employed a thermally induced benzannulation, our synthetic plan was to effect a photoinduced olefin isomerization/6π-electrocyclization sequence of triene 6.8 Triene 6 would in turn emerge from the addition of an indole nucleophile to α,β−unsaturated aldehyde 7. Intermediate 7 was traced back to known9 trans-decalin C-16 epimers 8a and 8b from which both configurations of the C-16 stereocenter could be accessed, leading to the synthesis of the xiamycin and oridamycin natural product families. Compounds 8a and 8b are accessible through a known four-step sequence from inexpensive and commercially available (R)-carvone (9).
Our synthetic studies commenced with the construction of the key trans-decalin system (Scheme 2). Following the precedent of Omura and Nagamitsu,9 keto-alcohols 8a and 8b were prepared in four steps from (R)-carvone (9) in multigram quantities. Evans reduction10 of keto-alcohol 8a using tetramethylammonium triacetoxyborohydride (Me4NBH(OAc)3) gave the desired equatorial alcohol 10 as a single diastereomer, which was unambiguously confirmed by X-ray crystallographic analysis.11 Following acetonide formation, addition of MeLi to the α,β-unsaturated ketone group yielded the corresponding tertiary alcohol as a single isomer. Subsequent allylic oxidation using SeO2 formed aldehyde 7, which was coupled with 2-lithiated N-(phenylsulfonyl)indole12 reagent 11 to produce an inconsequential mixture of diastereomeric alcohols in almost quantitative yield. The elimination of both hydroxy groups proved challenging. Under several conditions (e.g., using the Burgess reagent, MsCl/DIPEA, or SOCl2/py), only elimination of the secondary hydroxy group was observed. Fortunately, Martin’s sulfurane cleanly effected double dehydration at room temperature affording triene 6 in gram quantities.
Scheme 2.
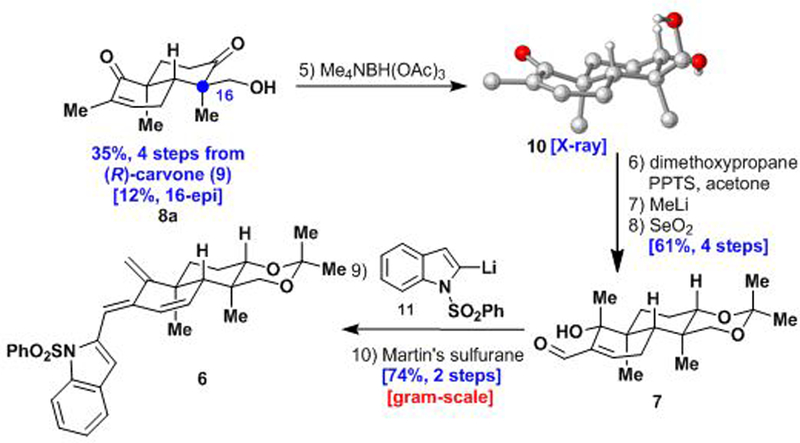
Synthesis of the 6π-electrocyclization precursor 6.
Having established an efficient route to precursor 6, we began to explore our proposed benzannulation sequence to forge the 2,3-fused carbazole.13 Due to the geometric constraint imposed by the trisubstituted (E)-double bond in 6, a thermal 6π-electrocyclization/aromatization sequence under aerobic conditions, as demonstrated in the Li and Trotta syntheses of related molecules, was not successful. This prompted us to attempt a photochemical benzannulation14,15 wherein E/Z isomerization of the double bond could be effected (Table 1). To our surprise, irradiation of 6 with UVB light (310 nm) in degassed benzene led to the direct isolation of desulfonylated carbazole 12 in 28% yield as the only detectable product (entry 1).16 After extensive experimentation, we found that irradiation with UVA light (350 nm) in aq. EtOH increased the yield of 12 to 44% (entry 2). The solubility of the starting material, as well as the yield, were further increased by adding THF to the reaction medium (entry 3). The use of other polar protic solvents (MeOH, iPrOH, HFIP) proved inferior. Polar aprotic solvents such as MeCN led only to isolation of trace amounts of 12 (entry 4). The addition of water was found to be beneficial in this case (entry 5) whereas the addition of radical scavengers (1,4-cyclohexadiene, entry 6) to possibly capture a potentially formed sulfonyl radical and prevent polymerization did not improve the yields. Attempts to buffer the resulting sulfinic acid byproduct by the addition of base (Na2CO3, entry 7) only led to a decrease in yield. Only scant reports of the photochemical desulfonylation of amines and indoles are known.17 The existing methods rely on an initial electron transfer from an appropriate donor (e.g., amines, electron-rich aromatics) to the excited state indole moiety. Applying these known conditions, which use DABCO (entry 8) and NEt3 (entry 9) as amine sources or anisole (entry 10), gave 12 in only low (5–33%) yield. While irradiation with blue LED light gave comparable yields to the optimized conditions (entry 11), longer wavelength visible light did not induce carbazole formation (entry 12).18
Table 1.
Optimization of the key photocyclization/desulfonylation reaction.
 | |||
|---|---|---|---|
| entry | wavelength (nm)a | conditionsd | yielde |
| 1 | 310/350 | PhH,1 h | 28%/33% |
| 2 | 310/350 | 5% aq. EtOH, 1 h | 30%/44% |
| 3 | 350 | 10% aq. EtOH/THF, 1 h | 46% |
| 4 | 350 | MeCN, 1 h | <5% |
| 5 | 350 | 50% aq. MeCN, 0.5 h | 23% |
| 6 | 310 | PhH, 1,4-CHD, 0.2 h | 30% |
| 7 | 350 | 5% aq. EtOH, Na2C03 0.5 h | <5% |
| 8 | 350 | 5% aq. MeOH, DABCO, 0.5 h | <5% |
| 9 | 350 | NEt3, nBu3SnH, MeCN, 0.5 h | <5% |
| 10 | 350 | 5% aq. EtOH, anisole, NaBH4 1.5 h | 33% |
| 11 | 400b | 5% aq. EtOH, 1.5 h | 30% |
| 12 | 500-800c | 5% aq. EtOH, 40°C, 1.5 h | no reaction |
Luzchem photobox
Kessil blue LED
Sunlite tungsten lamp
Reactions were performed in pyrex glass tubes; 5% aq. EtOH refers to technical grade (95%)
Isolated yield.
With access to carbazole 12 using our best conditions (entry 3), hydrogenation of the styrenyl double bond and subsequent cleavage of the acetonide yielded diol 13 in 60% yield over two steps (Scheme 3). Unfortunately, oxidation of the primary hydroxy group to give aldehyde 14 led to substantial decomposition under standard conditions such as TEMPO/PIDA, presumably due to the free carbazole nitrogen. We therefore opted to install the aldehyde group first. Addition of 2-lithiated N-(phenylsulfonyl)indole 11 to aldehyde 7 gave diol 15, which was subjected to a one-pot double dehydration followed by acetonide cleavage using Martin’s sulfurane and 6M HCl, respectively, to give the corresponding triene diol (not shown). Oxidation of the primary alcohol group under TEMPO/PIDA conditions gave a hydroxy-aldehyde which underwent the key photocyclization/desulfonylation to give aldehyde 16 in 21% over 3 steps.19 Finally, Pinnick oxidation20 followed by hydrogenation with Pd/C reduced the styrenyl double bond to complete the synthesis of xiamycin A (1) in a total of 14 steps from (R)-carvone (9). The spectroscopic data obtained for 1 were identical to those reported in literature.4a
Scheme 3a.
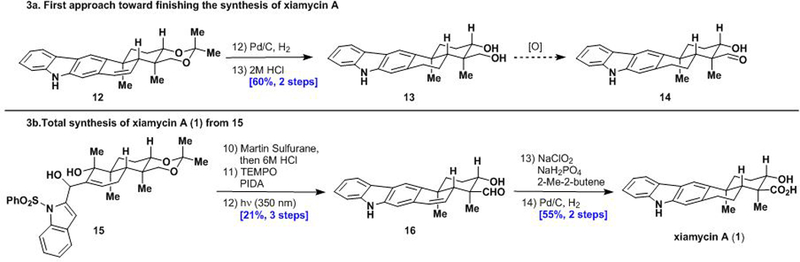
Failed endgame route. Scheme 3b. Completion of the total synthesis of xiamycin A (1).
We have also utilized the minor keto-alcohol diastereomer 8b in an analogous sequence to complete a total synthesis of oridamycin A (2, Scheme 4). Thus, subjecting minor diastereomer 8b to Narasaka–Prasad reduction21 conditions yielded boronate ester 17, which was cleaved by methanolysis on acidic silica to afford desired syn-diol 18 in 74% yield. The total synthesis of oridamycin A (2) was successfully completed by following the established route described above (i.e., steps 6–14; see the Supporting Information for details).
Scheme 4.
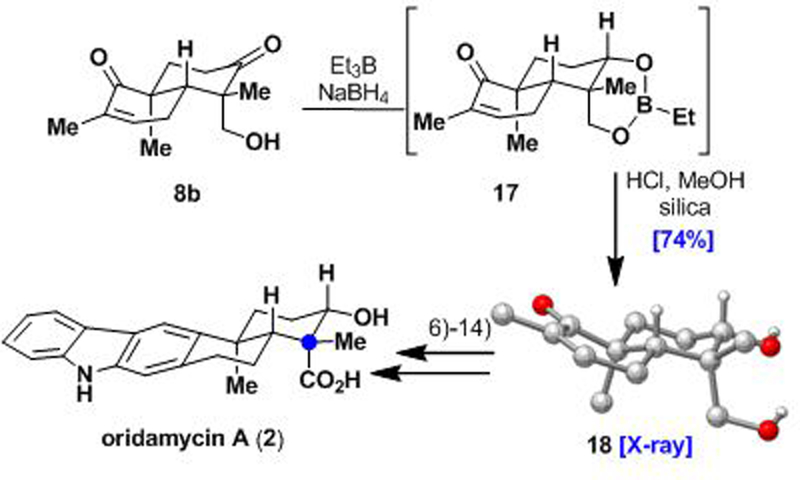
Total synthesis of oridamycin A(2)
In line with our initial plan, access to 16 has provided a common late-stage intermediate that we have applied to the synthesis of several other xiamycin congeners (Scheme 5). Specifically, we envisioned that the styrenyl double bond of 16 would serve as a handle for functionalization. Indeed, Mukaiyama hydration22 of 16 followed by Pinnick oxidation gave xiamycin C (3) and 19-epi-xiamycin C as a separable mixture of diastereomers, as well as xiamycin F (4). This sequence represents the first total syntheses of these natural products as well as the associated C-19 epimer of 3.
Scheme 5.
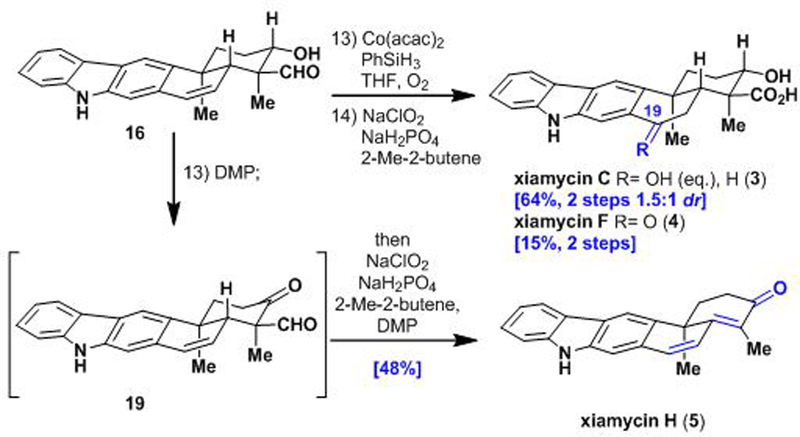
Completion of the syntheses of xiamycin C(4), F(5), and H(6) from common intermediate 16.
As a testament to the versatility of our route, we then sought to prepare the only known xiamycin congener that bears an alkene functional group (i.e., xiamycin H; 5). Oxidation of alcohol aldehyde 16 with Dess–Martin periodinane first yielded keto-aldehyde 19, which, in one-pot, underwent oxidative decarboxylation under Pinnick oxidation conditions to form xiamycin H (5) in 48% yield.
Bioactivity
Our syntheses of the xiamycins have also provided access to a number of synthetic intermediates which have been screened for bioactivity. Specifically, the in vitro fungitoxicity of these small molecules against three agriculturally relevant pathogens: wheat leaf blotch, rice blast (Pyricularia oryza) and corn smut (Ustilago maydis) was evaluated. At a concentration of 10 ppm, it was found that xiamycin H (5) demonstrated complete (100%) growth inhibition of wheat leaf blotch as well as partial (50% and 40%) inhibition of rice blast and corn smut, respectively. Compound 7 demonstrated selectivity toward the control of corn smut (50% growth inhibition) over other pathogens, whereas compound S-23 (from hydrogenation of 12) demonstrated some selectivity toward the control of rice blast (50% growth inhibition). These initial screening data provide a promising foundation for additional studies to identify even more potent derivatives of this class of natural products.
Conclusion
In summary, we have accomplished the divergent, enantiospecific total synthesis of the indolosesquiterpenoids xiamycin A (1), xiamycin C (3), xiamycin F (4), oridamycin A (2), as well as xiamycin H (5) in a maximum of ten steps from known compound 9. A key feature in the formation of the characteristic carbazole moiety is a photoinduced 6π-electrocyclization with concomitant desulfonylation, which represents a rare example of this type of transformation. These syntheses proceed in a total of 13–14 steps from carvone and are highly scalable, providing enough material for a preliminary bioactivity screen. Evaluation of the fungicidal activity of these compounds revealed that xiamycin H and some of the synthetic intermediates display notable inhibition of agriculturally relevant pathogens that could set the stage for the identification of new small molecules for crop protection.
Supplementary Material
Acknowledgements
Corteva Agriscience is acknowledged for support of this work. Partial financial support was provided to R.S. by the National Institutes of Health (NIGMS R35 GM130345). M.P. is grateful for a postdoctoral scholarship funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – PF 910/1–1. We thank Dr. Hasan Celik (UC Berkeley) for assistance with NMR experiments and Dr. Nicholas Settineri (UC Berkeley) for single-crystal X-ray diffraction studies. We also thank Jamie Lutz and Stacy Meyer of Corteva for collecting the biological data.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Ding L, Münch J, Goerls H, Maier A, Fiebig H-H, Lin W-H, Hertweck C, Bioorg. Med. Chem. Lett. 2010, 20, 6685–6687. [DOI] [PubMed] [Google Scholar]; b) Takada K, Kajiwara H, Imamura N, J. Nat. Prod. 2010, 73, 698–701. [DOI] [PubMed] [Google Scholar]; c) Ding L, Maier A, Fiebig H-H, Lin W-H, Hertweck C, Org. Biomol. Chem. 2011, 9, 4029–4031. [DOI] [PubMed] [Google Scholar]; d) Kim S, Ha T, Oh WK, Shin J, Oh D, J. Nat. Prod. 2016, 79, 51. [DOI] [PubMed] [Google Scholar]; e) Zhang Q, Li H, Yu L, Sun Y, Zhu Y, Zhu H, Zhang L, Li S, Shen Y, Tian C, Li A, Liu H, Zhang C, Chem. Sci. 2017, 8, 5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Meng Z, Yu H, Li L, Tao W, Chen H, Wan M, Yang P, Edmonds D, Zhong J, Li A, Nature Commun. 2015, 6, 6096. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rosen B, Werner E, O’Brian A, Baran P, J. Am. Chem. Soc. 2014, 136, 5571–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Feng J, Noack F, Krische M, J. Am. Chem. Soc. 2016, 138, 12364–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Trotta A, Org. Lett. 2015, 17, 3358–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Trotta A, J. Org. Chem. 2017, 82, 13500–13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Fones H, Gurr S, Fungal Genet. Biol. 2015, 79, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Poole NF, Arnaudin ME, Can. J. Plant Pathol. 2014, 36, 1–11. [Google Scholar]
- [4].Brotherton CE; Boger DL J. Org. Chem. 1984, 49, 4050–4055. [Google Scholar]
- [5].a) Weber M, Owens K, Masarwa A, Sarpong R, Org. Lett. 2015, 17, 5432–5435. [DOI] [PubMed] [Google Scholar]; b) Masarwa A, Weber M, Sarpong R, J. Am. Chem. Soc. 2015, 137, 6327–6334. [DOI] [PubMed] [Google Scholar]; c) Kuroda Y, Nicacio KJ, da Silva IA Jr, Leger PR, Chang S, Gubiani JR, Deflon VM, Nagashima N, Rode A, Blackford K, Ferreira AG, Sette LD, Williams DE, Andersen RJ, Jancar S, Berlinck RGS, Sarpong R, Nat. Chem. 2018, 10, 938–945. [DOI] [PubMed] [Google Scholar]; d) Finkbeiner P, Murai K, Röpke M, Sarpong R, J. Am. Chem. Soc. 2017, 139, 11349–11352. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kerschgens I, Rovira AR, Sarpong R, J. Am. Chem. Soc. 2018, 140, 9810–9813. [DOI] [PubMed] [Google Scholar]
- [6].a) Brill ZG, Condakes ML, Ting CP, Maimone TJ, Chem. Rev. 2017, 117, 11753–11795. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gaich T, Mulzer J, In Comprehensive Chirality; Carreira EM, Yamamoto H, Eds.; Elsevier Ltd: 2012; Vol. 2, pp 163–206. [Google Scholar]; c) Macaev FZ, In Studies in Natural Products Chemistry, Elsevier Ltd, 2013, Vol. 39, pp 233–267. [Google Scholar]
- [7].a) Chiou W-H; Ojima I In New Methodologies and Techniques for a Sustainable Organic Chemistry; Mordini A, Faigl F, Eds.; Springer: Dordrecht, 2008; Vol. 246, pp 55–83. [Google Scholar]; b) Brocksom TJ; Desiderá AL; de Carvalho Alves L; de Oliveira KT Curr. Org. Synth. 2015, 12, 496. [Google Scholar]
- [8].Binkley RW, Flechtner TW, In Synthetic Organic Photochemistry; Horspool WM, Eds.; Springer, 1984, Boston, pp 375–423. [Google Scholar]
- [9].a) Odani A, Ishihara K, Ohtawa M, Tomoda H, Omura S, Nagamitsu T, Tetrahedron 2011, 67, 8195–8203. [Google Scholar]; b) For another application of the trans-decalin in synthesis, see: Zhong Z, Zhao G, Xu D, Dong B, Song D, Xie X, She X, Chem. Asian J. 2016, 11, 1542–1547. [DOI] [PubMed] [Google Scholar]
- [10].a) Evans DA, Chapman KT, Tetrahedron Lett. 1986, 27, 5939–5942. [Google Scholar]; b) Reisman SE, Ready JM, Weiss MM, Hasuoka MH, Tamaki K, Ovaska TV, Smith CJ, Wood JL, J. Am. Chem. Soc. 2008, 130, 2087–2100. [DOI] [PubMed] [Google Scholar]
- [11].NMR comparison to known compound 7 (reference 9) showed several discrepancies (see supporting information for details) which led us to unambiguously confirm our result by X-ray analysis.
- [12].Volker I, Synthesis. 1979, 2, 136 [Google Scholar]
- [13].For a review on electrocyclization as a benzannulation strategy, see: Trauner D, Webster R, In Comprehensive Organic Synthesis: Second Edition Vol. 5, Elsevier Ltd, 2014, pp 783–826.For a recent review on carbazole syntheses, see: J. Roy, A. K. Jana, D. Ma, Tetrahedron, 2012, 68, 6099–6121. [Google Scholar]
- [14].:a) Bach T, Hehn JP, Angew. Chem. Int. Ed. 2011, 50, 1000–1045; Angew. Chem. 2011. 123, 1032–1077.b) Kärkäs MD, Porco JA Jr., Stephenson CRJ, Chem. Rev. 2016, 116, 9683–9747.c) Nicholls TP, Leonori D, Bissember AC, Nat. Prod. Rep 2016, 33, 1248–1254.
- [15].a) Bonesi SM, Erra-Balsells R, Photochemistry of carbazoles and its application in organic synthesis, in Trends in Heterocyclic Chemistry, 2009, 14, 39–77. [Google Scholar]; b) Choi S, Chatterjee T, Choi WJ, You Y, Cho EJ, ACS Catal. 2015, 5, 4796–4802. [Google Scholar]; c) Hernandez-Perez AC, Collins SK, Angew. Chem. Int. Ed. 2013, 52, 12696–12700. Angew. Chem. 2013, 125, 12928−12932. [DOI] [PubMed] [Google Scholar]; d) Hernandez-Perez A, Caron A, Collins SK, Chem. Eur. J, 2015, 21, 16673–16678. [DOI] [PubMed] [Google Scholar]
- [16].In general, full consumption of the starting material was observed. Broadened NMR-signals indicate the potential formation of unidentified polymerization side-products. No dihydroaromatic intermediates were observed by NMR of crude reaction mixtures.
- [17].a) Hamada T, Nishida A, Yonemitsu O, J. Am. Chem. Soc. 1986, 108, 140–145. [Google Scholar]; b) Art JF, Kestemont JP, Soumillion J, Tetrahedron Lett. 1991, 32, 1425–1428. [Google Scholar]; c) Hong K, Mejía-Oneto JM, France S, Padwa A, Tetrahedron Lett. 2006, 47, 2409–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kageyama Y, Ohshima R, Sakurama K, Fujiwara Y, Tanimoto Y, Yamada Y, Aoki S, Chem. Pharm. Bull. 2009, 57, 1257–1266. [DOI] [PubMed] [Google Scholar]
- [18].The UV/vis absorption spectrum of triene 6 and carbazole 12 showed absorption maxima at 328 and 320 nm, respectively. Product 12 was found stable under prolonged irradiation and was fully recovered.
- [19].We also found that the photocyclization also works on the stage of the β-hydroxy acid, forming the corresponding carbazole in 32% yield.
- [20].Bal B, Childers W, Pinnick H, Tetrahedron, 1981, 11, 2091–2096. [Google Scholar]
- [21].Kau-Ming C, Gunderson K, Hardtmann G, Prasad K, Repic O, Shapiro M, Chem. Lett. 1987, 16, 1923–1926. [Google Scholar]
- [22].Mukaiyama T, Yamada T, Bull. Chem. Soc. Jpn. 1995, 68, 17–35. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.