

Abstract
Cucurbitacin B (CuB), a highly cytotoxic constituent of the Cucurbitaceae plant, was identified to exhibit potent inhibitory activity against human cancer cells as well as normal cells. This disadvantage hampers the possibility of developing this compound into an anticancer drug candidate. In this work, several bioreductive prodrugs of CuB were designed to reduce toxicity to normal cells while maintaining the cytotoxic effect to cancer cells. Embedded with a bioreductive delivery and cleavable system in cancer tissues, cucurbitacin B-based prodrugs 1, 2, and 3 were synthesized and evaluated by in vitro and in vivo experiments. Compared with the parent CuB, prodrug 1 was found to significantly reduce the toxicity down to 310-fold lower against noncancerous cells. LC-MS analyses show that prodrug 1 efficiently releases the parent compound in the reductase-overexpressed MCF-7 cells. In addition, prodrug 1 shows satisfactory and comparable effectiveness in controlling tumor growth as that by tamoxifen in the 4T1 xenograft mice model.
Keywords: Cucurbitacin B, bioreductive prodrug, drug delivery, cytotoxicity
Development of natural products to new analogues with improved properties is highly desired in the drug discovery process.1 Targeted prodrug design for anticancer drugs is one of the interesting strategies in drug discovery.2−5 Many anticancer natural products are not target specific and might also be quite toxic to normal cells. To lower the toxicity while keeping the therapeutic property of active natural products, proper structure modification serves as an important and useful tool in drug discovery. Prodrug design has been proven one of the workable strategies to improve the physicochemical properties of a molecule and overcome unacceptable biopharmaceutical performance. Embedment of an additional molecular device, which is cleavable by a specific enzyme expressed predominantly in tumor cells, is a commonly applied method in contemporary drug design and development.6 A bioreductive prodrug can be designed to target a specific tumor followed by in situ release of the therapeutic drugs with the reductase (NAD(P)H:quinone oxidoreductase 1, NQO1) overexpressed in some tumor cells.7−9 Fortunately, the reductases are often reported to be overexpressed in a variety of cancer cells, such as breast cancer, ovarian cancer, thyroid cancer, adrenal cancer, and colon cancer.3,10−13
Trichosanthes cucumerina L. (Cucurbitaceae), distributed in many countries of Asia and well-known for its bitter taste, is a Thai medicinal herb. Cucurbitacins, the main components of T. cucumerina,14−17 exhibit a variety of pharmacological effects in vitro and in vivo, such as anticancer, hepatoprotective, cardiovascular, purgative, antiinflammatory, antimicrobial, anthelmintic, CNS effects, and antifertility activities.18−20 Our previous phytochemical study on T. cucumerina revealed that cucurbitacin B (CuB, Figure 1), the major constituent of the plant, shows potent inhibitory activities on human breast cancer cells (MCF-7, MDA-MB-231, and SKBR-3)16,21 and colon cancer cells (Caco-2 and SW620).22 Unfortunately, CuB is also highly toxic against normal cells. This disadvantage hampers the possibility of developing this compound into an anticancer drug. Lowering the cytotoxic effect of CuB against normal cells while maintaining the cytotoxic potency to cancer cells still remains a challenging task.
Figure 1.

Structure of cucurbitacin B.
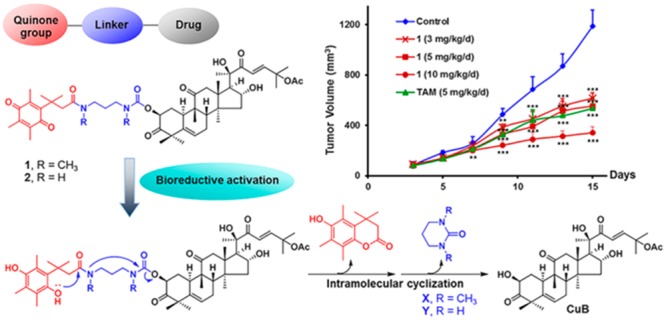
In this work, we explored the conversion of CuB into a prodrug design to improve the toxicity/safety index between breast cancer cells and noncancerous cells. Such a prodrug system is expected to be nontoxic or less toxic against normal cells until it reaches the tumor tissue and releases CuB. We rationally designed three prodrugs that were composed of a variety of linkers and the general quinone delivery system that has been successfully used in a number of pieces of prodrug research.3,12,13,23,24 The presence of the overexpressed reductase in breast tumor cells is supposed to trigger the release of CuB and cause the death of cancer cells. Three prodrugs 1–3 have been synthesized, and their crucial properties and biological activities were evaluated and compared.
Cucurbitacin B (CuB, Figure 1) is the major triterpenoid isolated from the fruit fibers of T. cucumerina. The spectroscopic (IR, 1H NMR, 13C NMR, and mass spectra) data of CuB are consistent with those reported.25−27 CuB showed significant cytotoxic activities against various cancer cells, such as breast cancer,16,21 colon cancer,22 human nasopharynx carcinoma cells,28 nonsmall cell lung cancer, human hepatocellular cells,29 HeLa cells, and HepG2 cells.30 CuB upregulated DNA methyltransferase 1 and heavy methylation in the promoters of c-Myc, cyclin D1, and survivin, which consequently downregulated the expression of all these oncogenes, were observed.31 However, its nonselective cytotoxic actions against both cancerous and normal cells greatly limited its further development to a potential anticancer drug. Conversion of this cytotoxic natural product to a safe prodrug is of extreme interest for potential treatment of cancers.
For bioreductive prodrug design, quinone delivery systems are capable of delivering drugs to target a specific tumor and can be activated by overexpressed reductases in cancer cells.3,23,32 The steric hindrance imparted by the three methyl groups on the quinone moiety (the “trimethyl lock”) has been shown to induce intramolecular cyclization to release the active drugs from the prodrugs in the target sites (Figure 2).24,33 At the beginning of the design of the prodrug(s), we found that upon acetylation of CuB, the 2-hydroxy group was more readily acetylated than the 16-hydroxy group, whereas the 20-hydroxy group was relatively inert under normal experimental conditions. The result indicated that attachment of the bioreductive unit at the 16-hydroxy group would need two extra synthetic steps: protection and deprotection of the more active 2-hydroxy group. We therefore decided to attach the bioreductive unit at the 2-hydroxy position. Following such a bioreductive prodrug concept, CuB was converted into prodrug molecules 1, 2, and 3, in which the quinone system was introduced through the carbamate and ester linkages (Figure 2).
Figure 2.

Rational design of CuB-based bioreductive prodrugs 1–3.
The synthesis of prodrugs 1, 2, and 3 is depicted in Schemes 1–3. First of all, reaction between trimethylhydroquinone (4) and 3,3-dimethylacrylic acid in the presence of dry methanesulfonic acid provided 5, which was further treated with N-bromosuccinimide (NBS) in aqueous acetone to afford 6. The 1,3-diaminopropanes 7a and 7b were treated with di-tert-butyldicarbonate (Boc2O) in DCM, yielding the mono-Boc-protected linkers 8a and 8b (Scheme 1).
Scheme 1. Synthetic Procedures for Compounds 5–8.

Reagents and conditions: (a) 3,3-dimethylacrylic acid, CH3SO3H, 70 °C; (b) NBS, acetone, water; (c) Boc2O, DCM.
Scheme 3. Synthesis of Prodrug 3.

Reagents and conditions: (a) 6, EDCI, cat. DMAP, DCM.
Connection of the quinone bioreductive unit 6 to CuB is summarized in Scheme 2. CuB was treated with 4-nitrophenyl chloroformate in DCM in the presence of Et3N to furnish compound 9. The attachment of the leaving group at the 2-position was evident from large downfield shifts of the NMR chemical shifts from CuB (δH 4.39 and δC 71.6) to 9 (δH 5.39 and δC 77.7) and was further confirmed by HMBC experiments (see Supporting Information). Parallel condensation of 9 with the linkers 8a and 8b, respectively, provided 10a and 10b. Treatment of 10a and 10b with 10% TFA gave amine intermediates 11a and 11b, in parallel. Finally, quinone unit 6 was coupled to amines 11a and 11b using EDCI and a catalytic amount of DMAP to yield the prodrugs 1 and 2, respectively (Scheme 2). Following a similar procedure, quinone 6 was directly conjugated to CuB to afford prodrug 3 (Scheme 3). The purity of the prodrugs 1–3 was checked by HPLC (see Figure S1).
Scheme 2. Synthesis of Prodrugs 1 and 2.

Reagents and conditions: (a) 4-nitrophenyl chloroformate, Et3N, DCM, 0 °C; (b) compounds 8a and 8b, Et3N, DCM; (c) 10% TFA, DCM; (d) compound 6, EDCI, DMAP, DCM.
The stability of the prodrug is an important parameter for further bioassays in vitro and in vivo. The stability of prodrugs 1, 2, and 3 was examined in the cell culture medium, DMEM with 10% FBS for a period of 72 h by using HPLC. The results show that all prodrugs were reasonably stable under the experiment conditions, and more than 90% of the tested molecules remained after 24 h. Chemically, the carbamate functionality applied in the prodrug linkage of 1 and 2 is expected to be more stable than the corresponding ester functionality of prodrug 3 to bind CuB with the quinone moiety. The experiment confirmed that prodrugs 1 and 2 were more stable than prodrug 3 (see Figure S2). In addition, it was also observed that the 1,3-di-N-methylaminopropyl linkage of 1 showed better stability than the unsubstituted 1,3-diaminopropyl linker employed in compound 2.
CuB and the prodrugs 1, 2, and 3 were evaluated for their cytotoxicity against breast cancer (MCF-7) and noncancerous Vero (African green monkey kidney) cells using the MTT assay. Tamoxifen (TAM), a well established breast cancer drug,34 was used as a positive control. As shown in Table 1, CuB exhibited potent activity toward MCF-7 cells (IC50 12.0 μM) and it was highly toxic toward the noncancerous Vero cells (IC50 0.04 μM). The prodrugs 1, 2, and 3 exhibited better cytotoxicity over TAM (IC50 22.6 μM) against MCF-7 cells with IC50 values of 18.1, 15.4, and 16.6 μM, respectively. Compared with CuB, the prodrugs 1, 2, and 3 showed lower cytotoxic activity toward the Vero cells, with an IC50 values of 12.4, 1.87, and 0.07 μM, respectively. They are approximately 310-, 47-, and 2-fold less toxic than CuB, respectively. Based on the above, significant decrease of the cytotoxic action against the normal Vero cells while keeping its cytotoxic effect against the cancerous MCF-7 cells mentions that the bioreductive prodrug design of CuB works by aid of the reductase overexpressed in the cancer cells. To further verify the role of NQO1 in the drug release process in the cells, we also performed MTT assays for MCF-7 cells pretreated with dicoumarol (DIC), an inhibitor of NQO1.35 Upon pretreatment with DIC, the cell inhibition decreased significantly (Table 1), suggesting that the prodrugs exerted their cytotoxicity depending on NQO1 bioreductive activation. We also evaluated whether the prodrugs could be efficiently reduced by NQO1 and could specifically release CuB3 (see Figure S3).
Table 1. In Vitro Cytotoxicity Activity of CuB and Prodrugs.
| IC50a (μM) |
||||
|---|---|---|---|---|
| Compound | MCF-7 | Verob | MCF-7+DIC | CIc |
| CuB | 12.0 ± 0.4 | 0.04 ± 0.02 | 9.72 ± 1.08 | |
| 1 | 18.1 ± 0.4 | 12.4 ± 0.5 | >100 | 310 |
| 2 | 15.4 ± 0.2 | 1.87 ± 0.24 | >100 | 46.8 |
| 3 | 16.6 ± 0.9 | 0.07 ± 0.02 | >100 | 1.75 |
| TAM | 22.6 ± 0.3 | |||
| Ellipticine | 4.06 ± 0.58 | |||
Each value was reproduced in three experiments.
African green monkey kidney.
Cytotoxicity index, IC50 of prodrug in Vero compared with IC50 of CuB parent in Vero.
To get an insight into how the prodrug releases drug (CuB) or other active species with the reductases in cancer cells, cellular uptake study was performed using MCF-7 cells, typical reductase-overexpressing cancer cells.12 First, MCF-7 cells were treated with 25 μM prodrugs, and the cells were lysed after 24 h incubation. The concentrations of prodrug in the culture medium and cell lysate were determined by HPLC with a calibration curve.2 As shown in Figure 3A, the concentrations of 1 and 3 in culture medium decreased to 8.39 and 6.21 μM after 24 h incubation, respectively. However, the concentration of 2 in the culture medium was not detected. This might be caused by better cell permeability of prodrug 2 compared with those of prodrugs 1 and 3.
Figure 3.

Cellular uptake experiments of prodrugs 1, 2, and 3. (A) Concentration of prodrugs 1 and 3 in cell culture media after 24 h incubation. aNot detected by HPLC. **P < 0.01 (n = 3). (B), (C), (D) HRMS analyses for prodrugs 1, 2, and 3 in cell lysate after 24 h incubation, respectively.
Next, we examined whether the prodrugs could release therapeutic drug CuB in the cells. As shown in Figure 3, prodrugs 1, 2, and 3 efficiently released CuB in the cell lysate, while the intermediates 11a and 11b were also detected in the cell lysate of 1 and 2, respectively. Very interestingly, 1, 2, and 3 in the cell lysate were not detected by HPLC. This highly mentions that the transformation of prodrugs to CuB is very efficient in the cancer cells by aid of the corresponding reductases. Furthermore, observation of the anticipated compounds or intermediates released from the prodrugs by high resolution ESI-TOFMS analysis confirmed such a conclusion. For the prodrug 1, the corresponding ions including CuB ([M + Na]+ at m/z = 581.3102), 11a ([M + H]+ at m/z = 687.4216), and the lactone 5 ([M + H]+ at m/z = 235.1334) were detected (Figure 3B). In addition, the ions of CuB and the lactone 5 were also detected by HRMS for prodrugs 2 and 3 (Figure 3C and 3D, respectively). Moreover, the [M + H]+ ion at m/z 659.3896 indicated the presence of intermediate 11b in the cell lysate of 2 (Figure 3C). It is noteworthy that the byproducts cyclic ureas X and Y (see Figure 2) were undetectable in the cell lysate under the HPLC conditions, because they are not UV-detectable. However, both compounds showed low-intensity ion peaks [M + H]+ ion at m/z 129.1025 and [M + H]+ ion at m/z 101.0695 in the HRMS analysis (see Figures 3B and 3C, respectively). It should also be noted that the ion peaks of the prodrugs 1, 2, and 3 at the extended m/z range to 1000 were not detected (see Figure S4). The cytotoxicity of the lactone 5 and cyclic ureas X and Y against MCF-7 and Vero cells using the MTT assay was evaluated, and they were found to be nontoxic (see Table S1).
The possibility of deactivation of prodrugs by thiol-containing species36 was also investigated. The prodrugs 1–3 were treated with glutathione (GSH), but no reaction was observed (Supporting Information).
Since the prodrug 1 exhibited significantly decreased toxicity against normal cells, it was therefore selected for in vivo antitumor study using the BALB/c mice model with breast cancer cells (4T1),37,38 and CuB and TAM were used as comparison controls. The antitumor efficacy of different dosages of the prodrug 1 (3, 5, and 10 mg/kg/d), CuB (3 mg/kg/d), and TAM (5 mg/kg/d) were tested in the animal model. As shown in Figure 4A, all concentrations of 1 showed significant inhibitory effects on tumor growth. The tumor growth inhibition (TGI) rate of 1 at a dose of 5 mg/kg/d was 53.8%, which was comparable to that of TAM (55.0%, 5 mg/kg/d) (Table S2). The excised tumors from the control animals ranged from 500 to 700 mg, while all concentrations of the prodrug 1-treated tumors weighed less than 400 mg (Figure 4B, 4C, and 4D). In addition, both dosages of 1 (3 and 5 mg/kg/d) showed increase in body weight (6.46 and 5.27%, respectively) compared with the control group (−1.48%) and TAM (4.99%). The prodrug 1 at 10 mg/kg/d group exhibited significant body weight loss −2.49% (Figure S5 and Table S2), which could mainly be ascribed to the tumor weight being inhibited. It is noteworthy that, when a dose of 3.0 mg/kg/d of CuB was used in vivo, all treated mice died after 1 day of CuB administration for its high in vitro cytotoxicity. These data clearly mention that the prodrug 1 is a successful design to reduce the in vivo toxicity of CuB, and it might be a therapeutically promising and less toxic agent for potential cancer treatment.
Figure 4.

In vivo antitumor activity of prodrug 1 on tumor growth against 4T1 xenograft in BALB/c mice. (A) Growth curves of tumor volume and days were plotted. (B) Xenografted mice after being sacrificed. (C) Mean tumor weight, and (D) tumor photographs after mice were sacrificed. aAll mice died, and the tumor weight of the CuB (3 mg/kg/d) treated group was not determinable. **P < 0.01, ***P < 0.001 vs control group; Student’s t test (n = 6).
In summary, we present a new successful example to convert a highly toxic natural product into potentially useful and less toxic anticancer compounds using cellular degradable prodrug design. Three bioreductive prodrugs (1, 2, and 3) were synthesized from CuB, the major constituent of T. cucumerina. Our study showed that these prodrugs significantly reduced toxicity against noncancerous cells compared to CuB and maintained the original actions against cancer cells. The experiments also confirmed that the prodrugs could efficiently release CuB in the reductase-overexpressing MCF-7 cells. Among them, the prodrug 1 exhibited significant toxicity reduction in both in vitro and in vivo studies and showed a comparable tumor growth inhibition in the 4T1 xenograft mice model at a dose of 5 mg/kg/d by comparison with tamoxifen (TAM).
Acknowledgments
This work was supported by The Thailand Research Fund (TRF, Nos. DBG 5980003 and DBG 6180030, to AS), Center of Excellence for Innovation in Chemistry, Office of the Higher Education Commission (to AS), National Natural Science Foundation of China (Nos. 21761142001 and 21532002, to ZJY), and the Royal Golden Jubilee (RGJ) Ph.D. Program of TRF (No. PHD 57K0084, to PS).
Glossary
Abbreviations
- CuB
cucurbitacin B
- TAM
tamoxifen
- NBS
N-bromosuccinimide
- Boc2O
di-tert-butyldicarbonate
- DCM
dichloromethane
- TFA
trifluoroacetic acid
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- DMAP
N,N-dimethyl-4-aminopyridine
- DIC
dicoumarol
- DMEM
Dulbecco’s Modified Eagle Medium
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00161.
Experimental section, HPLC purity analyses of prodrugs 1, 2, and 3; Stability examination of prodrugs 1, 2, and 3 in culture medium; In vitro cytotoxic activity of lactone 5 and cyclic ureas X and Y; HPLC assay for drug release studies; Drug release studies as a function of time in the presence of NQO1 and NADPH in PBS; Stability of prodrugs under the GSH activation; The original HRMS analysis of prodrugs 1, 2, and 3 in cell lysate after 24 h incubation; In vivo antitumor activity of prodrug 1 on mice body weight; In vivo effects of prodrug 1 and CuB. General; Isolation of cucurbitacin B; Synthesis of compounds 5, 6, 8a, 8b, 9, 10a, 10b, 11a, and 11b and spectroscopic data; Synthesis of prodrugs 1, 2, and 3 and spectroscopic data; and NMR spectral copies of all the compounds shown in Schemes 1–3 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mishra B. B.; Tiwari V. K. Natural product: an evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. 10.1016/j.ejmech.2011.07.057. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Tang K.; Wang H.; Liu Y.; Bao B.; Fang Y.; Zhang X.; Lu W. Design, synthesis, and biological evaluation of new cathepsin B-sensitive camptothecin nanoparticles equipped with a novel multifunctional linker. Bioconjugate Chem. 2016, 27, 1267–1275. 10.1021/acs.bioconjchem.6b00099. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Li X.; Li Z.; Wu X.; Wu Y.; You Q.; Zhang X. An NAD(P)H: quinone oxidoreductase 1 responsive and self-immolative prodrug of 5-fluorouracil for safe and effective cancer therapy. Org. Lett. 2018, 20 (12), 3635–3638. 10.1021/acs.orglett.8b01409. [DOI] [PubMed] [Google Scholar]
- Liu Y. W.; Shia K. S.; Wu C. H.; Liu K. L.; Yeh Y. C.; Lo C. F.; Chen C. T.; Chen Y. Y.; Yeh T. K.; Chen W. H. Targeting tumor associated phosphatidylserine with new zinc dipicolylamine-based drug conjugates. Bioconjugate Chem. 2017, 28, 1878–1892. 10.1021/acs.bioconjchem.7b00225. [DOI] [PubMed] [Google Scholar]
- Niu M.; Naguib Y. W.; Aldayel A. M.; Shi Y. C.; Hursting S. D.; Hersh M. A.; Cui Z. Biodistribution and in vivo activities of tumor-associated macrophage-targeting nanoparticles incorporated with doxorubicin. Mol. Pharmaceutics 2014, 11, 4425–4436. 10.1021/mp500565q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautio J.; Meanwell N. A.; Di L.; Hageman M. J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discovery 2018, 17, 559–587. 10.1038/nrd.2018.46. [DOI] [PubMed] [Google Scholar]
- Jaffar M.; Williams K. J.; Stratford L. J. Bioreductive and gene therapy approaches to hypoxic diseases. Adv. Drug Delivery Rev. 2001, 53, 217–228. 10.1016/S0169-409X(01)00228-9. [DOI] [PubMed] [Google Scholar]
- Saneyoshi H.; Yamamoto Y.; Kondo K.; Hiyoshi Y.; Ono A. Conjugatable and bioreduction cleavable linker for the 5′-functionalization of oligonucleotides. J. Org. Chem. 2017, 82, 1796–1802. 10.1021/acs.joc.6b02527. [DOI] [PubMed] [Google Scholar]
- Mooring S. R.; Jin H.; Devi N. S.; Jabbar A. A.; Kaluz S.; Liu Y.; Meir E. G. V.; Wang B. Design and synthesis of novel small-molecule inhibitors of the hypoxia inducible factor pathway. J. Med. Chem. 2011, 54, 8471–8489. 10.1021/jm201018g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begleitera A.; Leitha M. K.; Dohertya G. P.; Digbya T. J.; Pan S. Factors influencing the induction of DT-diaphorase activity by 1,2-dithiole-3-thione in human tumor cell lines. Biochem. Pharmacol. 2001, 61, 955–964. 10.1016/S0006-2952(01)00537-8. [DOI] [PubMed] [Google Scholar]
- Siegel D.; Ross D. Immunodetection of NAD(P)H:quinone oxidoreductase1 (NQO1) in human tissues. Free Radical Biol. Med. 2000, 29, 246–253. 10.1016/S0891-5849(00)00310-5. [DOI] [PubMed] [Google Scholar]
- Ramji S.; Lee C.; Inaba T.; Patterson A. V.; Riddick D. S. Human NADPH-cytochrome P450 reductase overexpression does not enhance the aerobic cytotoxicity of doxorubicin in human breast cancer cell lines. Cancer Res. 2003, 63, 6914–6919. [PubMed] [Google Scholar]
- Volpato M.; Abou-Zeid N.; Tanner R. W.; Glassbrook L. T.; Taylor J.; Stratford L.; Loadman P. M.; Jaffar M.; Phillips R. M. Chemical synthesis and biological evaluation of a NAD(P)H: quinone oxidoreductase-1-targeted tripartite quinone drug delivery system. Mol. Cancer Ther. 2007, 6, 3122–3130. 10.1158/1535-7163.MCT-07-0519. [DOI] [PubMed] [Google Scholar]
- Duyfjes B. E. E.; Pruesapan K. The genus Trichosanthes L. (Cucurbitaceae) in Thailand. Thai For. Bull. (Bot.) 2004, 32, 76–109. [Google Scholar]
- Sandhya S.; Chandrasekhar J.; Banji D.; Rao K. N. V. Pharmacognostical study on the leaf of Trichosanthes cucumerina Linn. Arch. Appl. Sci. Res. 2010, 2 (5), 414–421. [Google Scholar]
- Duangmano S.; Sae-lim P.; Suksamrarn A.; Patmasiriwat P.; Domann F. E. Cucurbitacin B causes increased radiation sensitivity of human breast cancer cells via G2/M cell cycle arrest. J. Oncol. 2012, 2012, 1–8. 10.1155/2012/601682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhya S.; Vinod K. R.; Sekhar J. C.; Aradhana R.; Nath V. S. An updated review on Trichosanthes cucumerina L. Pharm. Sci. Rev. Res. 2010, 1, 56–60. [Google Scholar]
- Chen J. C.; Chiu M. H.; Nie R. L.; Cordell G. A.; Qiu S. X. Cucurbitacins and cucurbitane glycosides: structures and biological activities. Nat. Prod. Rep. 2005, 22, 386–399. 10.1039/b418841c. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhao G. X.; Xu L. H.; Liu K. P.; Pan H.; He J.; Cai J. Y.; Ouyang D. Y.; He X. H. Cucurbitacin IIb exhibits anti-inflammatory activity through modulating multiple cellular behaviors of mouse lymphocytes. PLoS One 2014, 9 (2), 1–12. 10.1371/journal.pone.0089751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Militão G. C. G.; Dantas I. N. F.; Ferreira P. M. P.; Alves A. P. N. N.; Chaves D. C.; Monte F. J. Q.; Pessoa C.; Moraes M. O. D.; Costa-Lotufo L. V. In vitro and in vivo anticancer properties of cucurbitacin isolated from Cayaponia racemosa. Pharm. Biol. 2012, 50 (12), 1479–1487. 10.3109/13880209.2012.684691. [DOI] [PubMed] [Google Scholar]
- Duangmano S.; Sae-lim P.; Suksamrarn A.; Domann F. E.; Patmasiriwat P. Cucurbitacin B inhibits human breast cancer cell proliferation through disruption of microtubule polymerization and nucleophosmin/B23 translocation. BMC Complementary Altern. Med. 2012, 12 (185), 1–12. 10.1186/1472-6882-12-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promkan M.; Dakeng S.; Suebsakwong P.; Suksamrarn A.; Patmasiriwat P. Alterations of cellular proliferation, apoptosis and autophagy by cucurbitacin B treatment in colon cancer cells. Ann. Oncol. 2015, 26 (9), 151–152. 10.1093/annonc/mdv533.15. [DOI] [Google Scholar]
- Blanche E. A.; Maskell L.; Colucci M. A.; Whatmore J. L.; Moody C. J. Synthesis of potential prodrug systems for reductive activation. Prodrugs for anti-angiogenic isoflavones and VEGF receptor tyrosine kinase inhibitory oxindoles. Tetrahedron 2009, 65, 4894–4903. 10.1016/j.tet.2009.04.014. [DOI] [Google Scholar]
- Naughton D. P. Drug Targeting to hypoxic tissue using self-inactivating bioreductive delivery systems. Adv. Drug Delivery Rev. 2001, 53, 229–233. 10.1016/S0169-409X(01)00229-0. [DOI] [PubMed] [Google Scholar]
- Ryu S. Y.; Lee S. H.; Choi S. U.; Lee C. O.; No Z.; Ahn J. W. Antitumor activity of Trichosanthes kirilowii. Arch. Pharmacal Res. 1994, 17, 348–353. 10.1007/BF02974175. [DOI] [Google Scholar]
- Afifi M. S.; Ross S. A.; Elsohly M. A.; Naeem Z. E.; Halaweish F. T. Cucurbitacins of Cucumis prophetarum. J. Chem. Ecol. 1999, 25, 847–859. 10.1023/A:1020801002471. [DOI] [Google Scholar]
- Ayyad S. E. N.; Lateff A. A.; Basaif S. A.; Shier T. Cucurbitacins-type triterpene with potent activity on mouse embryonic fibroblast from Cucumis prophetarum, Cucurbitaceae. Pharmacogn. Res. 2011, 3, 189–193. 10.4103/0974-8490.85006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiratchariyakul W.; Kummalue T.. Experimental therapeutics in breast cancer cells. Breast Cancer-Current and Alternative Therapeutic Modalities; Gunduz E., Rijeka, Eds.; University Campus STeP Ri, Croatia, 2011; pp 243–269. [Google Scholar]
- Chen C.; Qiang S.; Lou L.; Zhao W. Cucurbitane-type triterpenoids from the stems of Cucumis melo. J. Nat. Prod. 2009, 72, 824–829. 10.1021/np800692t. [DOI] [PubMed] [Google Scholar]
- Bartalis J.; Halaweish F. T. In vitro and QSAR studies of cucurbitacins on HepG2 and HSC-T6 liver cell lines. Bioorg. Med. Chem. 2011, 19, 2757–2766. 10.1016/j.bmc.2011.01.037. [DOI] [PubMed] [Google Scholar]
- Dittharot K.; Dakeng S.; Suebsakwong P.; Suksamrarn A.; Patmasiriwat P.; Promkan M. Cucurbitacin B induces hypermethylation of oncogenes in breast cancer cells. Planta Med. 2019, 85, 370–378. 10.1055/a-0791-1591. [DOI] [PubMed] [Google Scholar]
- Maskell L.; Blanche E. A.; Colucci M. A.; Whatmore J. L.; Moody C. J. Synthesis and evaluation of prodrugs for anti-angiogenic pyrrolylmethylidenyl oxindoles. Bioorg. Med. Chem. Lett. 2007, 17, 1575–1578. 10.1016/j.bmcl.2006.12.108. [DOI] [PubMed] [Google Scholar]
- Naughton D. P.; Ferrer S.; Threadgill M. D. Drug targeting using bioreductive derivery systems. Proc. Indian Natl. Sci. Acad. 2002, 4, 371–378. [Google Scholar]
- Ma G.; He J.; Yu Y.; Xu Y.; Yu X.; Martinez J.; Lonard D. M.; Xu J. Tamoxifen inhibits ER-negative breast cancer cell invasion and metastasis by accelerating twist1 degradation. Int. J. Biol. Sci. 2015, 11, 618–628. 10.7150/ijbs.11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Bian J.; Li X.; Wu X.; Dong Y.; You Q. 2-Substituted 3,7,8-trimethylnaphtho[1,2-b]furan-4,5-diones as specific L-shaped NQO1-mediated redox modulators for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2017, 138, 616–629. 10.1016/j.ejmech.2017.06.028. [DOI] [PubMed] [Google Scholar]
- Whang C.-H.; Yoo E.; Hur S. K.; Kim K. S.; Kim D.; Jo S. A highly GSH-sensitive SN-38 prodrug with an ‘‘OFF-to-ON’’ fluorescence switch as a bifunctional anticancer agent. Chem. Commun. 2018, 31, 9031–9034. 10.1039/C8CC05010D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Xie Y.; Li J.; Peng Z. H.; Sheinin Y.; Zhou J.; Oupicky D. Tumor-penetrating nanoparticles for enhanced anticancer activity of combined photodynamic and hypoxia-activated therapy. ACS Nano 2017, 11, 2227–2238. 10.1021/acsnano.6b08731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman D. S.; Viglianti B. L.; Zennadi R.; Fels D.; Boruta R. J.; Yuan H.; Dreher M. R.; Grant G.; Rabbani Z. N.; Moon E. Sickle erythrocytes target cytotoxics to hypoxic tumor microvessels and potentiate a tumoricidal response. PLoS One 2013, 8, 1–11. 10.1371/journal.pone.0052543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.