

Abstract
Inflammasomes are cytosolic protein complexes, which orchestrate the maturation of active IL‐1β by proteolytic cleavage via caspase‐1. Although many principles of inflammasome activation have been described, mechanisms that limit inflammasome‐dependent immune responses remain poorly defined. Here, we show that the thiol‐specific peroxidase peroxiredoxin‐4 (Prdx4) directly regulates IL‐1β generation by interfering with caspase‐1 activity. We demonstrate that caspase‐1 and Prdx4 form a redox‐sensitive regulatory complex via caspase‐1 cysteine 397 that leads to caspase‐1 sequestration and inactivation. Mice lacking Prdx4 show an increased susceptibility to LPS‐induced septic shock. This effect was phenocopied in mice carrying a conditional deletion of Prdx4 in the myeloid lineage (Prdx4‐ΔLysMCre). Strikingly, we demonstrate that Prdx4 co‐localizes with inflammasome components in extracellular vesicles (EVs) from inflammasome‐activated macrophages. Purified EVs are able to transmit a robust IL‐1β‐dependent inflammatory response in vitro and also in recipient mice in vivo. Loss of Prdx4 boosts the pro‐inflammatory potential of EVs. These findings identify Prdx4 as a critical regulator of inflammasome activity and provide new insights into remote cell‐to‐cell communication function of inflammasomes via macrophage‐derived EVs.
Keywords: caspase‐1, extracellular vesicle, IL‐1β, inflammasome, Prdx4
Subject Categories: Immunology
Introduction
Inflammation is the physiologic response to infection or injury and aims to restore cellular and tissue integrity. Multimeric protein complexes termed “inflammasomes” are key mediators of acute and chronic inflammatory responses. They assemble in response to cellular stress and regulate the maturation and secretion of IL‐1‐like cytokines, which induce a potent pro‐inflammatory host response (Schroder & Tschopp, 2010). Pathologic conditions that lead to loss of control of IL‐1β processing and secretion are associated with various inflammatory diseases including hereditary periodic fever syndromes, gout, atherosclerosis (Ridker et al, 2017), and type 2 diabetes (Neven et al, 2004; Martinon & Tschopp, 2005; Dinarello et al, 2010; Duewell et al, 2010). The NLRP3 (NOD‐like receptor pyrin domain containing 3) inflammasome is the prototypical and best‐studied inflammasome and is strongly expressed in myeloid cells (Manji et al, 2002). The sensor and scaffolding protein NLRP3 and pro‐IL‐1β are induced in the presence of LPS (lipopolysaccharide), other TLR or NLR agonists, or certain cytokines such as TNF‐α or IL‐1β (Bauernfeind et al, 2009; Franchi et al, 2009). Following this priming step, NLRP3 is activated by a drop in intracellular K+ concentrations (Munoz‐Planillo et al, 2013) or by reactive oxygen species (Gaidt et al, 2016; Gross et al, 2016) commonly caused by various endogenous and exogenous danger signals like extracellular ATP‐induced purinergic receptor P2X7 (P2X7R) activation (Ferrari et al, 1997), monosodium urate, bacterial‐derived pore‐forming toxins, or nigericin (Kanneganti et al, 2006; Mariathasan et al, 2006). Upon activation, NLRP3 oligomerizes and forms a molecular platform by recruiting the adapter protein ASC (apoptosis‐associated speck‐like protein containing a CARD) and pro‐caspase‐1 (Martinon et al, 2009). Clustering of pro‐caspase‐1 molecules leads to proximity‐induced auto‐proteolysis into p20 and p10 subunits, which in turn cleave pro‐IL‐1β to generate active IL‐1β (Dinarello, 1998). Mature IL‐1β is released into the extracellular space alongside active caspase‐1 and oligomeric particles of the NLRP3 inflammasome (Baroja‐Mazo et al, 2014). Ever since an alternative secretory pathway for the leaderless IL‐1β has been reported (Rubartelli et al, 1990), the exact manner of release remains matter of debate. Suggested mechanisms include exocytosis via secretory lysosomes (Andrei et al, 1999, 2004), secretion by microvesicle shedding (MacKenzie et al, 2001), release of multivesicular bodies that may contain exosomes (Qu et al, 2007), an autophagy‐based secretory pathway (Dupont et al, 2011), gasdermin D‐dependent secretion via pores (Evavold et al, 2018) and a loss of membrane integrity leading to passive IL‐1β release that occurs in parallel with pyroptotic death of the secreting cell (Shirasaki et al, 2014; Martin‐Sanchez et al, 2016).
We have previously shown that the 2‐Cys oxidoreductase peroxiredoxin‐4 (Prdx4) is induced in response to microbial danger signals, particularly downstream of the innate immune receptor NOD2 and that Prdx4 negatively regulates NF‐κB signaling (Weichart et al, 2006). Here, we report that Prdx4 limits inflammasome activity by thiol‐mediated inactivation of caspase‐1. Mechanistically, we provide evidence that Prdx4 and caspase‐1 interact in the cytosol and form a redox‐sensitive regulatory complex via caspase‐1 cysteine 397 and a high‐molecular‐weight (HMW) complex of Prdx4. Furthermore, we show that Prdx4 is co‐localized with components of the inflammasome in extracellular vesicles (EVs). Within EVs, loss of Prdx4 resulted in increased levels of cleaved caspase‐1 and IL‐1β maturation. Importantly, EVs, derived from inflammasome‐activated macrophages, were able to transmit an IL‐1β‐dependent immune response to recipient cells, whereby Prdx4 deficiency boosted the pro‐inflammatory potential of EVs. We thus define a critical role for Prdx4 in the post‐translational and post‐secretional regulation of inflammasome activation and induction of inflammatory responses.
Results
Prdx4 protects from LPS‐induced septic shock
To determine how Prdx4 influences inflammatory responses in vivo, we generated Prdx4‐knockout (KO) mice (Appendix Fig S1). Mice were fertile and showed no spontaneous phenotype. To investigate the role of Prdx4 during inflammation, we challenged mice with sub‐lethal doses of LPS. We found that Prdx4‐deficient mice had increased body weight loss and delayed restoration of weight compared to their wild‐type (WT) littermates (Fig 1A). Consistent with the increased body weight loss, Prdx4 KO mice had significant higher Cxcl1, TNF‐α and IL‐1β levels in serum and peritoneal lavages at 24 h post‐LPS injection (Fig 1B–D). As IL‐1β is a major mediator of LPS‐induced systemic immune responses, we next blocked IL‐1β‐mediated signaling using the interleukin‐1‐receptor antagonist (IL‐1RA) Anakinra. In all IL‐1RA‐treated animals, weight loss was attenuated in response to LPS administration and no differences were found between Prdx4 KO and WT littermates (Fig 2A). Also, excessive serum Cxcl1, TNF‐α, and IL‐1β levels in LPS‐treated Prdx4 KO mice were significantly lowered upon the injection of IL‐1RA (Fig 2B). Thus, we concluded that loss of Prdx4 results in an aggravated inflammatory response, which involves increased IL‐1β signaling.
Figure 1. Prdx4 protects from LPS‐induced septic shock.

-
APercent body weight of male Prdx4 WT and KO mice over the 72 h course of LPS (4.5 mg/kg BW) or PBS injection (i.p.). Each circle represents a mean of n = 7 mice; vertical lines indicate SEM. *P < 0.05; **P < 0.01; ***P < 0.001 (two‐way‐ANOVA, Bonferroni post‐test).
-
B–DCytokine concentration in Prdx4 WT and KO mice in response to LPS injection. (B) Cxcl1 levels in serum (left) or peritoneal lavage (right) at indicated time points after LPS or PBS injection. (C) TNF‐α levels in serum (left) or peritoneal lavage (right) at indicated time points after LPS or PBS injection. (D) IL‐1β levels in serum (left) or peritoneal lavage (right) at indicated time points after LPS or PBS injection. Each dot represents an individual mouse. Horizontal lines indicate mean. **P < 0.01; ***P < 0.001; n.s. not significant (two‐tailed t‐test). Data are representative of two independent experiments.
Figure 2. Role of IL‐1 receptor blockade and myeloid‐specific ablation of Prdx4 in the endotoxin‐shock model.

- Percent body weight of male Prdx4 WT and KO mice over the 48 h course of LPS (4.5 mg/kg BW) injection (i.p.) and treatment with IL‐1 receptor antagonist (IL‐1RA) Anakinra (200 μg/mouse) or control. Arrows indicate time point of Anakinra injection. Each circle represents a mean of n = 5 mice; vertical lines indicate SEM. ***P < 0.001 (two‐way‐ANOVA, Bonferroni post‐test).
- Serum concentration of Cxcl1, TNF‐α, and IL‐1β in Prdx4 WT and KO mice injected with LPS, LPS, and IL‐1RA or control. Each dot represents an individual mouse. Horizontal lines indicate mean. *P < 0.05; **P < 0.01; ***P < 0.001; n.s. not significant (two‐tailed t‐test).
- Percent body weight of male Prdx4‐flox and Prdx4‐ΔLysMCre mice over the 48 h course of 4.5 mg/kg BW LPS (i.p.). Each circle represents a mean of n = 7 mice; vertical lines indicate SEM. *P < 0.05; ***P < 0.001 (two‐way‐ANOVA, Bonferroni post‐test).
- Serum concentration of Cxcl1, TNF‐α, and IL‐1β in Prdx4‐flox and Prdx4‐ΔLysMCre mice injected with LPS. Each dot represents an individual mouse. Horizontal lines indicate mean. *P < 0.05; ***P < 0.001; n.s. not significant (two‐tailed t‐test). Data are representative of two independent experiments.
Prdx4‐deficient macrophages display elevated cytokine responses and inflammasome activation
We next sought to determine the major cellular source of the increased IL‐1β generation. As myeloid cells have been described as critical producers of pro‐inflammatory cytokines in LPS‐induced septic responses (Dinarello et al, 1974; Baracos et al, 1983), we crossed floxed Prdx4 mice to a LysMCre deleter strain in order to obtain mice that specifically lack Prdx4 in cells of myeloid origin, hereafter referred to as Prdx4‐ΔLysMCre (Appendix Fig S2A). Knockout of Prdx4 was confirmed by Western blot analysis of bone marrow‐derived macrophages (BMDMs) with antibodies against Prdx4 (Appendix Fig S2B). Since the results from the whole‐body knockout mice showed the largest difference in body weight loss between 40 and 60 h after LPS injection, Prdx4‐ΔLysMCre and floxed littermates were monitored for 48 h post‐LPS injection. Comparable to Prdx4 KO mice, Prdx4‐ΔLysMCre mice showed a significantly increased body weight loss starting from 36 h after LPS injection until the end point (Fig 2C). Also, we found higher Cxcl1, TNF‐α, and IL‐1β levels in the serum of Prdx4‐ΔLysMCre mice compared to floxed littermates (Fig 2D). Collectively, these results suggest a critical role of the myeloid compartment for the Prdx4‐mediated protection during endotoxin shock.
Because Prdx4 deficiency led to increased cytokine responses following LPS challenge in vivo, we used BMDMs from Prdx4 WT and KO mice to characterize the altered responses to LPS in more detail. In a time course of LPS stimulation, we confirmed that LPS‐induced release of Cxcl1 and TNF‐α was significantly increased in Prdx4‐deficient BMDMs (Fig 3A). Importantly, we found that the absence of Prdx4 also led to a time‐dependent release of IL‐1β. This is of interest since LPS stimulation alone is usually not sufficient to trigger significant IL‐1β release in WT BMDMs (Hagar et al, 2013; Kayagaki et al, 2013). Thus, we next induced IL‐1β release by activation of the inflammasome. We confirmed that loss of Prdx4 leads to excessive release of IL‐1β in BMDMs that were primed with LPS to induce expression of inflammasome components (Bauernfeind et al, 2009) followed by a time course of ATP treatment (Fig 3B). Accordingly, we detected increased levels of mature IL‐1β in the supernatant of Prdx4‐deficient BMDMs (Fig 3C). Next, we used HEK293 cells that were forced to secrete IL‐1β by pro‐IL‐1β/caspase‐1 overexpression. In line with our previous findings, co‐expression of Prdx4 decreased levels of mature IL‐1β (Appendix Fig S3). Since previous reports demonstrated a role for Prdx4 in the redox‐dependent regulation of NF‐κB activation (Jin et al, 1997; Weichart et al, 2006; Yu et al, 2010) and reactive oxygen species (ROS) also contribute to NF‐κB‐dependent NLRP3 priming (Bauernfeind et al, 2011), we hypothesized that Prdx4 deficiency might affect inflammasome priming leading to the observed differences in IL‐1β levels. Unexpectedly, we did neither find genotype‐dependent differences on Nlrp3 protein levels or stability, nor on Nlrp3 or Il1b mRNA levels in response to LPS‐induced priming or on other inflammasome components or redox proteins related to inflammasome activation (Fig EV1). To investigate whether the formation of ASC specks downstream of inflammasome activation is affected by Prdx4, BMDMs were stimulated with nigericin after LPS priming or left untreated. We did not find differences in ASC speck formation (Fig 3D), indicating that increased IL‐1β levels in Prdx4 KO BMDMs do not result from increased ASC speck formation. However, we detected increased levels of cleaved caspase‐1 in the supernatant of Prdx4 KO BMDMs after nigericin‐induced inflammasome activation (Fig 3E), indicating that Prdx4 negatively influences caspase‐1 activation. In order to validate whether unrestrained caspase‐1 activity accounts for the IL‐1β hypersecretion in Prdx4‐deficient BMDMs, we used the selective caspase‐1 inhibitor YVAD. We found that YVAD completely reduced the elevated IL‐1β levels in the supernatant of Prdx4‐deficient BMDMs (Appendix Fig S4), confirming that Prdx4‐dependent IL‐1β hypersecretion is dependent on caspase‐1. Next, we investigated the impact of Prdx4 on canonical caspase‐1 inflammasome activation and IL‐1β release. We found that loss of Prdx4 led to increased IL‐1β release compared to WT BMDMs in response to canonical inflammasome activation induced by either ATP and nigericin (NLRP3 inflammasome), double‐stranded DNA (AIM2 inflammasome), or flagellin (NLRC4 inflammasome), although the highest fold change was found for ATP and nigericin stimulation (Fig 3F). Interestingly, the ATP‐, nigericin‐, and flagellin‐induced LDH release was affected by Prdx4 as well (Fig 3G). We therefore concluded that Prdx4 negatively regulates caspase‐1‐dependent inflammasome responses in myeloid cells.
Figure 3. Prdx4‐deficient macrophages display elevated cytokine responses and inflammasome activation.

- Concentration of Cxcl1, TNF‐α, and IL‐1β in the supernatants of Prdx4 WT and KO BMDMs in response to a time course of LPS stimulation (100 ng/ml LPS, time points indicated).
- IL‐1β release of Prdx4 WT and KO BMDMs, untreated, or primed for 6 h with LPS (100 ng/ml) and then pulsed for indicated time points with ATP (5 mM).
- Western blot analysis of IL‐1β in cell lysates and supernatants of Prdx4 WT and KO BMDMs, primed with LPS (100 ng/ml), and pulsed with ATP (5 mM) for 4 h or left untreated. Dashed line indicates vertical slice.
- Immunofluorescence microscopy of ASC speck formation in Prdx4 WT and KO BMDMs in response to nigericin (10 μg/ml) stimulation for 45 min of LPS‐primed cells. Cells were stained with an antibody to ASC, and nuclei were counterstained using DAPI. Scale bar indicates 20 μm. ASC speck‐positive cells were counted and expressed as percentage of total cells. Bars represent a mean of n = 4 mice; vertical lines indicate SD. n.s. not significant (two‐tailed t‐test).
- Western blot analysis of caspase‐1 cleavage in the supernatant of Prdx4 WT and KO BMDMs in response to nigericin (10 μg/ml) stimulation for 1 h after priming with LPS (100 ng/ml), LPS priming alone or without stimulation. Whole‐cell lysates were analyzed for pro‐caspase‐1 and Gapdh levels.
- IL‐1β release in Prdx4 WT and KO BMDMs, untreated, or primed for 6 h with LPS (100 ng/ml) and then pulsed for 3 h with ATP (5 mM) or nigericin (10 μg/ml) or transfected for 3 h with poly(dA:dT) or flagellin (1 μg/ml each) or treated with transfection agent only.
- Quantification of cell death by LDH release in Prdx4 WT and KO BMDMs, untreated, or primed for 6 h with LPS (100 ng/ml) and then pulsed for 3 h with ATP (5 mM) or nigericin (10 μg/ml) or transfected for 3 h with poly(dA:dT) or flagellin (1 μg/ml each) or treated with transfection agent only.
Figure EV1. Prdx4 does not impact priming of inflammasome components or associated factors (relates to Fig 3).

- qRT–PCR analysis of Nlrp3, Il1b, Il18, ASC, caspase‐1, Nlrp1, Trxnip, and Nos2 relative to Gapdh mRNA in Prdx4 WT and KO BMDMs, primed for 6 h with LPS or left untreated.
- Western blot analysis of NLRP3, pro‐caspase‐1, ASC, pro‐IL‐1β, Prdx4, and β‐actin (loading control) in Prdx4 WT and KO BMDMs at 6 h after LPS stimulation.
- Western blot analysis of NLRP3, Prdx4, and β‐actin (loading control) in Prdx4 WT and KO BMDMs at 6 h after LPS stimulation and CHX treatment for the time points indicated.
Prdx4 interacts with C397 of caspase‐1 to block its function
In order to investigate the molecular mechanism by which Prdx4 negatively regulates caspase‐1‐dependent inflammasome activation, we hypothesized that Prdx4 directly interacts with caspase‐1 to limit its downstream cleavage and activation. To test this hypothesis, we assessed whether Prdx4 and caspase‐1 interact in vitro using active forms of recombinant human PRDX4 (rPRDX4) and human caspase‐1 (rCASP‐1). Under physiologic conditions and depending on the redox environment, Prdx4 is known to form oligomeric high‐molecular‐weight (≥ 250‐kDa) structures, with a high abundance of decamers consisting of five disulfide‐linked dimers (Tavender et al, 2008). We therefore co‐incubated rPRDX4 with rCASP‐1 and analyzed the proteins under non‐reducing conditions, in order to preserve disulfide bridges and to detect the presence of disulfide‐linked complexes. Notably, we detected rCASP‐1 at approx. 250 kDa, corresponding to the size of the described PRDX4 decamer with a concurrent reduction of rCASP‐1 p10 levels (Fig 4A). This suggested a direct interaction of rCASP‐1 with the PRDX4 high‐molecular‐weight complex (HMWC). In order to verify this finding by an independent approach, we performed HPLC‐MS analysis from cut‐out bands of Coomassie‐stained SDS–PAGE under non‐reducing conditions. Within the gel bands corresponding to the size of the Prdx4 decamer/HMWC, we detected peptides corresponding to the p10 as well as to the p20 subunit of rCASP‐1 (Fig EV2, Table EV1). Additionally, we observed that co‐incubation of rCASP‐1 with rPRDX4 resulted in a decrease of the Prdx4 band intensity at approx. 250 kDa and the appearance of an additional band at approx. 50 kDa (longer exposure of WB), which corresponds to the molecular weight of a Prdx4 dimer. To rule out that caspase‐1 catalytically cleaves Prdx4, we searched for putative caspase‐1 cleavage sites in the mature Prdx4 protein using MEROPS (Rawlings et al, 2018) (www.ebi.ac.uk/merops/) and ExPaSy (Artimo et al, 2012) (www.expasy.org/) databases, which did not result in predicted target motifs. Moreover, overexpression of the catalytically inactive p20 C285S active‐site mutant did not terminate the shift of p20 into the Prdx4 decamer/HMWC, nor the occurrence of the weaker band corresponding to the molecular weight of the Prdx4 dimers (Appendix Fig S5A), arguing against a caspase‐1‐mediated cleavage of Prdx4. We therefore hypothesized that Prdx4 controls caspase‐1 function in a thiol‐specific manner resulting in the integration of caspase‐1 into the Prdx4 decamer leading to its inactivation. Thus, we next tested whether the integration of caspase‐1 by Prdx4 decamers depends on cysteine residues of caspase‐1. We alkylated rCaspase‐1 (CA), rPrdx4 (PA), or both proteins (CA+PA) with iodoacetamide before their co‐incubation, thereby disabling the formation of intermolecular disulfide bonds. Under all conditions tested, alkylation terminated the integration of rCASP‐1 into the rPRDX4 decamer/HMWC (Fig 4B), implying that caspase‐1 and Prdx4 interact via disulfide bridges. We then determined whether caspase‐1 function is specifically altered by non‐reduced Prdx4 complexes and measured caspase‐1 activity in the presence of non‐reduced or the reduced form of Prdx4. Indeed, we found that non‐reduced rPRDX4 significantly inhibited caspase‐1 activity whereas the reduced form of rPRDX4 had no significant effect (Fig 4C). In order to control for specificity, we co‐incubated rPRDX4 with rGAPDH, which contains an active‐site cysteine, known to be redox‐sensitive (Nakajima et al, 2009). However, co‐incubation with non‐reduced rPRDX4 had no impact on the oligomeric structure of the homotetrameric rGAPDH (Appendix Fig S5B), pointing toward a specific disulfide bond exchange between caspase‐1 and Prdx4. Together, this confirmed that (i) caspase‐1 and Prdx4 interact in a disulfide‐dependent manner and (ii) a high‐molecular‐weight complex of Prdx4 controls caspase‐1 activity.
Figure 4. Prdx4 interacts with C397 of caspase‐1 to block its function.

- Western blot analysis of rPRDX4, rCASP‐1, and co‐incubated rPRDX4 and rCASP‐1 after non‐reducing SDS–PAGE. Black arrows at 250 kDa indicate the corresponding molecular weights of rCASP‐1 and rPRDX4 oligomers and decrease of the Prdx4 band intensity upon co‐incubation with rCASP‐1. Black arrow at approx. 50 kDa indicates the appearance of an additional rPRDX4 band which corresponds to the molecular weight of a Prdx4 dimer.
- Western blot analysis of rPRDX4 (=P) and rCASP‐1 (=C) after alkylation (=A) and non‐reducing SDS–PAGE. Black arrows indicate the termination of the integration of rCASP‐1 into the rPRDX4 decamer/HMWC upon alkylation.
- Rate of caspase‐1 activity in the presence of non‐reduced decameric rPrdx4, reduced dimeric and monomeric rPRDX4, YVAD, or control.
- Fold change in IL‐1β concentration in supernatants of HEK293 cells transfected with plasmids for NLRP3, ASC, IL‐1β, and caspase‐1 WT or Cys‐to‐Ser mutants C362S or C397S and co‐transfected with Prdx4 or control. Cells were stimulated with 2.5 mM ATP for 30 min before analysis.
- Western blot analysis of non‐reducing SDS–PAGE of cell lysates from HEK293 cells transfected with caspase‐1 WT, C362S, C397S, or C362S plus C397S mutants and co‐transfected with Prdx4‐GFP or GFP as control.
- Western blot analysis of co‐immunoprecipitation using HA‐magnetic beads from cell lysates of HEK293 cells transfected with HA‐tagged caspase‐1 WT, C362S, C397S, or C362S plus C397S mutants and co‐transfected with Prdx4‐GFP or GFP as control.
- IL‐1β concentration in supernatants of HEK293 cells transfected with plasmids for NLRP3, ASC, IL‐1β, and caspase‐1 and co‐transfected with Prdx4 WT or Cys‐to‐Ala mutants C51A, C124A, C245A, or DM C124A/C245A or control. Cells were stimulated with 2.5 mM ATP for 30 min before analysis.
- Western blot analysis of co‐immunoprecipitation using HA‐magnetic beads from cell lysates of HEK293 cells transfected with HA‐tagged caspase‐1 WT or control and co‐transfected with Prdx4 WT or Cys‐to‐Ala mutants C51A, C124A, C245A, or DM C124A/C245A.
Figure EV2. Analysis of caspase‐1 HMW complex by mass spectrometry (relates to Fig 4).

-
A, BSDS–PAGE and mass spectrometry analyses of rPRDX4, rCASP‐1, or co‐incubated rPRDX4+rCASP‐1. (A) rPRDX4, rCASP‐1, or co‐incubated rPRDX4+rCASP‐1 as well as rPRDX4 and rCASP‐1 treated with DTT were loaded onto the gel, and SDS–PAGE was performed on a 4% polyacrylamide stacking gel followed by a 10% gel for separation. The gray box depicts a schematic illustration of Prdx4 decamers/multimers, dimers and monomers. Excised spots (1–24), analyzed by in‐gel digestion and LC‐ESI MS, are indicated (red rectangles). Gel spots were digested with pepsin, and peptide extracts were analyzed by LC‐ESI MS with an Orbitrap Velos mass spectrometer. Both HCD and ETD spectra were acquired. Caspase‐1 was identified by three peptides together with Prdx4 in‐gel spot 1 (asterisk). (B) SDS–PAGE was performed on a 4–20% gradient gel. Excised spots (1–24), analyzed by in‐gel digestion and LC‐ESI MS, are indicated (red rectangles). Gels spots were digested with pepsin, and peptide extracts were analyzed on two LC‐ESI MS platforms. HCD spectra were acquired with a QExactive MS, and both HCD and ETD spectra were acquired using an Orbitrap Velos MS instrument. Spots where at least two caspase‐1 peptides could be detected on Prdx4 gel spots are indicated by asterisk.
Source data are available online for this figure.
Next, we wanted to know whether this can be attributed to a specific cysteine residue of caspase‐1. The cysteines C362 and C397 have previously been found to be modified by glutathione and their mutation into serine resulted in increased caspase‐1 activity (Meissner et al, 2008). We thus hypothesized that the Prdx4‐mediated decrease of caspase‐1 activity would be lost in a Cys‐to‐Ser mutant that displays sensitivity toward Prdx4. We therefore overexpressed the gain‐of‐function Cys‐to‐Ser mutants C362S, C397S, or the C285S active‐site mutant together with either Prdx4 or a control and analyzed subsequent IL‐1β secretion. We confirmed that C362S and C397S mutants exhibited increased caspase‐1 activity compared to WT caspase‐1 when Prdx4 was not co‐expressed (Fig 4D), whereas the C285S active‐site mutant, as expected, showed no effect. In the presence of Prdx4, caspase‐1 C362S activity was decreased compared to the control, whereas it remained unaltered in the C397S mutant. This indicated that the cysteine 397 of caspase‐1 is responsive to Prdx4 and mediates the Prdx4‐induced caspase‐1 inhibition. To underscore this finding, we analyzed the interaction of caspase‐1 with the high‐molecular‐weight form of Prdx4 under non‐reducing conditions. To this end, cells were transfected with caspase‐1 WT, C362S, C397S, or C362S plus C397S mutants in the presence or absence of co‐transfected Prdx4. We found that overexpression of the C397S as well as the C362S plus C397S mutants strongly decreased the formation of the Prdx4‐caspase‐1 high‐molecular‐weight complex (Fig 4E), indicating that the C397 of caspase‐1 forms the disulfide bridge with Prdx4. To confirm this result, we performed co‐immunoprecipitation from cells that were transfected with caspase‐1 WT, C362S, C397S, or C362S plus C397S mutants in the presence or absence of co‐transfected Prdx4. Notably, Prdx4 co‐precipitated with either caspase‐1 WT or caspase‐1 C362, confirming the interaction between Prdx4 and caspase‐1 (Fig 4F). However, Prdx4 co‐precipitation was drastically reduced with the caspase‐1 C397S or the C362S plus C397S mutants, underscoring the necessity of caspase‐1 C397 for the thiol‐dependent interaction with Prdx4. In order to reciprocally map the cysteine residues of Prdx4, we used catalytic (C124, C245) and conformational (C51) Cys‐to‐Ala mutants of Prdx4 that lack the ability to form functional decamers or multimers as previously described (Tavender et al, 2008, 2010). Notably, unlike WT Prdx4, all mutants tested failed to decrease the caspase‐1‐induced IL‐β secretion (Fig 4G) and showed loss of caspase‐1 interaction (Fig 4H). Since the proper structure and function of the high‐molecular‐weight form of Prdx4 is compromised in all mutants tested (Tavender et al, 2008, 2010), we conclude that rather than a particular cysteine of Prdx4, the high‐molecular‐weight conformation of Prdx4 is required for the interaction with the redox‐sensitive C397 of caspase‐1 to block its function.
Prdx4 is secreted upon activation of the NLRP3 inflammasome and co‐localizes with caspase‐1 in MVBs
In a next step, we explored whether and where endogenous Prdx4 and caspase‐1 co‐localize in cells under physiological conditions, which is a prerequisite for their functional interaction. Several compartments have been shown to be involved in inflammasome/caspase‐1‐mediated IL‐1β maturation and secretion, which may differ between cell types and activation states. In monocytes and macrophages, evidence points to primary processing of pro‐IL‐1β in the cytosol, while the lysosomal secretory pathway seems less important (Singer et al, 1995; Brough & Rothwell, 2007). Prdx4 is described to be mainly localized within the ER (Tavender et al, 2008; Zito et al, 2010; Kakihana et al, 2013) or can be secreted via classical exocytosis (Matsumoto et al, 1999; Okado‐Matsumoto et al, 2000). Several systematic analyses of subcellular protein localization (Itzhak et al, 2016; Thul et al, 2017), however, have suggested that a pool of Prdx4 might be present in the cytosol, too. We used cellular fractionation methods to address the question where Prdx4 is localized under baseline conditions and whether activation of the inflammasome has an influence on the cellular compartmentalization of Prdx4. To this end, we stimulated BMDMs with LPS or LPS+ATP and monitored levels of Prdx4 in the cytosol, the insoluble fraction containing all membranous compartments (including ER and Golgi) by Western blot [using a modified version of the protocol by Song et al (2006)]. We show that (i) Prdx4 can be found, as expected, in membranous compartments as well as in the cytosolic fraction, where also caspase‐1 and Gapdh are present. (ii) Upon LPS and LPS+ATP stimulation, levels of Prdx4 increased in both compartments (Fig 5A). LPS stimulation of BMDMs in vitro led to a significant release of Prdx4 into the supernatant, which was further increased by adding ATP (Fig 5B). LPS injection in mice in vivo resulted in significantly elevated Prdx4 levels between 3 h and 24 h post‐LPS challenge (Fig 5C). Since high serum levels of Prdx4 have been associated with disease severity in human sepsis patients (Schulte et al, 2011), we sought to investigate the link between Prdx4 secretion and inflammasome activation in more detail. Thus, we next used YVAD to block caspase‐1 activity and to monitor Prdx4 release. We found that YVAD significantly reduced levels of extracellular Prdx4 after LPS/ATP stimulation (Fig 5D), indicating that Prdx4 secretion follows caspase‐1 activation. In order to evaluate the mode of Prdx4 release upon inflammasome activation in more detail (Fig 5E), we next tested whether Prdx4 is passively lost in response to pyroptotic membrane rupture. We found that pretreatment with the cytoprotective agent glycine had no effect on Prdx4 release (Fig 5F). In contrast, both blockage of the pyroptotic pore‐forming protein gasdermin D (GSDMD) by its direct chemical inhibitor necrosulfonamide (NSA) (Rathkey et al, 2018) and inhibition of extracellular vesicle shedding by GW4869 (Kosaka et al, 2010; Mittelbrunn et al, 2011) significantly lowered Prdx4 secretion. Together, these data suggest that Prdx4 is not passively lost in response to inflammasome activation and its release involves GSDMD‐dependent mechanisms and also the formation of extracellular vesicles. Of note, GW4869, which inhibits the ceramide‐mediated inward budding of multivesicular bodies (MVBs) and release of mature extracellular vesicles from MVBs (Trajkovic et al, 2008), also significantly diminished Prdx4 and caspase‐1 release into the medium. Although these experiments do not provide definitive evidence for cytosolic Prdx4, we reasoned that cytosolic Prdx4 together with cytosolic caspase‐1 might be sorted into MVBs and released via EVs from the cell. To further confirm the presence of Prdx4 and caspase‐1 in MVBs, we next performed density gradient ultracentrifugation of LPS and ATP‐treated BMDMs to fractionate membranous compartments, including MVBs. We found that Prdx4 was enriched in fraction I‐III. Moreover, fraction I and II were exclusively positive for CD63, a reported marker for MVBs (Kobayashi et al, 2000) (Fig 5G), suggesting that Prdx4 is present in MVBs. The ER/Golgi protein Gosr1 was enriched in fractions III and VI, whereas mitofilin, a marker for mitochondria, was enriched in fractions V and VI. Fractions I‐III, and to a lesser extent fraction IV, were positive for the secretory protein beta‐2 microglobulin. When we investigated the fractionation of caspase‐1 and other inflammasome components, we found that Asc was highly abundant in fraction I and II, pro‐caspase‐1 was enriched in fraction II and III and as well as in fraction V and VI and that pro‐IL‐1β was present in all fractions. Together, the data show the complexity of the compartmentalization of pro‐IL1β, caspase‐1, Prdx4, and the inflammasome apparatus. The results suggest that Prdx4 and caspase‐1 along with Asc and pro‐IL‐1β are co‐present in MVBs and led to the hypothesis that inflammasome activation may trigger the shedding of MVB‐derived extracellular vesicles.
Figure 5. Prdx4 is secreted upon activation of the NLRP3 inflammasome and co‐localizes with caspase‐1 in MVBs.

- Western blot analysis of Prdx4, pro‐caspase‐1, Gapdh, and E‐Cadherin from the cytosolic and insoluble cell fraction of LPS and/or ATP‐stimulated BMDMs or untreated controls.
- Prdx4 concentration in supernatants of Prdx4 WT or KO BMDMs, primed for 6 h with LPS, and pulsed for indicated time points with 5 mM ATP. Each circle represents a mean of n = 3 mice; vertical lines indicate SD. **P < 0.01; n.s. not significant (two‐tailed t‐test).
- Concentration of Prdx4 in the serum of WT mice, injected with LPS (4.5 mg/kg BW) for the time points indicated. Each dot represents an individual mouse. Horizontal lines indicate mean. *P < 0.05; **P < 0.01; ***P < 0.001; n.s. not significant (two‐tailed t‐test).
- Concentration of Prdx4 in supernatants of Prdx4 WT and KO BMDMs. Cells were primed with LPS (100 ng/ml) for 6 h, followed by pretreatment with 20 μM YVAD or DMSO as control for 30 min and stimulated with 5 mM ATP for 4 h or no further stimulation. Each bar represents a mean of n = 3 mice; vertical lines indicate SD. **P < 0.01; n.s. not significant (two‐tailed t‐test).
- Schematic illustration of selected mechanisms that were targeted by either glycine, necrosulfonamide (NSA), or GW4869 to study LPS+ATP‐induced Prdx4 secretion.
- Relative levels of Prdx4 secretion in response to LPS+ATP stimulation and pretreatment with either glycine, NSA, or GW4869 and relative levels of caspase‐1 secretion in response to LPS+ATP stimulation and pretreatment with in response to LPS+ATP stimulation and pretreatment with GW4869. Each bar represents a mean of n = 3 biological with two technical replicates; vertical lines indicate SD. *P < 0.05; **P < 0.01; n.s. not significant (two‐tailed t‐test).
- Western blot analysis of subcellular organelle fractions. OptiPrep density gradient ultracentrifugation was used to fractionate subcellular organelles from Prdx4 WT BMDMs that were primed with LPS (100 ng/ml) for 12 h and stimulated with 5 mM ATP for 4 h.
ATP‐induced NLRP3 inflammasome activation leads to secretion of distinct EVs from BMDMs
The data described above indicate that a proportion of the inflammasome and Prdx4 are co‐secreted upon inflammasome activation from macrophages, most likely originating from MVBs. To examine whether vesicle shedding occurs in response to caspase‐1 activation, we isolated and purified extracellular vesicles from the supernatant of BMDMs that were untreated or LPS‐primed in the presence or absence of either ATP, nigericin, poly(dA:dT), or flagellin to induce caspase‐1‐dependent inflammasome activation. Subsequently, we applied several methods to comprehensively characterize the EV isolates (Fig 6A). Our analyses demonstrated a significant increase in protein concentration upon stimulation with all inflammasome activators (Fig 6B), whereas the number of EVs was exclusively enhanced after ATP‐ and nigericin stimulation (Fig 6C). Interestingly, TEM data revealed the presence of EVs with an average diameter of approximately 50 nm in all isolates except in EVs derived from LPS+ATP‐stimulated BMDMs, which exhibited an average diameter of approximately 110 nm (Fig 6D and E). To validate this finding, we independently determined the size distribution profile by performing dynamic light scattering (DLS) measurements. We obtained similar results using DLS particle analysis, confirming that the average size of particles isolated from LPS+ATP‐stimulated BMDMs was higher compared to particles derived from unstimulated BMDMs (Fig EV3A and B), thus demonstrating a shift in EV size in response to ATP‐induced NLRP3 inflammasome activation. Moreover, we found that NLRP3 inflammasome activation led to significantly increased levels of Prdx4, caspase‐1, and IL‐1β in EV lysates compared to EVs from unstimulated or LPS‐primed BMDMs (Fig 6F and G). IL‐1β, however, was also found to be significantly increased in EV lysates from poly(dA:dT)‐ or flagellin‐treated BMDMs. Analysis of EV protein lysates from LPS and ATP‐stimulated BMDMs confirmed the presence of inflammasome components, Prdx4, as well as positive and negative markers for EVs (Fig EV3C). In order to investigate whether Prdx4 and caspase‐1 are co‐localized within the same vesicle, we used confocal microscopy as previously described for EVs (Athman et al, 2015). We observed a speckled co‐localization of Prdx4 and caspase‐1 on microscopy slides, which were coated with isolated EVs derived from LPS+ATP‐stimulated BMDMs. Counterstain with a lipophilic dye suggested that the co‐localization is confined to lipid‐containing structures, which most likely represent the membranes of EVs (Fig EV4). Collectively, these data indicate that distinct EV populations are released in response to NLRP3 inflammasome activation, containing constituents of the inflammasome as well as Prdx4.
Figure 6. ATP‐induced NLRP3 inflammasome activation leads to secretion of distinct EVs from BMDMs.
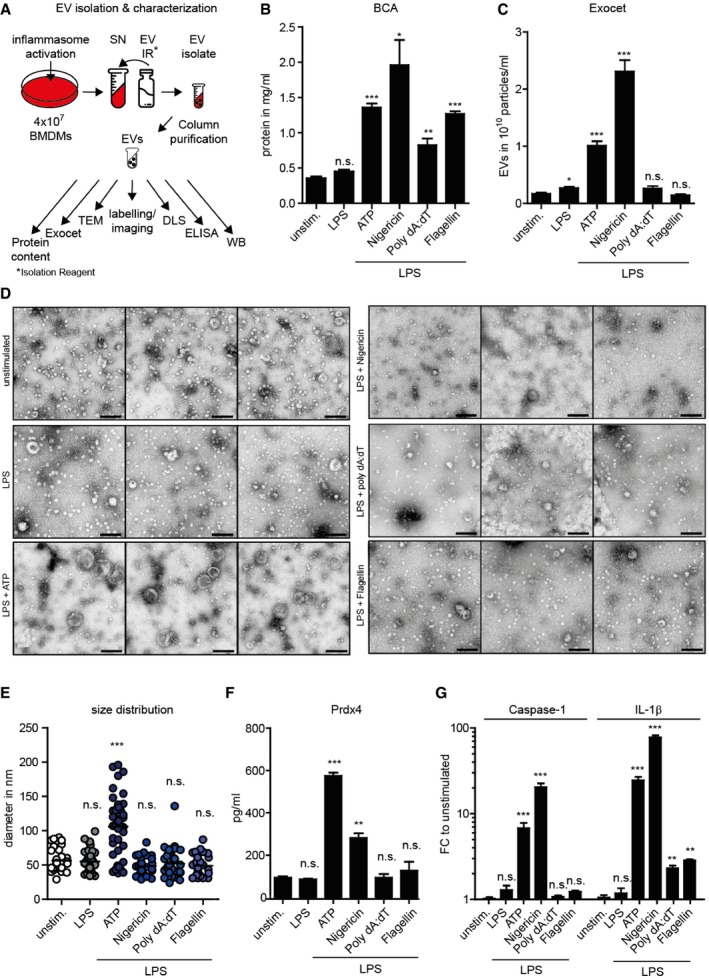
- Schematic illustration of workflow for EV isolation and characterization (panels B–G) from BMDMs that were left untreated or LPS‐primed in the presence or absence of either ATP, nigericin, poly(dA:dT), or flagellin.
- BCA analysis of EV protein concentration in EVs isolates. Each bar represents a mean of n = 3 technical replicates; vertical lines indicate SD. *P < 0.05; **P < 0.01; ***P < 0.001; n.s. not significant (two‐tailed t‐test).
- Exocet quantification assay of EVs particle numbers. Each bar represents a mean of n = 3 technical replicates; vertical lines indicate SD. *P < 0.05; ***P < 0.001; n.s. not significant (two‐tailed t‐test).
- Transmission electron microscopy (TEM) of EV isolates. Three representative pictures are displayed. Scale bar indicates 200 nm.
- Analysis of size distribution of EVs. Each dot indicates the diameter in nm of an individual vesicle. SD. ***P < 0.001; n.s. not significant (one‐way ANOVA, followed by a Tukey multiple comparison test).
- Prdx4 concentration in EV lysates. Each bar represents a mean of n = 2 technical replicates; vertical lines indicate SD. **P < 0.01; ***P < 0.001; n.s. not significant (two‐tailed t‐test).
- Caspase‐1 and IL‐1β concentration in EV lysates. Each bar represents a mean of n = 2 technical replicates; vertical lines indicate SD. **P < 0.01; ***P < 0.001; n.s. not significant (two‐tailed t‐test).
Figure EV3. Characterization of EV isolates, obtained from inflammasome‐activated BMDMs (relates to Fig 6).

- Dynamic light scattering (DLS) measurements from EVs, isolated from the supernatant of unstimulated or LPS+ATP‐treated BMDMs.
- Transmission electron microscopy (TEM) and high magnification of EVs, isolated from the supernatant of unstimulated or LPS+ATP‐treated BMDMs. Scale bar indicates 100 nm.
- Reducing SDS–PAGE (left panel) or non‐reducing SDS–PAGE (right panel) followed of EV and whole‐cell lysates followed by Western blot analysis using antibodies to Prdx4, inflammasome components (NLRP3, pro‐caspase‐1, pro‐IL‐1β, and ASC), CD63 as a positive EV marker, Grp94, mitofilin, and cytochrome c as negative markers and β‐actin as control.
Source data are available online for this figure.
Figure EV4. Prdx4 and caspase‐1 co‐localize in EVs (relates to Fig 6).

-
A–CFluorescence microscopy of EVs isolated from the supernatant of LPS+ATP‐treated (A) or unstimulated (B) BMDMs. EV membrane was stained with CellVue Burgundy, EVs were then fixed and stained with antibodies to Prdx4 (green) or caspase‐1 (red) (A, B) or second antibodies only as control (C).
Prdx4 controls caspase‐1 cleavage and IL‐1β maturation in extracellular vesicles
The observation, that highest levels of Prdx4 were found in EVs from LPS+ATP‐treated cells and that Prdx4 and caspase‐1 were co‐localized in the same EV particle, prompted us to investigate the consequences of Prdx4 deletion for EV function. We therefore isolated EVs from the supernatant of either untreated or LPS+ATP‐stimulated BMDMs from Prdx4 WT and KO as well as from Asc‐deficient mice. Upon LPS and ATP treatment, we detected Prdx4, NLRP3, Asc, pro‐caspase‐1, and pro‐IL‐1β in CD63+ EVs (Fig 7A). Using immunoprecipitation against caspase‐1, we found increased levels of cleaved caspase‐1 p10 in EVs derived from Prdx4 KO BMDMs compared to the WT. Together, this demonstrates that also within EVs, caspase‐1 activation is limited by Prdx4, resulting in reduced caspase‐1 cleavage in the presence of Prdx4. Because we were unable to detect mature IL‐1β in EVs by immunoblotting, we asked whether we can determine IL‐1β levels by detection of IL‐1β‐mediated signaling. Given that the presence or absence of Prdx4 within EVs determines the intensity of caspase‐1 cleavage and thus levels of IL‐1β, we reasoned that Prdx4 influences the ability of EVs to induce IL‐1β‐immune responses in recipient cells. In order to test our hypothesis, we followed a three‐tiered approach (Fig 7B). First, we used HEK‐blue IL‐1R reporter cells and stimulated them with EVs isolated from Prdx4 WT or Prdx4 KO BMDMs. Induction of secreted alkaline phosphatase (SEAP) activity was determined as a measure for IL‐1R activation. Second, caspase‐1‐deficient BMDMs were used as recipient cells to exclude IL‐1R activation from endogenously derived IL‐1β. Third, C57Bl/6N mice were injected with EVs from Prdx4 WT and KO BMDMs using EV protein content as a measure to control for the relative dose of administered EVs. Serum Cxcl1 levels were determined 3 h post‐injection. Following our first approach, EVs from LPS and ATP‐stimulated BMDMs induced a significant increase in SEAP activity compared to EVs from either unstimulated or LPS‐treated BMDMs (Appendix Fig S6). Furthermore, in comparison with EVs from LPS and ATP‐stimulated WT donors, EVs derived from Prdx4 KO BMDMs led to significantly higher SEAP activity. In caspase‐1‐deficient cells, we found that EVs derived from LPS‐ as well as from LPS and ATP‐stimulated BMDMs induced Cxcl1 secretion compared to EVs from unstimulated cells (Fig 7C). Again, EVs from Prdx4‐deficient BMDMs induced a significantly higher Cxcl1 response compared to EVs from WT donors. Compared to this finding, inhibition of IL‐1R by Anakinra significantly reduced Cxcl1 levels, confirming the involvement of IL‐1R activation. We finally injected EVs from Prdx4 WT or KO mice as well as from ASC KO mice in C57Bl/6N recipient mice. Analysis of serum cytokine levels showed that Prdx4‐deficient EVs induced a higher Cxcl1 response compared to EVs from Prdx4 WT or ASC KO mice (Fig 7D). Together, we found that (i) EVs, derived from inflammasome‐activated cells, are able to actively release IL‐1β and (ii) the presence of Prdx4 in EVs lowers the potency of IL‐1β‐mediated pro‐inflammatory responses in recipient cells or mice.
Figure 7. Prdx4 controls caspase‐1 cleavage and IL‐1β maturation in extracellular vesicles.

- Western blot analysis of Prdx4, NLRP3, ASC, caspase‐1, IL‐1β, and CD63 from EV lysates (upper panel) or caspase‐1 after immunoprecipitation against caspase‐1 (lower panel).
- EVs isolated from donor BMDMs were transferred to recipient cells or mice followed by subsequent readout of inflammatory response markers.
- Cxcl1 concentration in supernatants of caspase‐1‐deficient BMDMs stimulated with EVs from LPS, LPS, and ATP or control‐treated Prdx4 WT or KO BMDMs, as well as caspase‐1‐deficient BMDMs pre‐treated with Anakinra and stimulated with EVs from LPS and ATP‐treated Prdx4 WT or KO BMDMs. Each bar represents a mean of n = 3 biological with two technical replicates; vertical lines indicate SD. *P < 0.05; **P < 0.01; n.s. not significant (two‐tailed t‐test).
- Serum Cxcl1 in C57Bl6/N mice injected with either PBS or EVs from LPS and ATP or control‐treated Prdx4 WT or KO or ASC KO BMDMs. Each dot represents an individual mouse. Horizontal lines indicate mean.
Discussion
Cellular release of IL‐1β is a tightly regulated process and critical to maintain immune homeostasis. Aberrantly high IL‐1β levels have been implicated in several inflammatory diseases, including rheumatoid arthritis, osteoarthritis, gout, hereditary periodic fever, and type II diabetes (Dinarello, 2009). Inflammasomes are multiprotein complexes that—after their regulated assembly in response to danger signals—orchestrate caspase‐1 activation and cleavage of pro‐IL‐1β into its active form. Mechanisms that are required to prime and activate inflammasomes are well described (reviewed in He et al, 2016), yet little is known about endogenous factors that negatively regulate caspase‐1 activity and thus may limit the pro‐inflammatory cascade (Poudel & Gurung, 2018).
Our study unveils Prdx4 as a critical modulator of caspase‐1 function. We show that mice lacking Prdx4 are highly sensitive to endotoxic shock. Prdx4 KO BMDMs release increased amounts of IL‐1β upon inflammasome activation. The in vivo phenotype could be fully abolished by IL‐1RA treatment and was phenocopied by a conditional deletion of Prdx4 in the myeloid compartment.
Two salient observations emerge from the experiments presented here: First, we show that the 2‐Cys oxidoreductase Prdx4 directly regulates caspase‐1 function in a redox‐sensitive manner. Several findings had already put peroxiredoxins in the context of inflammatory signaling (Li et al, 2007; Yang et al, 2007; Lee et al, 2017). Members of the Prdx family were shown to reduce intracellular ROS levels and to modulate cell death induced by pro‐inflammatory stimuli (Rao et al, 2017). Whereas our initial assumption was that the antioxidant properties of Prdx4 would affect ROS‐dependent inflammasome priming (Zhou et al, 2011), our data showed no differences in transcriptional regulation of inflammasome components, or ASC speck formation between Prdx4 KO and WT mice. Instead, we present experimental evidence that caspase‐1 functionally interacts with Prdx4 via a redox‐sensitive mechanism. Regulation of the proteolytic activity of caspase‐1 via an altered cellular redox potential has been described previously. Its function is impaired by reversible oxidation via intracellular superoxide (Meissner et al, 2008), which can be abrogated by hypoxic conditions or addition of exogenous DTT generation (Tassi et al, 2009). Two cysteines in caspase‐1 (Cys362 and Cys397) have been proposed as redox‐sensitive residues and are regulated by glutathionylation (Meissner et al, 2008). Our data show that the presence of high‐molecular‐weight oligomers of Prdx4 leads to inhibition of caspase‐1 activity under non‐reducing conditions. In such a milieu, Prdx4 has been shown to form stable oligomers, with a preponderance of decamers consisting of five disulfide‐linked Prdx4 dimers (Tavender et al, 2008; Cao et al, 2011). We show that recombinant caspase‐1 (p10 and p20 subunit) co‐migrates at the expected molecular weight of the recombinant Prdx4 decamer in a non‐reducing gel. Alkylation of either partner leads to an absence of caspase‐1 in the high‐molecular‐weight fraction. The data suggest that caspase‐1 is first integrated into the high‐molecular‐weight complex of Prdx4 in a disulfide‐dependent manner, forming a redox‐active complex with Prdx4, which finally leads to the inactivation of caspase‐1. Consistently, we find in a mutational analysis of cysteines in caspase‐1 that replacement of the cysteine at position 397 by a serine leads to an overall increase of caspase‐1 activity. Importantly, this effect is associated with a complete loss of the blocking activity of Prdx4, a strong reduction of the mutated caspase‐1 in the high‐molecular‐weight fraction and ultimately loss of physical interaction between caspase‐1 and Prdx4, indicating the necessity of the cysteine residue for the observed functional interaction. Vice versa, Cys‐to‐Ala mutants of Prdx4, that lack the ability to form functional high‐molecular‐weight oligomers (Tavender et al, 2008, 2010), showed a loss of the caspase‐1 interaction and failed to inhibit caspase‐1‐induced IL‐1β secretion. Altogether, our findings highlight the critical role of high‐molecular‐weight oligomers of Prdx4 for a novel redox‐dependent regulatory mechanism of caspase‐1 activity.
The second important observation is related to the extracellular compartment of the functional interaction of Prdx4 with caspase‐1. In line with other studies, we show that Prdx4 is upregulated and secreted upon induction of inflammation (Matsumoto et al, 1999; Okado‐Matsumoto et al, 2000; Wong et al, 2000). Strikingly, we find that extracellular Prdx4 is located in extracellular vesicles, where it co‐localizes with caspase‐1. An extracellular role of the inflammasome and its components has already been suggested (Baroja‐Mazo et al, 2014; Franklin et al, 2014; Mitra et al, 2015). In particular, ASC specks have been shown to accumulate in the extracellular space, where they promoted IL‐1β maturation (Franklin et al, 2014). Moreover, IL‐1β, caspase‐1, and other inflammasome components have been described to localize to exosomes (Qu et al, 2007) and it was suggested from a meta‐analysis of proteomic and protein interaction data that caspase‐1 cleaves its substrates to propagate inflammation to neighboring and remote cells in extracellular vesicles (Wang et al, 2016). Yet, the exact role of their presence in the compartment remained unclear. Our data critically expand and underscore these observations by demonstrating that inflammasome‐containing extracellular vesicles (EVs) induce an IL‐1β‐dependent pro‐inflammatory signal in recipient cells. The loss of Prdx4 boosted the potential of EVs to transmit the immune response in vitro and in vivo, thereby defining a critical role for Prdx4 in the regulation of inflammasome‐mediated responses. These findings unveil a novel long‐range effect of inflammasomes via transport in macrophage‐derived EVs.
In conclusion, we propose that the transmission of inflammasome components and mature IL‐1β by EVs constitutes a mechanism for the propagation of inflammation in remote cells and organs. At the same time, the net inflammatory potential of EVs is influenced by the presence of the redox‐active constituent Prdx4 that negatively regulate caspase‐1 activity. A targeted modulation of the redox balance would therefore open new avenues for anti‐inflammatory strategies. In particular in patients suffering from septicemia, high Prdx4 serum concentrations were associated with increased disease severity (Schulte et al, 2011). It is unclear in how far these elevated Prdx4 levels reflect the physiological attempt to dampen excessive inflammation or the pathological condition suppressing systemic immune functions. We suggest, that the spatiotemporal control of the redox environment within EVs plays a key role in the regulation of inflammasome activity, where under hypoxic conditions—such as in local infections or tumors—inflammasomes in EVs would be biased to secrete IL‐1β. Future studies are needed to translate these findings to humans and carefully evaluate intervention strategies, which could exploit this principle in inflammatory conditions.
Materials and Methods
Mice
Prdx4 constitutive and conditional knockout mice were generated by a commercial supplier (GenOway) and were back‐crossed onto C57Bl/6N background for at least 10 generations. Exon 1 of Prdx4 was flanked by LoxP sites to enable its excision by Cre recombinase. Deletion of exon 1 resulted in the deletion of the ATG initiation codon and thus absence of transcription. To obtain constitutive knockout mice, Prdx4 flox/flox mice were crossed to ubiquitous Cre‐deleter mice. Mice were housed under specific pathogen‐free (SPF) conditions in individual‐ventilated cages (IVCs) in a 12‐h light–dark cycle and were provided with a standard rodent diet and food and water ad libitum. Male mice, aged 8–12 weeks, were used for in vivo experiments. Bone marrow‐derived macrophages (BMDMs) were generated from age‐matched males or females from 8 to 20 weeks of age. Hemizygous Prdx4 KO and WT F1 littermates were obtained by crossing heterozygous females to WT C57Bl/6N males. For inflammasome‐related studies, the following strains were used: caspase‐1‐KO (Casp1tm2.1Flv) (Blazejewski et al, 2017) and ASC KO (B6.129S2‐ASC tm1Sesh) (Ozoren et al, 2006) mice. All experiments were carried out according to the German Animal Protection Law and in accordance with the guidelines for Animal Care of the University of Kiel Votes No.: V242‐2904/2019 (18‐2/19), V242‐7224.121‐33 (99‐7/13) and (156‐11/13).
Reagents and antibodies
Nigericin (trlrl‐nig), poly(dA:dT) dsDNA (tlrl‐patn), ultrapure flagellin (tlrl‐pstfla5), and ultrapure LPS (tlrl‐peklps) were from Invivogen. Deep‐rough LPS (Kdo2‐lipid A) from E. coli F515 or KPM53 (Mamat et al, 2009) was prepared according to published protocols (KPM53: Ranf et al, 2015); F515: Zahringer et al, 2001). The triethylamine salt of the deep‐rough LPS was formed (Zahringer et al, 2001) prior to its use in any conducted experiment. Fugene 6 was from Promega. ATP and DMSO were from Sigma. Active recombinant human Prdx4 was from Abcam (ab93947); active recombinant human caspase‐1 was from Enzo Life Sciences (ALX‐201‐056). Caspase‐1 fluorometric assay kit was from Enzo (ALX‐850‐212‐KI01). Caspase‐1 inhibitor Ac‐YVAD‐cmk was from Invivogen (inh‐yvad). IL‐1R‐antagonist Anakinra (Kineret®) was from Swedish Orphan Biovitrum. The following antibodies were used: rabbit antibody to mouse Prdx4 (ab59542; Abcam), mouse antibody to human Prdx4 (ab16943; Abcam), rabbit antibody to mouse caspase‐1 (sc‐514; Santa Cruz Biotechnology), goat antibody to mouse IL‐1β (AF 401‐NA; R&D Systems), rabbit antibody to ASC (AG‐25B‐0067; AdipoGen), rabbit antibody to CD63 (EXOAB‐CD63A‐1; SBI System Bioscience), mouse antibody to β‐actin (A‐5441; Sigma), mouse antibody to DDK (TA50011‐100; Origene), rabbit antibody to turboGFP (AB513; evrogen). All HRP‐conjugated secondary antibodies were obtained from TH Geyer. Mouse cytokines were determined in culture supernatants or serum with ELISA Kits from R&D Systems Cxcl1 (DY453), Life Technologies: IL‐1β (CMC0813) and cloud clone: Prdx4 (SEF754HU).
Cell culture, plasmids, and transfection
HEK cells were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ) and maintained in RPMI medium containing 10% (v/v) fetal calf serum (FCS) at 37°C with 5% CO2. Transfections were performed at 24 h post‐seeding using Fugene6 according to the manufacturer's instructions (Roche). Myc‐DDK and GFP‐tagged plasmids encoding the full length coding sequences of human ASC/PYCARD, CASPASE‐1, IL‐1β, PRDX4, and empty controls were purchased from Origene (pCMV6‐Entry‐PYCARD, RC215592; pCMV6‐Entry‐CASP1, RC218364; pCMV6‐Entry‐IL1B, RC202079; pCMV6‐AC‐GFP PRDX4, RG203330; pCMV6‐Entry, PS100001; pCMV6‐AC‐GFP; PS1000010). HA‐tagged caspase‐1 p10, p20 WT, and p20 C285S mutant were described in Keller et al (2008). Further HA‐tagged caspase‐1 Cys‐to‐Ser mutants were described in Meissner et al (2008).
LPS‐induced sub‐lethal endotoxic shock
Mice 8–12 weeks of age were injected intraperitoneally with a dose of 4.5 mg E. coli F515 LPS per kg body weight or NaCl as control. Mice were weighed and monitored for signs of endotoxemia every 6 h over the time course of the experiment. A drop of weight below 20% of initial body weight and/or signs of a severely impaired state of health led to the exclusion from the experiment. For analysis of serum cytokines, blood was obtained by cardiac puncture after ketamine/xylazine anesthesia. Cytokines were determined by ELISA. Mice were killed by cervical dislocation, and spleens were removed and weighed.
Isolation and generation of murine bone marrow‐derived macrophages (BMDMs)
Femur and tibia were removed, and bone marrow was isolated under sterile conditions. BMDMs were cultivated for 7 days on 145‐mm‐diameter Petri dishes in BMDM medium consisting of macrophage SFM medium and DMEM (Gibco) in a one‐to‐one ratio and containing 10% FCS (Biochrom) and 1% penicillin/streptomycin (Gibco) plus 1% Fungizone® (Thermo), supplemented with 20 ng/ml recombinant murine macrophage colony‐stimulating factor (rm M‐CSF, Immunotools).
Inflammasome assays
BMDMs were plated in 96‐ to 6‐well plates according to assay requirements in BMDM medium consisting of macrophage SFM medium and DMEM (Gibco) in a one‐to‐one ratio and containing 10% FCS (Biochrom) and 1% penicillin/streptomycin (Gibco) plus 1% Fungizone® (Thermo), supplemented with 20 ng/ml recombinant murine macrophage colony‐stimulating factor (rm M‐CSF, Immunotools). FCS was omitted from the media when supernatants were used for concentration and subsequent Western blotting. Cells were primed with ultrapure LPS for 6 h and treated with inflammasome activators for 0.5–6 h. Fugene 6 was used to transfect poly (dA:dT) or flagellin (1 μg/ml each). Transfection complexes were incubated for 1 h at room temperature and added onto cells followed by centrifugation of 4 min at 300 ×g. As a control, cells were treated with transfection agent only. All stimulations were at least performed in triplets, and cytokine production was monitored by ELISA. For Western blot analysis, cells were lysed in 1% SDS‐containing lysis buffer in the presence of protease inhibitors or supernatants were precipitated using chloroform/methanol extraction as described in Gross (2012) or concentrated using Amicon ultra‐0.5 or 2 ml centrifugal for protein purification (Merck). LDH release into the cell culture supernatant was quantified using Pierce LDH Cytotoxicity Assay (Thermo Scientific).
SDS–PAGE & MS
Lyophilized recombinant human active caspase‐1 (Enzo Life Sciences; ALX‐201‐056) was dissolved in HEPES buffer (20 mM, pH 7.2) to a concentration of 0.25 μg/μl (0.625 U/μl). Recombinant human Prdx4 (Abcam; AB93947) was diluted with water to a concentration of 0.5 μg/μl. Aliquots of 4 μl rCaspase‐1 solution were incubated with 1.1 μg of rPrdx4 protein. All samples were diluted to a volume of 10 μl by adding water and then incubated at 37°C for 3 h.
Afterward, single protein samples, as well as mixed samples, were separated by SDS–PAGE using a 4% polyacrylamide stacking gel above a 10% resolving gel for separation or a 4–20% precast gradient gel (Bio‐Rad; Mini‐PROTEAN TGX gel), respectively. SDS–PAGE was performed according to the standard protocol, under non‐reducing conditions using a Mini‐PROTEAN Tetra Cell as described in the instructions of the manufacturer (Bio‐Rad). Briefly, the samples were mixed with 5 μl of Laemmli sample buffer with or without DTT (5%) for reducing or non‐reducing condition, respectively. Samples including DTT were heated for 5 min at 60°C. Gels used for Western blotting were loaded with 1:10 of the protein concentration. Samples were loaded into the gel wells, along with a molecular weight marker, and then separated by first applying a constant voltage of 60 V for 15 min and then 100 V until the running front reached the bottom of the gel. Gels were stained by Coomassie and protein bands were excised for subsequent in‐gel digestion and LC‐MS analysis. Alternatively, proteins were transferred onto membrane for Western blot. After adding 10 μl (0.5 μg) of trypsin in 0.1 M TEAB, the samples were incubated for 16 h at 37°C. The digestion was stopped by adding 3 μl of FA, lyophilized to dryness, and reconstituted in 15 μl of HPLC loading buffer (3% ACN, 0.05% TFA in water).
LC‐MS analysis
Digested peptide samples were analyzed by LC‐MS. A Dionex U3000 HPLC system was coupled to a Q Exactive Plus mass spectrometer or the Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). The digested samples were analyzed in duplicate. Samples were injected on a C18 PepMap 100 μ‐precolumn (column dimensions: 300 μm i.d. × 5 mm; Thermo Scientific) with a flow rate of 30 μl/min, trapped and desalted for 2 min, and then separated on an Acclaim PepMap RSLC column (column dimension: 75 μm i.d. × 50 cm; Thermo Scientific) over a gradient of eluent A (0.05% aqueous FA) and eluent B (80% ACN, 0.04% FA) with a flow rate of 0.3 μl/min. Peptides were eluted using a gradient from 5% eluent B to 20% eluent B in 100 min, then to 40% B in 80 min, followed by an increased to 90% eluent B in 8 min. After isocratic elution at 90% eluent B for 10 min, the column was equilibrated for 15 min with 5% eluent B. After each sample LC run, the column was washed using a blank run, injecting 5 μl of loading buffer. The LC‐system was directly coupled to the Q Exactive Plus mass spectrometer. Full MS scans were acquired from 4 min to 72 min in positive ion mode with a resolution of 70,000, an AGC target of 1e5, maximum injection time of 50 ms with a scan range for 350 to 1,400 m/z. Data‐dependent MS/MS spectra of the ten most intense precursor ions were acquired with a resolution of 7,500; scan parameters were set to an isolation window of 1.2 m/z, a normalized collision energy of 27, the AGC target of 1e5 and a maximum injection time of 100 ms. Precursors with a charge states < 2 and > 6 as well as isotopes were excluded and precursors were excluded from subsequent isolation for 10 s. MS raw files were searched against a database containing E. coli (the expression system), the two recombinant human proteins and common contaminants using Sequest search algorithm and the Proteome discoverer software (Thermo Fisher Scientific). Peptide spectrum matches and protein identifications were restricted to a false discovery rate below 1%.
Alkylation assay
For alkylation of Prdx4 or caspase‐1 cysteines, disulfide bonds were reduced with 10 mM DTT at 56°C for 30 min. Free cysteine residues were then alkylated with 20 mM iodoacetamide for 20 min at room temperature in the dark. The reaction was quenched using 1% formic acid.
Immunoprecipitation, co‐immunoprecipitation, and Western blotting
For immunoprecipitation of WT and mutant caspase‐1 and their co‐incubation with rPrdx4, HEK293 cells were transfected as indicated with plasmids for HA‐tagged caspase‐1 p10 plus p20 WT and p10 plus p20 C285S active‐site mutant or empty HA as control. For co‐immunoprecipitation of WT and Cys‐to‐Ser mutants C362 or C397 of caspase‐1 with Prdx4, HEK293 cells were transfected as indicated with plasmids for HA‐tagged caspase‐1 WT, caspase‐1 C362S, caspase‐1 C397S, caspase‐1 C362S plus C397S, or an empty HA control and co‐transfected with either Prdx4‐GFP or empty GFP as control. At 24 h after transfection, cells were lysed in RIPA Buffer (50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 0.25% Na‐deoxycholate, 1% NP‐40) in the presence of PMSF inhibitor and proteins were captured by Anti‐HA Magnetic Beads (Thermo Scientific) following the manufacturer's instructions. Eluates were incubated for 1 h with recombinant Prdx4 or left untreated or precipitates were washed, eluted, and separated by SDS–PAGE. For immunoprecipitation of endogenous caspase‐1 from EVs, EVs were lysed in RIPA buffer (50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 0.25% Na‐deoxycholate, 1% NP‐40) in the presence of PMSF inhibitor and caspase‐1 was captured using caspase‐1 antibodies and Dynabeads (Thermo Scientific) according to the manufacturers’ protocol. For non‐reducing SDS–PAGE, DTT was omitted from loading buffer and samples were not boiled. After transfer onto polyvinylidene difluoride membranes (Millipore), membranes were blocked with 5% non‐fat dried milk and probed with primary antibodies as indicated, washed, and incubated with peroxidase‐conjugated secondary antibodies. Proteins were visualized using chemiluminescent substrates (ECL, Amersham Biosciences) and exposure to X‐ray films (Hyperfilm, Amersham).
ASC speck visualization
1 × 105 BMDMs were seeded on 8‐wells Nunc Lab‐Tek II Chamber Slides (Thermo Scientific) and allowed to sit for 1 day. Cells were then pre‐treated with 100 ng/ml ultrapure LPS for 6 h or left untreated, followed by stimulation with 10 μg/ml nigericin for 45 min or no further stimulation. Cells were washed, fixed in 4% paraformaldehyde‐PBS, blocked for 1 h in 1% BSA‐PBS and incubated with primary (1:300 of anti‐ASC, AG‐25B‐0067, AdipoGen) and secondary (1:500 of AF488 donkey anti‐rabbit; A21206, Thermo Scientific) antibodies for 1 h. DAPI was used for DNA counterstaining. Images were acquired using a Zeiss AxioImager.Z1 apotome fluorescence microscope and the AxioVision Imaging software (Carl Zeiss MicroImaging Inc.). For each genotype, ASC specks were counted and calculated as % of ASC speck+ cells.
Caspase‐1 activity assay
Activity of recombinant human caspase‐1 was determined in the presence or absence of Prdx4 using the fluorogenic substrate YVAD‐AFC (ENZO). A final concentration of 1 U/μl of rCaspase‐1 was prepared in caspase‐1 reaction buffer consisting of 50 mM Hepes, pH 7.2, 50 mM sodium chloride, 0.1% Chaps, 10 mM EDTA, and 5% glycerol. Recombinant Prdx4 was added in a final concentration of 10 ng/μl and was either reduced with 1 mM DTT or left untreated. As control, YVAD (Invivogen)) was used at a concentration of 20 μM to block caspase‐1 activity. Fluorescence was quantified using a fluorescence microtiter plate reader at 505 nm.
Isolation of extracellular vesicles
For isolation of extracellular vesicles, the Total Exosome Isolation Reagent from cell culture media (Thermo Scientific) was used. After differentiation, BMDMs were seeded in a density of 1 × 107 cells/10 ml dish. FCS supplement in BMDM media was replaced by exosome‐depleted FCS (Thermo Scientific). In order to avoid carryover of LPS after the LPS priming step, BMDMs were rinsed twice with pre‐warmed PBS before stimulation with ATP. After ATP stimulation, cell culture media was harvested and centrifuged at 2,000 ×g for 30 min at 4°C to remove cells and debris. The supernatant was transferred into a new tube and mixed with the reagent mixture well by vortexing. Samples were incubated overnight at 4°C. After incubation, samples were centrifuged at 10,000 ×g for 1 h at 4°C. The supernatant was carefully discarded. Extracellular vesicles were resuspended in PBS. To remove ATP and possible contaminants, Exosome Spin Columns (MW3000, Thermo Scientific) were used according to the manufacturer's protocol. The protein content of the EVs was determined using BCA protein assay (Pierce), and subsequent stimulations and injections were carried out using equal amounts of EV protein.
Cell fractionation
BMDMs were rinsed twice with PBS, pelleted in 250 μl ice‐cold RSB buffer (10 mM Tris HCl, pH 7.4, 10 mM NaCl, 1.5 mM MgCl, 10 mM NaF) containing protease and phosphatase inhibitors, incubated on ice for 5 min, and passed 15 times through a 26‐gauge needle. Lysates were centrifuged for 10 min at 1,000 ×g, and the supernatants were subsequently centrifuged for 30 min at 20,000 ×g. The cytosolic fraction was derived from the supernatant and the insoluble fraction was derived from re‐suspension of the pellet in 30 μl PBS. Protein concentrations were determined using a protein assay (Bio‐Rad) and equilibrated before loading.
Subcellular fractionation
For subcellular fractionation, a protocol described by Schmidt et al (2009) was used. In brief, a total number of approximately 1 × 108 BMDMs were used. BMDMs were stimulated with LPS (KPM53) for 12 h and pulsed with 2.5 mM ATP for 4 h. BMDMs were washed once with ice‐cold PBS and resuspended in 2.5 ml extraction buffer including a protease inhibitor cocktail (Sigma). Gradient media and buffers were purchased as a kit from Sigma. The cells were disrupted in a dounce glass homogenizer with a small clearance pestle using 25 strokes. For the initial enrichment of organelles, the homogenates were separated by centrifugation at 1,000 ×g for 10 min to pellet nuclei and remaining intact cells. The post‐nuclear supernatant was sedimented at 20,000 ×g for 20 min. The resulting pellet was adjusted to 19% (v/v) Optiprep® (Sigma), loaded in the middle of a non‐ionic, low osmotic discontinuous density gradient with 27, 22.5, 19, 16, 12, 8% Optiprep®, and subjected to an ultracentrifugation at 150,000 ×g for 5 h. The osmolarity was adjusted to 290 mOsm with 2.3 M sucrose. The subcellular fractions were collected from the top of the tube, washed, and concentrated with HB buffer (250 mM sucrose, 10 mM Hepes pH 7.3 and 0.3 mM EDTA) at 150,000 ×g for 20 min. All ultracentrifugation steps were carried out at 4°C in Ultra‐Clear centrifugation tubes in a swing‐out rotor (SW60Ti, Beckman Coulter). The protein content of the individual fractions was determined using a BCA protein assay (Pierce).
Transmission electron microscopy (TEM) and dynamic light scattering (DLS) spectroscopy of extracellular vesicles
TEM was performed as described before (Arnold et al, 2014). In brief, 5 μl EV solution was added to a previously negatively glow discharged carbon covered cooper grid (Science Service, Munich, Germany). After removal of the solution with filter paper, the grid was washed with half saturated uranyl acetate twice and then air‐dried. Images were taken on a JEM1400Plus (JEOL, Munich, Germany) operating at 100 kV using a 4kx4k digital camera (F416, TVIPS, Munich Germany) with a resolution of 4.58 Å/pixel. Diameter analysis was performed in EMMENU4 (TVIPS, Munich, Germany) using the measure tool.
DLS was measured in a laser spectroscatter 201 (RiNA Gmbh, Berlin, Germany) at 660 nm using a quartz cuvette. For each sample, eight repetitive measurements (5 s at 20°C) were conducted and the average is displayed.
Expression analysis
Total RNA was isolated using the RNeasy Mini kit (Qiagen). Reverse transcription was achieved using the Maxima H Minus First Strand cDNA Synthesis kit (Thermo Scientific). Quantitative real‐time PCR was performed using the TaqMan Gene Expression Master Mix (Applied Biosystems) according to the manufacturer′s protocol and analyzed by the 7900HT Fast Real‐Time PCR System (Applied Biosystems). Taqman assays were ordered from Applied Biosystems. Total RNA (1 μg) was reverse‐transcribed to cDNA according to the manufacturer's instructions (MultiScribe Reverse Transcriptase, Applied Biosystems). Reactions were carried out on the ABI PRISM Sequence 7700 Detection System (Applied Biosystems), and relative transcript levels were determined using GAPDH as a housekeeper marker using the standard curve method (Livak & Schmittgen, 2001).
Statistical analyses
GraphPad Prism 5 software was used for statistical analyses and visualization. Data were analyzed for normal distribution using Shapiro–Wilk normality test. Normally distributed data were analyzed for significant group differences using a two‐tailed unpaired Student's t‐test. Nonparametric Mann–Whitney t‐test (two‐tailed) was used for non‐normally distributed data. For repeated measures over time, two‐way analysis of variance (ANOVA) and Bonferroni post‐test were performed. P < 0.05 were considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001).
Author contributions
SL, SP, PA, CT, KA, HE, MF‐P, AF, JK, AL, and SB‐B performed experiments and analyzed the data. NG, GN, H‐DB, ML, TS, and SS provided reagents, plasmids, or mice. AT contributed MS instrumentation (Orbitrap and QExactive) and input on data interpretation. The manuscript was prepared by SL and PR. SL and PR conceived the study and supervised the work. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Sabine Kock, Melanie Nebendahl, Dorina Ölsner, Tanja Klostermeier, Katharina Göbel, Maren Reffelmann, Tatjana Schmidtke, Karina Greve, Stefanie Baumgarten, Birte Buske, and Ursula Schombel for their expert technical assistance. We thank Arturo Zychlinsky for providing plasmids of caspase‐1 Cys‐to‐Ser mutants. We thank Neil Bulleid for providing plasmids of Prdx4 Cys‐to‐Ala mutants. We thank Kathrin Boersch for her help with graphical illustrations. This work was supported by Deutsche Forschungsgemeinschaft (DFG), Clusters of Excellence “Inflammation at Interfaces” (EXC306) and Precision Medicine in Inflammation (EXC2167), Bundesministerium für Bildung und Forschung (BMBF) E:med consortium SysInflame through grant 012X1306F, the CRC1182 projects A1 and C2, the CRC877 projects A13, B9, and Z3, and the SH Excellence Chair Program (to P.R.). T.S. was supported by the Helmholtz Association (#VH‐NG‐933 to T.S.) and by the DFG (STR 1343/1).
The EMBO Journal (2019) 38: e101266
Contributor Information
Simone Lipinski, Email: s.lipinski@ikmb.uni-kiel.de.
Philip Rosenstiel, Email: p.rosenstiel@mucosa.de.
References
- Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A (1999) The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome‐related vesicles. Mol Biol Cell 10: 1463–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A (2004) Phospholipases C and A2 control lysosome‐mediated IL‐1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci USA 101: 9745–9750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold P, Himmels P, Weiss S, Decker TM, Markl J, Gatterdam V, Tampe R, Bartholomaus P, Dietrich U, Durr R (2014) Antigenic and 3D structural characterization of soluble X4 and hybrid X4‐R5 HIV‐1 Env trimers. Retrovirology 11: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E et al (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40: W597–W603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athman JJ, Wang Y, McDonald DJ, Boom WH, Harding CV, Wearsch PA (2015) Bacterial membrane vesicles mediate the release of Mycobacterium tuberculosis lipoglycans and lipoproteins from infected macrophages. J Immunol 195: 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracos V, Rodemann HP, Dinarello CA, Goldberg AL (1983) Stimulation of muscle protein degradation and prostaglandin E2 release by leukocytic pyrogen (interleukin‐1). A mechanism for the increased degradation of muscle proteins during fever. N Engl J Med 308: 553–558 [DOI] [PubMed] [Google Scholar]
- Baroja‐Mazo A, Martin‐Sanchez F, Gomez AI, Martinez CM, Amores‐Iniesta J, Compan V, Barbera‐Cremades M, Yague J, Ruiz‐Ortiz E, Anton J et al (2014) The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15: 738–748 [DOI] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes‐Alnemri T, Wu J, Monks BG, Fitzgerald KA et al (2009) Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V (2011) Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187: 613–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazejewski AJ, Thiemann S, Schenk A, Pils MC, Galvez EJC, Roy U, Heise U, de Zoete MR, Flavell RA, Strowig T (2017) Microbiota normalization reveals that canonical caspase‐1 activation exacerbates chemically induced intestinal inflammation. Cell Rep 19: 2319–2330 [DOI] [PubMed] [Google Scholar]
- Brough D, Rothwell NJ (2007) Caspase‐1‐dependent processing of pro‐interleukin‐1beta is cytosolic and precedes cell death. J Cell Sci 120: 772–781 [DOI] [PubMed] [Google Scholar]
- Cao Z, Tavender TJ, Roszak AW, Cogdell RJ, Bulleid NJ (2011) Crystal structure of reduced and of oxidized peroxiredoxin IV enzyme reveals a stable oxidized decamer and a non‐disulfide‐bonded intermediate in the catalytic cycle. J Biol Chem 286: 42257–42266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA, Goldin NP, Wolff SM (1974) Demonstration and characterization of two distinct human leukocytic pyrogens. J Exp Med 139: 1369–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (1998) Interleukin‐1 beta, interleukin‐18, and the interleukin‐1 beta converting enzyme. Ann N Y Acad Sci 856: 1–11 [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2009) Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol 27: 519–550 [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Donath MY, Mandrup‐Poulsen T (2010) Role of IL‐1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 17: 314–321 [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464: 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V (2011) Autophagy‐based unconventional secretory pathway for extracellular delivery of IL‐1beta. EMBO J 30: 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC (2018) The pore‐forming protein gasdermin D regulates interleukin‐1 secretion from living macrophages. Immunity 48: 35–44 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F (1997) Extracellular ATP triggers IL‐1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol 159: 1451–1458 [PubMed] [Google Scholar]
- Franchi L, Warner N, Viani K, Nunez G (2009) Function of Nod‐like receptors in microbial recognition and host defense. Immunol Rev 227: 106–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al‐Amoudi A et al (2014) The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat Immunol 15: 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid‐Burgk JL, Rapino F, Robertson AA, Cooper MA, Graf T, Hornung V (2016) Human monocytes engage an alternative inflammasome pathway. Immunity 44: 833–846 [DOI] [PubMed] [Google Scholar]
- Gross O (2012) Measuring the inflammasome. Methods Mol Biol 844: 199–222 [DOI] [PubMed] [Google Scholar]
- Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S et al (2016) K(+) efflux‐independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 45: 761–773 [DOI] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 341: 1250–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Hara H, Nunez G (2016) Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41: 1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, Borner GH (2016) Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5: e16950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DY, Chae HZ, Rhee SG, Jeang KT (1997) Regulatory role for a novel human thioredoxin peroxidase in NF‐kappaB activation. J Biol Chem 272: 30952–30961 [DOI] [PubMed] [Google Scholar]
- Kakihana T, Araki K, Vavassori S, Iemura S, Cortini M, Fagioli C, Natsume T, Sitia R, Nagata K (2013) Dynamic regulation of Ero1alpha and peroxiredoxin 4 localization in the secretory pathway. J Biol Chem 288: 29586–29594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanneganti TD, Ozoren N, Body‐Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P et al (2006) Bacterial RNA and small antiviral compounds activate caspase‐1 through cryopyrin/Nalp3. Nature 440: 233–236 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A et al (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: 1246–1249 [DOI] [PubMed] [Google Scholar]
- Keller M, Ruegg A, Werner S, Beer HD (2008) Active caspase‐1 is a regulator of unconventional protein secretion. Cell 132: 818–831 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Vischer UM, Rosnoblet C, Lebrand C, Lindsay M, Parton RG, Kruithof EK, Gruenberg J (2000) The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol Biol Cell 11: 1829–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T (2010) Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem 285: 17442–17452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Park JH, Han SB, Yoon DY, Jung YY, Hong JT (2017) Peroxiredoxin 6 overexpression attenuates lipopolysaccharide‐induced acute kidney injury. Oncotarget 8: 51096–51107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shoji W, Takano H, Nishimura N, Aoki Y, Takahashi R, Goto S, Kaifu T, Takai T, Obinata M (2007) Increased susceptibility of MER5 (peroxiredoxin III) knockout mice to LPS‐induced oxidative stress. Biochem Biophys Res Commun 355: 715–721 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- MacKenzie A, Wilson HL, Kiss‐Toth E, Dower SK, North RA, Surprenant A (2001) Rapid secretion of interleukin‐1beta by microvesicle shedding. Immunity 15: 825–835 [DOI] [PubMed] [Google Scholar]
- Mamat U, Schmidt H, Munoz E, Lindner B, Fukase K, Hanuszkiewicz A, Wu J, Meredith TC, Woodard RW, Hilgenfeld R et al (2009) WaaA of the hyperthermophilic bacterium Aquifex aeolicus is a monofunctional 3‐deoxy‐D‐manno‐oct‐2‐ulosonic acid transferase involved in lipopolysaccharide biosynthesis. J Biol Chem 284: 22248–22262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji GA, Wang L, Geddes BJ, Brown M, Merriam S, Al‐Garawi A, Mak S, Lora JM, Briskin M, Jurman M et al (2002) PYPAF1, a PYRIN‐containing Apaf1‐like protein that assembles with ASC and regulates activation of NF‐kappa B. J Biol Chem 277: 11570–11575 [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232 [DOI] [PubMed] [Google Scholar]
- Martinon F, Tschopp J (2005) NLRs join TLRs as innate sensors of pathogens. Trends Immunol 26: 447–454 [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27: 229–265 [DOI] [PubMed] [Google Scholar]
- Martin‐Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja‐Mazo A, Bagnall J, Spiller D, White M, Daniels MJ, Mortellaro A et al (2016) Inflammasome‐dependent IL‐1beta release depends upon membrane permeabilisation. Cell Death Differ 23: 1219–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Okado A, Fujii T, Fujii J, Egashira M, Niikawa N, Taniguchi N (1999) Cloning of the peroxiredoxin gene family in rats and characterization of the fourth member. FEBS Lett 443: 246–250 [DOI] [PubMed] [Google Scholar]
- Meissner F, Molawi K, Zychlinsky A (2008) Superoxide dismutase 1 regulates caspase‐1 and endotoxic shock. Nat Immunol 9: 866–872 [DOI] [PubMed] [Google Scholar]
- Mitra S, Wewers MD, Sarkar A (2015) Mononuclear phagocyte‐derived microparticulate caspase‐1 induces pulmonary vascular endothelial cell injury. PLoS ONE 10: e0145607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbrunn M, Gutierrez‐Vazquez C, Villarroya‐Beltri C, Gonzalez S, Sanchez‐Cabo F, Gonzalez MA, Bernad A, Sanchez‐Madrid F (2011) Unidirectional transfer of microRNA‐loaded exosomes from T cells to antigen‐presenting cells. Nat Commun 2: 282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz‐Planillo R, Kuffa P, Martinez‐Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38: 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Amano W, Kubo T, Fukuhara A, Ihara H, Azuma YT, Tajima H, Inui T, Sawa A, Takeuchi T (2009) Glyceraldehyde‐3‐phosphate dehydrogenase aggregate formation participates in oxidative stress‐induced cell death. J Biol Chem 284: 34331–34341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, Derfalvi B, Benjaponpitak S, Vesely R, Sauvain MJ et al (2004) Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell‐mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood 103: 2809–2815 [DOI] [PubMed] [Google Scholar]
- Okado‐Matsumoto A, Matsumoto A, Fujii J, Taniguchi N (2000) Peroxiredoxin IV is a secretable protein with heparin‐binding properties under reduced conditions. J Biochem 127: 493–501 [DOI] [PubMed] [Google Scholar]
- Ozoren N, Masumoto J, Franchi L, Kanneganti TD, Body‐Malapel M, Erturk I, Jagirdar R, Zhu L, Inohara N, Bertin J et al (2006) Distinct roles of TLR2 and the adaptor ASC in IL‐1beta/IL‐18 secretion in response to Listeria monocytogenes. J Immunol 176: 4337–4342 [DOI] [PubMed] [Google Scholar]
- Poudel B, Gurung P (2018) An update on cell intrinsic negative regulators of the NLRP3 inflammasome. J Leukoc Biol 26: 3MIR0917‐350R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Franchi L, Nunez G, Dubyak GR (2007) Nonclassical IL‐1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 179: 1913–1925 [DOI] [PubMed] [Google Scholar]
- Ranf S, Gisch N, Schaffer M, Illig T, Westphal L, Knirel YA, Sanchez‐Carballo PM, Zahringer U, Huckelhoven R, Lee J et al (2015) A lectin S‐domain receptor kinase mediates lipopolysaccharide sensing in Arabidopsis thaliana . Nat Immunol 16: 426–433 [DOI] [PubMed] [Google Scholar]
- Rao Z, Wang S, Wang J (2017) Peroxiredoxin 4 inhibits IL‐1beta‐induced chondrocyte apoptosis via PI3K/AKT signaling. Biomed Pharmacother 90: 414–420 [DOI] [PubMed] [Google Scholar]
- Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR et al (2018) Chemical disruption of the pyroptotic pore‐forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3: eaat2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46: D624–D632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131 [DOI] [PubMed] [Google Scholar]
- Rubartelli A, Cozzolino F, Talio M, Sitia R (1990) A novel secretory pathway for interleukin‐1 beta, a protein lacking a signal sequence. EMBO J 9: 1503–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Gelhaus C, Lucius R, Nebendahl M, Leippe M, Janssen O (2009) Enrichment and analysis of secretory lysosomes from lymphocyte populations. BMC Immunol 10: 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140: 821–832 [DOI] [PubMed] [Google Scholar]
- Schulte J, Struck J, Kohrle J, Muller B (2011) Circulating levels of peroxiredoxin 4 as a novel biomarker of oxidative stress in patients with sepsis. Shock 35: 460–465 [DOI] [PubMed] [Google Scholar]
- Shirasaki Y, Yamagishi M, Suzuki N, Izawa K, Nakahara A, Mizuno J, Shoji S, Heike T, Harada Y, Nishikomori R et al (2014) Real‐time single‐cell imaging of protein secretion. Sci Rep 4: 4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer II, Scott S, Chin J, Bayne EK, Limjuco G, Weidner J, Miller DK, Chapman K, Kostura MJ (1995) The interleukin‐1 beta‐converting enzyme (ICE) is localized on the external cell surface membranes and in the cytoplasmic ground substance of human monocytes by immuno‐electron microscopy. J Exp Med 182: 1447–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Hao Y, Sun A, Li T, Li W, Guo L, Yan Y, Geng C, Chen N, Zhong F et al (2006) Sample preparation project for the subcellular proteome of mouse liver. Proteomics 6: 5269–5277 [DOI] [PubMed] [Google Scholar]
- Tassi S, Carta S, Vene R, Delfino L, Ciriolo MR, Rubartelli A (2009) Pathogen‐induced interleukin‐1beta processing and secretion is regulated by a biphasic redox response. J Immunol 183: 1456–1462 [DOI] [PubMed] [Google Scholar]
- Tavender TJ, Sheppard AM, Bulleid NJ (2008) Peroxiredoxin IV is an endoplasmic reticulum‐localized enzyme forming oligomeric complexes in human cells. Biochem J 411: 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavender TJ, Springate JJ, Bulleid NJ (2010) Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J 29: 4185–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM et al (2017) A subcellular map of the human proteome. Science 356: eaal3321 [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M (2008) Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319: 1244–1247 [DOI] [PubMed] [Google Scholar]
- Wang L, Fu H, Nanayakkara G, Li Y, Shao Y, Johnson C, Cheng J, Yang WY, Yang F, Lavallee M et al (2016) Novel extracellular and nuclear caspase‐1 and inflammasomes propagate inflammation and regulate gene expression: a comprehensive database mining study. J Hematol Oncol 9: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichart D, Gobom J, Klopfleisch S, Hasler R, Gustavsson N, Billmann S, Lehrach H, Seegert D, Schreiber S, Rosenstiel P (2006) Analysis of NOD2‐mediated proteome response to muramyl dipeptide in HEK293 cells. J Biol Chem 281: 2380–2389 [DOI] [PubMed] [Google Scholar]
- Wong CM, Chun AC, Kok KH, Zhou Y, Fung PC, Kung HF, Jeang KT, Jin DY (2000) Characterization of human and mouse peroxiredoxin IV: evidence for inhibition by Prx‐IV of epidermal growth factor‐ and p53‐induced reactive oxygen species. Antioxid Redox Signal 2: 507–518 [DOI] [PubMed] [Google Scholar]
- Yang CS, Lee DS, Song CH, An SJ, Li S, Kim JM, Kim CS, Yoo DG, Jeon BH, Yang HY et al (2007) Roles of peroxiredoxin II in the regulation of proinflammatory responses to LPS and protection against endotoxin‐induced lethal shock. J Exp Med 204: 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Mu Y, Ao J, Chen X (2010) Peroxiredoxin IV regulates pro‐inflammatory responses in large yellow croaker (Pseudosciaena crocea) and protects against bacterial challenge. J Proteome Res 9: 1424–1436 [DOI] [PubMed] [Google Scholar]
- Zahringer U, Salvetzki R, Wagner F, Lindner B, Ulmer AJ (2001) Structural and biological characterisation of a novel tetra‐acyl lipid A from Escherichia coli F515 lipopolysaccharide acting as endotoxin antagonist in human monocytes. J Endotoxin Res 7: 133–146 [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]
- Zito E, Melo EP, Yang Y, Wahlander A, Neubert TA, Ron D (2010) Oxidative protein folding by an endoplasmic reticulum‐localized peroxiredoxin. Mol Cell 40: 787–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7