

Abstract
Alexander disease (AxD) is an extremely rare neurodegenerative disorder caused by glial fibrillary acidic protein (GFAP) gene mutations. Compared with the cerebral type, which is characterized by infantile onset, the bulbospinal type and intermediate form are associated with a late onset, spanning from juveniles to the elderly, and more diverse clinical spectrum, suggesting the existence of factors contributing to phenotypic diversity. To build a foundation for future genetic studies of this rare disease, we obtained genomic data by whole exome-sequencing (WES) and DNA microarray derived from thirty-one AxD patients with the bulbospinal type and intermediate form. Using this data, we aimed to identify genetic variations determining the age at onset (AAO) of AxD. As a result, WES- or microarray-based association studies between younger (<45 years; n = 13)- and older (≥45 years; n = 18)-onset patients considering the predicted GFAP-mutation pathogenicity identified no genome-wide significant variant. The candidate gene approach identified several variants likely correlated with AAO (p < 0.05): GAN, SLC1A2, CASP3, HDACs, and PI3K. Although we need to replicate the results using an independent population, this is the first step towards constructing a database, which may serve as an important tool to advance our understanding of AxD.
Subject terms: Disease genetics, Neurodegeneration
Introduction
Alexander disease (AxD, OMIM #203450)1 is an extremely rare neurodegenerative disorder pathologically characterized by cytoplasmic inclusions called Rosenthal fibers in astrocytes. The prevalence of AxD is estimated to be one case per 2.7 million individuals2. Since mutations of glial fibrillary acidic protein (GFAP) were identified as the cause of AxD in 20013, AxD patients with a wide spectrum of clinical presentations have been reported. AxD can be classified into three subtypes: cerebral type, bulbospinal type, and intermediate form, based on the neurological symptoms and lesion location. Cerebral type is characterized by an infantile or juvenile onset with frontal-dominant white matter abnormalities on brain MRI and certain neurological symptoms, including convulsions, macrocephaly, and psychomotor developmental delay. Bulbospinal type is characterized by a juvenile or adult onset with an abnormal signal or atrophy of the medulla oblongata and/or cervical cord on MRI. The neurological symptoms of bulbospinal type include muscle weakness, spasticity, and bulbar symptoms, which occur in various combinations. The intermediate form is characterized by juvenile or adult onset, and by the coexistence of neurological features of both the cerebral and bulbospinal types.
To date, more than 100 GFAP mutations have been reported to be associated with AxD (http://www.waisman.wisc.edu/alexander-disease/). A total of 70% of patients with the cerebral type have amino acid mutations located in either residue R79, R88, or R2392. On the contrary, a wide distribution of AAO and many mutations without hot spots have been reported in bulbospinal type and intermediate form. Moreover, an identical mutation, such as R416W, was reported in cerebral type, bulbospinal type, and also intermediate form4. These findings suggest the presence of some modifying factors leading to the phenotypic variation in the bulbospinal type and intermediate form in addition to GFAP mutations, while the almost constant phenotype of the cerebral type may be determined mostly by GFAP mutations themselves.
Recent genome-wide association studies on Huntington disease and Duchenne muscular dystrophy, both of which are monogenic neurological diseases, identified some susceptible loci as modifier genes that are likely to contribute to the diversity of AAO and age at loss of ambulation, respectively5,6. However, the literature on extremely rare diseases including AxD mainly comprises case reports from different institutes. An association study involving a number of AxD patients has not been reported to date.
We collected blood samples from and clinical information on AxD patients referred from facilities throughout Japan, and obtained genomic data from 31 bulbospinal-type and intermediate-form AxD patients by whole-exome sequencing (WES) and microarray analysis. Then, we performed association analyses focusing on AAO in an attempt to identify genes responsible for the phenotypic diversity of the bulbospinal type and intermediate form. Although no genome-wide significant variant was detected, we identified genetic variations likely to be associated with AAO by focusing on AxD-related genes.
Results
Clinical information on subjects
Clinical information is presented in Table 1. Twenty-five patients had already been reported in the literature7–17. The distribution of AAO and clinical classification (bulbospinal type or intermediate form) are shown in Fig. 1. The AAO was distributed widely (5 to 72 years) and bimodally: one peak was in those in their teens to thirties and the other peak was around 60 years, and the boundary was approximately 45 years. Thus, we defined the subjects with an onset at younger than 45 years as a “younger-onset” group (n = 13) and those with an onset at 45 years or older as an “older-onset” group (n = 18). Although the exact AAO of patient 12 was unknown, she was included in the younger-onset group because she had been diagnosed at 32 years. All patients in the older-onset group had the bulbospinal type, whereas most of the younger-onset patients had intermediate form (92%). Gait disturbance was the most frequent initial symptom in both the younger (62%)- and older (33%)-onset patients. In the older-onset patients, bulbar symptoms such as dysarthria and dysphagia were the second most frequent initial symptoms (22%).
Table 1.
Clinical characteristics of the patients.
| No. | GFAP mutation | Sex | Age at diagnosis, years | Age at onset, years | Initial symptom | Ref |
|---|---|---|---|---|---|---|
| 1 | R79H | F | 51 | 5 | Gait disturbance | 7 |
| 2a | R126_L127dup | M | 12 | 10 | Gait disturbance | 8 |
| 3 | D360N | F | 36 | 14 | Dysarthria | |
| 4a | L357P | F | 24 | 18 | Gait disturbance | 8 |
| 5 | E243dup | F | 27 | 22 | Blurred vision | 9 |
| 6 | A268D | M | 47 | 30 | Gait disturbance | 8 |
| 7 | A244V | F | 54 | 32 | NA | 8 |
| 8 | R79H | M | 37 | 36 | Gait disturbance | 7, 10, 11 |
| 9 | R79H | F | 40 | 38 | Dementia | 7, 12 |
| 10a | R416W | M | 39 | 38 | Gait disturbance | 8 |
| 11a | Y242N | F | 65 | 40 | Gait disturbance | 8, 13 |
| 12 | R276L | F | 32 | NA | Mental retardation | |
| Bulbospinal type | ||||||
| 13 | E362G | M | 58 | 12 | Gait disturbance | 7 |
| 14 | R416W | M | 49 | 45 | Limb weakness | 8 |
| 15 | L123P | F | 51 | 45 | Weakness | 8 |
| 16 | R124_L125insE | F | 47 | 46 | Gait disturbance | 8, 14 |
| 17 | M74T | M | 56 | 51 | Limb clumsiness | 7, 10, 15 |
| 18 | G301D | F | 52 | 51 | Gait disturbance | 8 |
| 19 | R258C | M | 59 | 55 | Gait disturbance | |
| 20 | M74T | M | 60 | 56 | Limb weakness | |
| 21 | E210K | F | 65 | 58 | Incontinence | 8, 16 |
| 22 | N386S | M | 60 | 59 | Dysarthria | 8 |
| 23 | R258H | M | 65 | 60 | Limb weakness | 8, 16 |
| 24 | N386S | M | 68 | 61 | NA | |
| 25 | N386S | M | 65 | 62 | Gait disturbance | 8, 16 |
| 26 | N386S | M | 66 | 63 | Dysarthria | 8 |
| 27 | R70W | M | 67 | 64 | Dysphagia | 7, 10 |
| 28 | R70W | F | 65 | 64 | Dysarthria | 8, 16 |
| 29 | R70W | M | 66 | 65 | Limb weakness | |
| 30 | N102K | F | 71 | 66 | Gait disturbance | 8 |
| 31 | N386S | M | 72 | 72 | Gait disturbance | 17 |
aThese patients had been diagnosed with the bulbospinal type8, but they were subsequently classified with the intermediate form based on strict re-evaluation in this study.
Ref = reference; F = female; M = male; NA = not available. Phenotypic differences in patients with the same genotype may be due to genetic modifiers.
Figure 1.
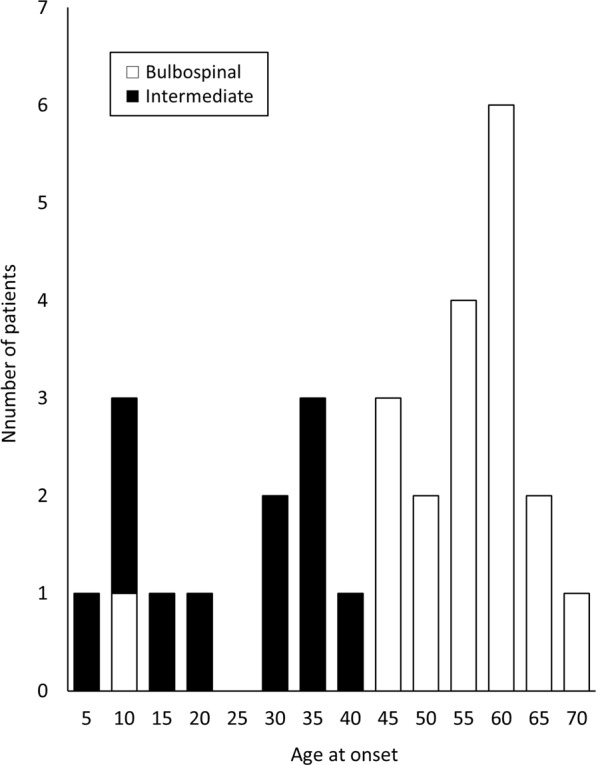
Distribution of age at onset of patients with Alexander disease. The age at onset (AAO) was distributed widely and bimodally: one peak was in those in their teens to thirties, and the other peak was at around 60 years. Most of the younger-onset patients presented with intermediate form, whereas older-onset patients presented with bulbospinal type. The boundary of both AAO and the phenotype was around 45 years. White and black bars indicate bulbospinal type and intermediate form, respectively.
GFAP genotype- phenotype correlation
A total of 21 unique GFAP mutations were identified in the 31 patients. They were distributed widely including head, rod, and tail domains, without hot spots (Fig. 2). As for 5 mutations shared by multiple patients, patients with R70W, M74T, and N386S all belonged to the older-onset group, and those with R79H belonged to the younger-onset group, whereas those with R416W were distributed in both younger- and older-onset groups. The remaining sixteen mutations were singletons. These findings suggested the difficulty of analyzing genotype-phenotype correlations for each mutation. Alternatively, we divided the mutations into two groups: “Neutral” and “Deleterious”, based on the prediction tool PROVEAN and compared AAO between the two groups (Fig. 3). Nine patients had “Neutral” and 22 patients had “Deleterious” mutations. The mean AAO was significantly different between “Neutral” (58.0 ± 8.8 years) and “Deleterious” (38.9 ± 8.8 years) groups (p < 0.01, t-test).
Figure 2.

Schematic localization of GFAP mutation and age at onset. Age at onset (longitudinal axis) and amino acid number of GFAP mutations (horizontal axis) of each patient are plotted. White and black dots indicate bulbospinal Alexander disease and intermediate form, respectively. GFAP protein consists of head, rod, and tail domains. The rod domain includes four alpha-helical subdomains (white boxes).
Figure 3.

The predicted pathogenicity of GFAP mutations and age at onset. The pathogenicity of GFAP mutations was divided into “Neutral” and “Deleterious” using the prediction tool PROVEAN. The mean age at onset of the “Deleterious” group (38.9 ± 8.8 years) was significantly younger than that of the “Neutral” group (58.0 ± 8.8 years) (p = 0.00999, t-test). White and black dots indicate bulbospinal Alexander disease and intermediate form, respectively.
WES/Microarray analysis
WES was performed to identify exonic functional variants, and microarray was used for genome-wide analysis. In WES, the mean depth (defined as “total number of reads mapped to the target regions/total number of target regions”) was 169, and the mean coverage (defined as “number of target regions covered over 50 reads/total number of target regions”) was 90.7%. In microarray analysis, the average call rate was 99.8%.
An association study with logistic regression was performed by considering the predicted pathogenicity of the GFAP mutations described above. The number of variants investigated in WES and microarray analyses were 38,679 and 266,925, respectively (Fig. 4). Forty-six variants (37 genes) had a p-value < 0.01 in WES (Supplementary Table S1) and 645 variants (188 gene loci) met this value in microarray analysis (Supplementary Table S2). Of the forty-six variants with a p-value < 0.01 in WES data, thirty-five variants (76%) were reproducible in microarray data. The p-value of the recessive model tended to be unavailable or high mainly due to the absence or too small a number of minor allele homozygotes. No variant had a p-value below the significance level after Bonferroni correction using either analysis (WES-based significance: p < 1.29 × 10−6, microarray-based significance: p < 1.87 × 10−7).
Figure 4.

Flowchart of association analysis. We performed WES- and microarray-based association analysis (upper) and also adopted a candidate gene approach (lower). Initially, an association study between younger- and older-onset patients considering predicted GFAP mutation pathogenicity was conducted. There was no variant reaching genome-wide significance. Next, we focused on variants in candidate genes related to the pathophysiology of Alexander disease. (a–d) Lists of the variants or genes are presented in Supplementary Tables S1(a), S2(b), S5(c) and 2(d). WES = whole-exome sequencing; Chr = chromosome.
We additionally searched for modifiers solely in 19 patients with mutations that cause the bulbospinal type, because their symptoms are comparatively mild and the effect of these GFAP mutations might not be severe (Supplementary Materials S3 and S4). Sixteen and 22 variants showed p-values below 1.0 × 10−5 in WES and microarray analysis, respectively. One variant, p.Ala155Ala in TRABD2B, reached WES-based significance (p = 2.05 × 10−7). However, after excluding the outlier with a markedly early age at onset, no variant showed p-values below 1.0 × 10−5.
Candidate gene approach
The lack of WES- or microarray-based significant variants may be expected due to the small sample size and weaker modifier effect than GFAP mutations. To identify likely associated variants in AxD-related genes, we employed a candidate gene approach. Among 33 genes reported to have an association with AxD in the literature (Supplementary Table S5), the variants of 5 genes had a p-value < 0.05 using next-generation sequencing data (Table 2): upstream insertion variant of HDAC4; intron single nucleotide polymorphisms (SNPs) of CASP3 and GAN; and missense variants of PIK3CG and PIK3C2G. The variants of 7 genes were identified with a p-value < 0.05 using microarray data (Table 2): intron SNPs of HDAC9, PIK3AP1, SLC1A2, PIK3C2G, GAN and PIK3R5; 3’UTR SNP of HDAC9; and missense variants of PIK3CG and PIK3C2G. The missense variants in PIK3CG and PIK3C2G were identified with a p-value < 0.05 using both next-generation sequencing and microarray data.
Table 2.
Summary of next-generation sequencing and microarray candidate gene analysis in association with age at onset.
| Chr | Position | Minor/ Major Allele | MAF | Location (Effect) | Gene Name | Genotypea | p-value | Odds Ratiob | |
|---|---|---|---|---|---|---|---|---|---|
| Younger Onset | Older Onset | ||||||||
| Next-generation sequencing | |||||||||
| 2 | 240323905 | T/TG | 0.27 | upstream | HDAC4 | 0/1/12 | 1/11/6 | 0.018 | 0.06 |
| 4 | 185559487 | G/C | 0.21 | intron | CASP3 | 0/1/12 | 0/8/10 | 0.009 | 0.04 |
| 7 | 106509331 | A/C | 0.15 | exon (missense) | PIK3CG | 0/7/6 | 0/3/15 | 0.033 | 11.94 |
| 12 | 18649057 | T/C | 0.31 | exon (missense) | PIK3C2G | 0/5/8 | 1/13/4 | 0.030 | 0.14 |
| 16 | 81398520 | G/A | 0.29 | intron | GAN | 0/8/5 | 0/2/16 | 0.014 | 18.22 |
| Microarray | |||||||||
| 7 | 18328833 | C/T | 0.18 | intron | HDAC9 | 1/0/12 | 0/9/9 | 0.035 | 0.08 |
| 7 | 18979516 | G/A | 0.27 | intron | HDAC9 | 2/7/4 | 0/6/12 | 0.031 | 5.26 |
| 7 | 19035920 | G/A | 0.39 | 3’ UTR | HDAC9 | 0/4/9 | 4/12/2 | 0.008 | 0.07 |
| 7 | 106509331 | A/C | 0.16 | exon (missense) | PIK3CG | 0/7/6 | 0/3/15 | 0.033 | 11.94 |
| 10 | 98395083 | T/C | 0.44 | intron | PIK3AP1 | 0/6/7 | 7/7/4 | 0.022 | 0.17 |
| 10 | 98403893 | A/G | 0.45 | intron | PIK3AP1 | 4/7/2 | 2/9/7 | 0.046 | 5.14 |
| 11 | 35399110 | A/G | 0.23 | intron | SLC1A2 | 1/9/3 | 0/3/15 | 0.009 | 11.87 |
| 12 | 18641138 | A/G | 0.32 | intron | PIK3C2G | 0/5/8 | 1/13/4 | 0.030 | 0.14 |
| 12 | 18649057 | T/C | 0.32 | exon (missense) | PIK3C2G | 0/5/8 | 1/13/4 | 0.030 | 0.14 |
| 12 | 18651702 | T/C | 0.39 | intron | PIK3C2G | 1/4/8 | 3/12/3 | 0.018 | 0.06 |
| 12 | 18787403 | G/A | 0.37 | intron | PIK3C2G | 1/9/3 | 3/6/9 | 0.036 | 7.28 |
| 16 | 81361294 | A/C | 0.31 | intron | GAN | 3/7/3 | 0/6/12 | 0.029 | 5.59 |
| 16 | 81363643 | C/A | 0.42 | intron | GAN | 6/6/1 | 0/8/10 | 0.014 | 15.30 |
| 16 | 81372725 | A/C | 0.32 | intron | GAN | 4/6/3 | 0/6/12 | 0.024 | 5.56 |
| 16 | 81395603 | T/C | 0.40 | intron | GAN | 6/6/1 | 0/7/11 | 0.011 | 17.61 |
| 16 | 81397804 | T/C | 0.16 | intron | GAN | 0/8/5 | 0/2/16 | 0.014 | 18.22 |
| 17 | 8834595 | A/G | 0.45 | intron | PIK3R5 | 3/9/1 | 3/7/8 | 0.020 | 16.98 |
The variants with a p-value < 0.05 in either additive, dominant, or recessive models are shown.
aThe number of patients with minor allele homozygous/heterozygous/major allele homozygous is shown.
bOdds ratio represents the odds of a younger onset with the minor compared with major allele.
Chr = chromosome; MAF = minor allele frequency; UTR = untranslated region.
These results were verified by Sanger sequencing of the SNP with the lowest p-value in each gene (Supplementary Fig. S6–1). In all patients, genotypes of the 9 representative SNPs determined by WES/microarray were consistent with those determined by the Sanger method, except for PIK3C2G SNP (position chr12:18651702, rs2305220) in one patient. In this particular patient, rs2305220 was T/T based on microarray; however, it was T/C based on Sanger sequencing. This discrepancy may have been due to another heterozygous SNP, rs191094996, located next to rs2305220 (Supplementary Fig. S6−2), suggesting a genotyping error caused by mismatched hybridization in microarray. When using corrected genotyping data, the p-value of PIK3C2G rs2305220 remained at < 0.05 (p = 0.0151).
Discussion
Our study is the first genome-wide analysis focusing on genetic modifiers in extremely rare monogenic diseases including AxD. The predicted pathogenicity of GFAP mutation was significantly correlated with AAO. In the association study considering GFAP mutation pathogenicity, we detected several genes likely to be associated with AAO, which may contribute to the pathophysiology of AxD, although no genome-wide variants with significance after Bonferroni correction were detected.
In this study, we obtained clinical information linked with genomic data for the first time for AxD, being helpful to elucidate phenotype-genotype associations. Because of the extreme rarity of AxD, independent patients in different hospitals were recruited. To standardize the format of their clinical data, we used a questionnaire including AAO, initial symptoms, family history, neurological examination, brain/spine MRI findings, physiological testing, and degree of disability. Furthermore, we re-evaluated original MRI data based on unified standards.
In our study analyzing patients with bulbospinal type and intermediate form of AxD, AAO was distributed widely from 5 to 72 years, and older-onset patients (AAO ≥ 45) accounted for 58%, which is a very large number of older-onset patients compared with a previous study where the mean AAO was around 21 years old18. That is, AxD, which belongs to monogenic neurological diseases, has a broader AAO than previously thought, suggesting the existence of modifier genes other than causative GFAP mutations contributing to the diversity of AAO, especially in the bulbospinal type and intermediate form. We also demonstrated that the GFAP mutation pathogenicity predicted by sequence conservation was significantly correlated with AAO. The mean AAO was 20 years younger in patients with “Deleterious” compared with “Neutral” mutations. We applied this finding to association analysis.
In the WES- or microarray-wide association study, each variant was analyzed considering the predicted pathogenicity of GFAP mutations as a covariate. A WES-based association study is a novel approach, validated with microarray-based analysis. A total of 76% of the variants based on the WES-based association study with a p-value < 0.01 were reproducible in microarray-based analysis, suggesting the reliability of the WES-based association study. Such a WES-based association study is expensive compared with microarray analysis, but it has the merit of identifying functional variants directly. A WES-based association study would be particularly useful if WES data already exist.
We could not detect any variants reaching WES- or microarray-based significance. This is mainly due to not only the small sample size but also the effect of GFAP mutations. Intermediate-type patients showed a more severe phenotype and a high frequency of GFAP mutations predicted as deleterious (11 out of 12, 92%). On the other hand, bulbospinal-type patients showed a mild phenotype and their GFAP mutations included both deleterious (11 out of 19, 58%) and neutral (8 out of 19, 42%) forms. These results suggest the weaker effect of GFAP mutations in the bulbospinal type. In the quantitative trait locus (QTL) analysis of bulbospinal-type patients, although the influence of one outlier was not negligible, comparatively lower p-values were obtained. Increasing the number of patients, especially with the bulbospinal type, will be critical to identify genetic modifiers in the future.
In rare monogenic disorders, a small number of samples will lead to a weak statistical power, but it is rational to focus on the pathophysiology related to causative genes playing a primary role. With a focus on variants in candidate genes selected from the literature, five significant candidate genes with p-values under 0.05 were identified. GAN encodes gigaxonin which plays a role in the neurofilament architecture. Gigaxonin is mutated in giant axon neuropathy (OMIM #256850), in which abnormal GFAP aggregation occurs. A recent report indicated that gigaxonin targets GFAP for proteasomal degradation19. Interestingly, the decreased proteasomal function in astrocytes would contribute to increase the toxic effects of mutant GFAPs20, suggesting that the dysfunction of gigaxonin may be involved in accelerating GFAP aggregation. SLC1A2 encodes the major glutamate transporter of astrocytes, GLT-121. Reductions of GLT-1 protein and mRNA were exacerbated in AxD mouse models22. The alteration of GLT-1 expression may affect the susceptibility to glutamate excitotoxity and neuron death. CASP3 encodes a protease, caspase 3, which plays a central role in cell apoptosis. Caspase 3 activation was reported to be correlated with mutant GFAPs in the C-terminal domain of GFAP, leading to the loss of astrocyte viability23. HDACs encode histone deacetylases, which play a role in the regulation of gene expression. Some HDACs were reported to control GFAP expression in primary human astrocytes24. In an exome next-generation sequencing study of two half-siblings with adult-onset AxD, the HDAC6 variant was shown to modulate a motor neuron disease-like phenotype25. PI3K encodes phosphatidylinositol-4,5-bisphosphate-3-kinase that is upstream of the key events leading to GFAP expression26, suggesting that the dysregulation of GFAP expression due to PI3K mutations may influence an abnormal GFAP aggregation process. Mutations in components of the PI3K-AKT pathway were found in patients with megalencephaly27, which is one of the major symptoms of intermediate form.
We validated the results of the candidate gene approach by Sanger sequencing to rule out latent genotype errors caused by WES/microarray methods. Surprisingly, ~5% errors caused by NGS in cancer samples were reported28. Of 273 genotyping data (9 variants of 31 patients) by Sanger sequencing, one genotype mismatched with that determined by microarray. This was probably due to another adjacent variant identified by the Sanger method. We should verify genotyping data by the Sanger method, especially when positive results are obtained by WES/microarray.
Recently, research involving a Drosophila AxD model identified a number of modifier genes29. Interestingly, of the 188 genes (p-value < 0.01) identified by our microarray analysis (Fig. 4), 9 genes (ABAT, MYCBP2, OPCML, PRKG1, TRIO, CDKAL1, CACNA2D3, GUCY1A2, and GPC5) corresponded to the modifier genes identified in the Drosophila model29.
Our study has several limitations. First, because the patients were all Japanese, our results may not be applicable to other ethnicities. Second, the number of patients was too small to detect variants with a weak effect. It was difficult to collect an adequate number of patients due to the markedly low prevalence of AxD: one case per 2.7 million individuals, of which bulbospinal type and intermediate form were estimated to collectively comprise 65%2. Finally, we could not collect a sufficient number of patients with identical GFAP mutations. Alternatively, we applied the in silico prediction of GFAP mutation pathogenicity. Of the prediction tools based on phylogenic conservation such as PROVEAN, PolyPhen-2, SIFT, and Mutation Assessor, we selected PROVEAN because it covers insertion/deletion mutations. To improve the prediction of pathogenicity, effects of mutations on the structure or stability of GFAP protein and annotations other than conservation should be considered. Combined Annotation–Dependent Depletion (CADD) is a prediction method to objectively integrate many diverse annotations into a single measure30. We compared predictions by PROVEAN, PolyPhen-2, SIFT, Mutation Assessor, and CADD (Supplementary Table S7). For some mutations, predictions were not consistent among the tools. In addition, we considered predicted pathogenicity based on the protein structure or stability reported previously31,32 (Supplementary Table S7). These findings suggest that prediction method used in this study is not definite. The use of multiple prediction tools and consideration of the protein structure or stability may be necessary in future analyses. To overcome these limitations in the future, recruiting larger numbers of AxD patients is indispensable. The identification of significant genome-wide genes will help elucidate the pathophysiology and therapeutic targets of AxD.
In conclusion, we conducted genome-wide analyses for the first time in AxD, which is an extremely rare monogenic disease, by employing two methods: WES and microarray. The association study in consideration of GFAP pathogenicity identified several variants of candidate genes with a p-value < 0.05. The results of the WES-based association study were comparable to those generated by microarray-based analysis. It is challenging to identify modifier genes with genome-wide significance for extremely rare monogenic disorders. Efforts toward establishing a genomic database of AxD may be important for confirming the results of the present study as well as identifying other potential modifier genes.
Methods
Participants
Forty-two Japanese AxD patients of 40 families with heterozygous GFAP mutations were diagnosed between 2004 and 2016, all of whom were referred to our facility from other hospitals throughout Japan for GFAP gene analysis because of suspected AxD. Clinical information, including AAO, family history, initial symptom, neurological findings, and MRI data, were obtained by survey questionnaire form. To strictly classify the clinical types of patients, brain and spinal MRI findings were re-evaluated by experienced neurologists (RY, TY) using original image data. Four patients who gave written informed consent for genetic testing, but did not consent to participate in further research, were excluded from this study. From the 38 patients who gave written consent of this study, we excluded five patients with cerebral type because the history and prognosis of cerebral type are almost constant for each case, and thus AAO of this form may be almost exclusively determined by GFAP mutations. Of the remaining 33 patients from 31 families, who met the diagnostic criteria of bulbospinal type or intermediate form, 31 probands were included. All participants provided written informed consent. This study was performed in accordance with the declaration of Helsinki and approved by the Ethical Review Board of Kyoto Prefectural University of Medicine.
WES and microarray analysis
We extracted genomic DNA from leukocytes and performed WES using standard protocols. DNA was captured with SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) and sequenced using Illumina HiScanSQ (Illumina, San Diego, CA, USA) with 108-bp paired-end reads in the NGS Core Facility at Kyoto Prefectural University of Medicine. After base calling with CASAVA v.1.8.2 (Illumina), reads were aligned to the human reference genome sequence (GRCh37/hg19) with Burrows-Wheeler Aligner v.0.7.1233. PCR duplicates were removed with Piacard v1.119. Single nucleotide variants and indels were called with the Genome Analysis Toolkit UnifiedGenotyper v.1.6-1334. Called variants were annotated with SnpEff v.4.0e35. Autosomal variants whose effect-impact was predicted as “High”, “Moderate”, or “Low” (except for synonymous variants) were analyzed. The bases without variant calls were considered to be identical genotypes to the reference allele. To ensure the accuracy of genotyping, variants with a minimum depth of 10 or lower were excluded.
In microarray analysis, we genotyped 551,839 genetic markers with InfiniumCoreExome-24 v.1.1 BeadChip (Illumina) and Genome Studio Software v.2011.1 (Illumina) according to the manufacturer’s instructions. The genotype data were obtained by using Genome Studio Software v.2011.1 (Illumina). A quality control filter was applied as: minor allele frequency >0 (i.e., monomorphic markers were excluded) and a call rate ≥95% for autosomal markers.
To validate the accuracy of WES/microarray genotyping, the SNP with the lowest p-value for each significant candidate gene was chosen and further PCR and Sanger sequencing were performed. The data were analyzed by Sequencher 4.10.1 (Gene Codes Corporation, Ann Arbor, MI, USA). The PCR/sequencing primers are described (Supplementary Table S8).
Statistical analysis
Firstly, GFAP mutations were divided into “Neutral” or “Deleterious” using the prediction tool PROVEAN36 based on sequence conservation. “Deleterious” means that the mutation is considered selectively deleterious because its alignment-based score among species is equal to or below a predefined threshold. “Neutral” means that the mutation is considered selectively neutral because its alignment-based score is above the threshold. The mean AAO was compared between patients with “Neutral” or “Deleterious” mutations using the t-test (R v.3.2.2)37.
Next, to identify variants related to AAO of AxD, we performed association analysis with logistic regression using WES and microarray data (Fig. 4 upper). The dependent variable was AAO as categorical data (a younger (<45 years) or older (≥45 years) onset). The independent variables were genotypes of each variant. The covariate was the predicted pathogenicity of GFAP mutations as binary categorical data (“Neutral” or “Deleterious”). Association analysis for additive, dominant, and recessive models and QTL analysis for age at onset were performed using PLINK v.1.07 software38. The additive model tested the effects of a minor allele dosage (0, 1, 2). Dominant and recessive models tested the effects of a minor allele between (DD, Dd) versus dd, and DD versus (Dd, dd), respectively (D and d are the minor and major alleles, respectively).
Finally, we focused on the candidate genes related to AxD pathophysiology (Fig. 4 lower). The p-values of all variations in the candidate genes were extracted from WES and microarray data. A list of candidate genes was created based on a search of PubMed using the keywords “Alexander disease” and “GFAP”. We also hand-searched relevant publications and articles (Supplementary Table S5).
Supplementary information
Acknowledgements
We thank all the participants. This work was supported by Intractable Disease Research Grants from the Ministry of Health, Labour and Welfare of the Government of Japan. We are grateful to the following doctors who provided valuable clinical data: Yoshiharu Takata (National Hospital Organization Okinawa Hospital), Hiroshi Kurisaki (National Hospital Organization Tokyo National Hospital), Yuichi Hayashi (Gifu University Graduate School of Medicine), Tadahiro Yonezu, Atsuhiko Sugiyama, and Setsu Sawai (Chiba University Graduate School of Medicine), Hirofumi Shii (Kokura Memorial Hospital), Akiko Nagaishi (National Hospital Organization Nagasaki Kawatana Medical Center), Takayuki Oka and Toshihiro Ide (Saga University), Yoshiaki Takahashi and Hisashi Narai (National Hospital Organization Okayama Medical Center), Yasuyoshi Kimura (Osaka University Graduate School of Medicine), Kwiyoung Park (National Hospital Organization Utano Hospital), Yasuo Ito (Saitama Medical University), Shotaro Haji (Hiroshima City Asa Hospital, Hiroshima), Ayako Sakoda (Kyusyu University Graduate School of Medicine), Tomohiro Hayashi (Toyama University), Yoshinari Kawai (Yokkaichi Municipal Hospital), Yuji Matsumoto (Ehime Prefectural Central Hospital), Tadanori Hamano (University of Fukui), Hana Ogura (St.Marianna University School of Medicine Hospital), Seitaro Nohara (Tsukuba University), Yuko Kawahara (Okayama University Graduate School of Medicine and Dentistry), Kentaro Maeda (Okazaki City Hospital), Eri Tanaka (Saiseikai Fukuoka General Hospital), Hiroaki Ishikawa (Teikyo University Chiba Medical Center), Tomoko Sekiya (Sumitomo Hospital).
Author Contributions
R.Y. performed experiments, analyzed data, and drafted the manuscript. R.S., H.A., Y.T., K.S. and J.M. performed experiments, and analyzed data. M. Nakano, T.Y. and I.M. designed the experiments and drafted the manuscript. M.Nakagawa, K.T. and T.M. designed the experiments and supervised the research. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rei Yasuda and Masakazu Nakano contributed equally.
Contributor Information
Tomokatsu Yoshida, Email: toyoshid@koto.kpu-m.ac.jp.
Kei Tashiro, Email: tashiro@koto.kpu-m.ac.jp.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-51390-8.
References
- 1.Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373–381. doi: 10.1093/brain/72.3.373. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida T, et al. Nationwide survey of Alexander disease in Japan and proposed new guidelines for diagnosis. J. Neurol. 2011;258:1998–2008. doi: 10.1007/s00415-011-6056-3. [DOI] [PubMed] [Google Scholar]
- 3.Brenner M, et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 2001;27:117–120. doi: 10.1038/83679. [DOI] [PubMed] [Google Scholar]
- 4.Li R, et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann. Neurol. 2005;57:310–326. doi: 10.1002/ana.20406. [DOI] [PubMed] [Google Scholar]
- 5.Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bello L, et al. Association Study of Exon Variants in the NF-kappaB and TGFbeta Pathways Identifies CD40 as a Modifier of Duchenne Muscular Dystrophy. Am. J. Hum. Genet. 2016;99:1163–1171. doi: 10.1016/j.ajhg.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida T, et al. Effects of a polymorphism in the GFAP promoter on the age of onset and ambulatory disability in late-onset Alexander disease. J. Hum. Genet. 2013;58:635–638. doi: 10.1038/jhg.2013.83. [DOI] [PubMed] [Google Scholar]
- 8.Yoshida T, Yasuda R, Mizuta I, Nakagawa M, Mizuno T. Quantitative Evaluation of Brain Stem Atrophy Using Magnetic Resonance Imaging in Adult Patients with Alexander Disease. Eur. Neurol. 2017;77:296–302. doi: 10.1159/000475661. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda R, Yoshida T, Mizuta I, Nakagawa M, Mizuno T. A novel three-base duplication, E243dup, of GFAP identified in a patient with Alexander disease. Hum Genome Var. 2017;4:17028. doi: 10.1038/hgv.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida T, et al. Glial fibrillary acidic protein mutations in adult-onset Alexander disease: clinical features observed in 12 Japanese patients. Acta Neurol. Scand. 2011;124:104–108. doi: 10.1111/j.1600-0404.2010.01427.x. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi Y, et al. Central hypothermia associated with Alexander disease. A case report. Clin. Neurol. Neurosurg. 2017;157:31–33. doi: 10.1016/j.clineuro.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Nagaishi A, Nakane S, Fukudome T, Matsuo H, Yoshida T. A case of Alexander disease suspected juvenile-onset and exacerbating after long stationary state. Rinsho Shinkeigaku (Clin Neurol) 2013;53:474–477. doi: 10.5692/clinicalneurol.53.474. [DOI] [PubMed] [Google Scholar]
- 13.Ogura H, Maki F, Sasaki N, Yoshida T, Hasegawa Y. Familial Adult-Onset Alexander Disease with a Novel GFAP Mutation. Mov. Disord. Clin. Pract. 2016;3:300–302. doi: 10.1002/mdc3.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawahara Y, et al. Type 2 Alexander disease with a novel glial fibrillary acidic protein gene mutation and its unique clinical features. Neurol. Clin. Neurosci. 2017;5:183–185. doi: 10.1111/ncn3.12146. [DOI] [Google Scholar]
- 15.Yonezu T, et al. A case of adult-onset alexander disease featuring severe atrophy of the medulla oblongata and upper cervical cord on magnetic resonance imaging. Case Rep. Neurol. 2012;4:202–206. doi: 10.1159/000345303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida T, et al. Characteristic abnormal signals in medulla oblongata—“eye spot” sign: Four cases of elderly-onset Alexander disease. Neurol Clin Pract. 2015;5:259–262. doi: 10.1212/CPJ.0000000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugiyama A, et al. Incidental diagnosis of an asymptomatic adult-onset Alexander disease by brain magnetic resonance imaging for preoperative evaluation. J. Neurol. Sci. 2015;354:131–132. doi: 10.1016/j.jns.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Prust M, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology. 2011;77:1287–1294. doi: 10.1212/WNL.0b013e3182309f72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin NH, et al. The role of gigaxonin in the degradation of the glial-specific intermediate filament protein GFAP. Mol. Biol. Cell. 2016;27:3980–3990. doi: 10.1091/mbc.E16-06-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in alexander disease astrocytes, and the small heat shock protein alphaB-crystallin reverses the inhibition. J. Biol. Chem. 2010;285:10527–10537. doi: 10.1074/jbc.M109.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messing, A. & Brenner, M. In Neuroglia (eds Helmut Kettenmann & Bruce R. Ransom) 884–895 (Oxford University Press, 2013).
- 22.Minkel HR, Anwer TZ, Arps KM, Brenner M, Olsen ML. Elevated GFAP induces astrocyte dysfunction in caudal brain regions: A potential mechanism for hindbrain involved symptoms in type II Alexander disease. Glia. 2015;63:2285–2297. doi: 10.1002/glia.22893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen YS, Lim SC, Chen MH, Quinlan RA, Perng MD. Alexander disease causing mutations in the C-terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability. Exp. Cell Res. 2011;317:2252–2266. doi: 10.1016/j.yexcr.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanski R, et al. Histone acetylation in astrocytes suppresses GFAP and stimulates a reorganization of the intermediate filament network. J. Cell Sci. 2014;127:4368–4380. doi: 10.1242/jcs.145912. [DOI] [PubMed] [Google Scholar]
- 25.Melchionda L, et al. Adult-onset Alexander disease, associated with a mutation in an alternative GFAP transcript, may be phenotypically modulated by a non-neutral HDAC6 variant. Orphanet J. Rare Dis. 2013;8:66. doi: 10.1186/1750-1172-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herrera F, Chen Q, Schubert D. Synergistic effect of retinoic acid and cytokines on the regulation of glial fibrillary acidic protein expression. J. Biol. Chem. 2010;285:38915–38922. doi: 10.1074/jbc.M110.170274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riviere JB, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xing R, et al. Whole-genome sequencing reveals novel tandem-duplication hotspots and a prognostic mutational signature in gastric cancer. Nature communications. 2019;10:2037. doi: 10.1038/s41467-019-09644-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, et al. Tissue and cellular rigidity and mechanosensitive signaling activation in Alexander disease. Nature communications. 2018;9:1899. doi: 10.1038/s41467-018-04269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z, Wang KK. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015;38:364–374. doi: 10.1016/j.tins.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim B, Kim S, Jin MS. Crystal structure of the human glial fibrillary acidic protein 1B domain. Biochem. Biophys. Res. Commun. 2018;503:2899–2905. doi: 10.1016/j.bbrc.2018.08.066. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/ (2015).
- 38.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.