

Abstract
We report a novel benzimidazole (BI) based DprE1 inhibitor that resulted from scaffold morphing of a 1,4-azaindole series. The clinical progression of the 1,4-azaindole series from our previous work validates the potential of exploring newer chemical entities with antimycobacterial activity driven via a noncovalent inhibition of the decaprenylphosphoryl-β-d-ribose-2′-epimerase (DprE1). The representative compounds from the new scaffold reported in this study exhibited an improved solubility and higher free plasma fraction, while retaining potent DprE1 inhibition and antimycobacterial activity. A representative compound from the benzimidazole series demonstrated good efficacy in a murine model of tuberculosis. Furthermore, molecular modeling of the BI scaffold suggests plausible modes of binding in the active site of DprE1 enzyme from Mycobacterium tuberculosis that can be used for further exploration of the series.
Keywords: DprE1, Mycobacterium tuberculosis, azaindole, benzimidazole
Tuberculosis (TB) continues to be a global emergency with the World Health Organization (WHO) reporting an estimated 10 million new cases and 1.3 million deaths in 2017.1 The emergence of multidrug-resistant tuberculosis (MDR-TB) and extremely drug-resistant tuberculosis (XDR-TB) makes TB treatment challenging and highlighted an urgent need to develop new TB drugs with a novel mechanism of action.1 Several published reports from our group as well as others have focused on targeting the decaprenylphosphoryl-β-d-ribose-2′-epimerase (DprE1) of Mycobacterium tuberculosis (Mtb), an essential enzyme involved in cell wall synthesis for the identification of promising antitubercular agents.2−8 DprE1 is involved in the conversion of decaprenylphosphoryl-β-d-ribose (DPR) to decaprenylphosphoryl-β-d-arabinofuranose (DPA), a precursor of the mycobacterial cell wall arabinan.8
The identification of both covalent (e.g., BTZ043, PBTZ-169) and noncovalent (e.g., TBA7371) inhibitors of DprE1 that are efficacious in various murine models of TB has provided pharmacological validation of this important target in the physiology of Mtb.2−10 Our earlier study reported 1,4 azaindoles—a novel chemical series with an excellent in vivo antimycobacterial activity achieved via noncovalent inhibition of DprE1,4−6 which has advanced to first in human clinical studies.7 This promising development mandates the need to explore newer chemical entities toward populating the TB drug pipeline.
Scaffold-morphing, a medicinal chemistry tool for molecular backbone replacements, is a rational drug-design strategy that has been used to build novel molecules with potentially improved properties and generating, new intellectual property (IP) space, while retaining the primary potency. Such drug-design strategies are limited only by synthetic feasibility of new scaffolds and are essentially chemistry-driven approaches. We utilized a scaffold morphing approach with the objective of identifying novel DprE1 inhibitors with diverse physicochemical properties. Our scaffold morphing efforts led to the identification of benzimidazole (BI) series, by the replacement of the azaindole core with an alternative 5,6 fused bicyclic heteroaromatics. This new scaffold retains potent DprE1 enzyme inhibition with potent antimycobacterial activity coupled with improved properties including aqueous solubility, similar logD, and increased free plasma fraction. A representative benzimidazole compound assessed in a murine model of TB demonstrated efficacy similar to the reported azaindoles.5In silico docking with DprE1 predicts different binding interactions between the benzimidazole and the azaindole scaffold. These new scaffolds have the potential to be developed for the treatment of drug sensitive and drug resistant tuberculosis therapy.
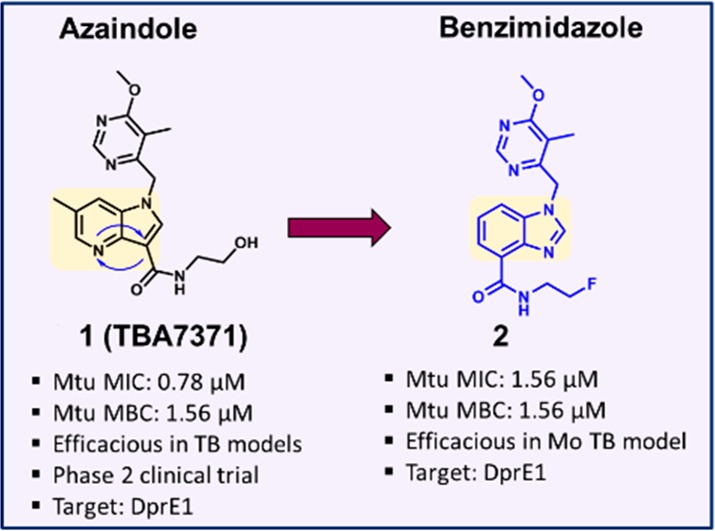
We designed several alternate scaffolds starting from the 1,4-azaindole series (1). One of them was envisaged by moving the N-4 nitrogen to the N-3 position, and subsequently transposing the amide side chain from C-3 to the C-4 position resulting in a new 5,6-fused bicyclic heteroaromatic core. The resulting benzimidazole (2) compound was synthesized and characterized for DprE1 enzyme inhibition and antimycobacterial activity (Figure 1). Although the azaindole core was altered, the hydrophobic group and the amide side chain were retained in the BI scaffold. The presence of a 6-methoxy-5-methylpyrimidine-4-yl group maintained the overall hydrophobic footprint and hydrogen bond acceptor nature, similar to the 1,4-azaindoles, while the amide side chain maintained an envisaged critical hydrogen bond donor–acceptor interaction with the DprE1 enzyme. The benzimidazole compound 2 retained antimycobacterial activity with potent enzymatic inhibition of the Mtb DprE1 enzyme, suggesting a similar mode of action. Thus, our scaffold-morphing medicinal chemistry design strategy has resulted in the successful identification of BI ring system as a potent DprE1 inhibitors.
Figure 1.
Scaffold morphing of azaindole to benzimidazole (BI).
Analogous to the azaindoles,4,5 we have also established a minimal inhibitory concentration (MIC)-based structure–activity relationship (SAR) against Mtb for BI. Three diversification points were maintained, namely, the amide side chain, hydrophobic group, and core ring substitutions (Table 1). The secondary amide of the amide side chain is critical for maintaining a potent MIC and is hypothesized to be involved in hydrogen bonding with the DprE1 enzyme (compounds 2–10, Table 1). Small hydrophobic amides such as fluoro-ethyl and difluoro ethyl amides are preferred for antimycobacterial activity (compounds 2–10). Based on the knowledge gained from azaindole SAR, the optimal hydrophobic groups were retained in the exploration of SAR with the BI series. Substitution at the C-6 position of the benzimidazole core with methyl or methoxy groups also enhanced antimycobacterial activity, as observed for azaindoles (compound 2 vs compounds 3–10). All resulting BI compounds exhibited potent DprE1 enzyme inhibition and potent Mtb MIC (Table 1).
Table 1. Structure–Activity Relationship of BI Analogues.

The synthesis of benzimidazole analogues is outlined in Schemes 1–3. Synthesis was initiated with the commercially available starting material, A. Benzimidazole was obtained using formic acid and iron to yield B, which was further alkylated with benzyl halide in the presence of cesium carbonate to give compound C. Further hydrolysis of C to acid D followed by amide coupling using HATU afforded target molecule 2 (Scheme 1). The synthesis of the 6-methyl group on the benzimidazole core involved the bromination of A to afford E followed by Suzuki reaction to yield intermediate F. Similar to Scheme 1, intermediate F was used to synthesize compounds 3–6 (Scheme 2). To introduce 6-methoxy functionality on benzimidazole core, the syntheses involved the acetylation of 2-amino-5-methoxybenzoic acid (G) to give intermediate H, which was subsequently hydrolyzed to yield N-acetyl benzoic acid I. Nitration of I using nitronium tetrafluoroborate yielded intermediate J, which was further deprotected with 6 N HCl to obtain K. Reduction followed by cyclization yielded benzimidazole core L, which on esterification gave an intermediate M. Similar to Scheme 1, intermediate M was used to obtain compounds 7–10 (Scheme 3).
Scheme 1. Synthesis of BI Analogues.

Reagents/conditions: (a) HCOOH, Fe, iPrOH, 80%; (b) 4-(chloromethyl)-6-methoxy-5-methylpyrimidine, CsCO3, DMF, 60%; (c) LiOH, MeOH, 80%; (d) HATU, 2-fluoroethanamine, NMP, 60%.
Scheme 3. Synthesis of 6-Substituted BI Analogues.

Reagents/conditions: (a) Ac2O, 90%; (b) H2O, 90%; (c) NO2BF4, CH3CN, 85%; (d) 6 N HCl, 100%; (e) HCOOH, Fe, iPrOH, 85%; (f) H2SO4, MeOH, 90%.
Scheme 2. Synthesis of 6-Substituted BI Intermediate.

Reagents/conditions: (a) AcOH, Br2 90%; (b) 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane, Pd(PPh3)2Cl2, K2CO3, DME, 80%.
The BI analogs and TBA7371 were docked (Glide6.1-Schrodinger)11,12 to the Mtb DprE1 enzyme active site using multiple published crystal structures.13−15 One of the potential binding modes for BI (3) from unconstrained docking is shown in Figure 2A (pdb ID 4KW5 binding site).15 The amide carbonyl oxygen forms an H-bond contact with Ser228 and the amide NH with the cofactor FAD carbonyl oxygen. The core benzimidazole ring is engaged with Trp230 and Tyr314 via CH−π contacts (Figure 2A). The terminal F atom is in close proximity with Asn385 side chain. The substituted pyrimidine is partly exposed to solvent. The hydrophobic contacts with Trp230, Leu263, and val365 may be influencing its conformation. An overlay with the binding mode of azaindole (Figure 2B) revealed a similar binding mode for the amide group. However, the relative disposition of the substituents between the two chemotypes is reflected in the shift of the core rings and conformational change in substituted pyrimidine ring. The presence of multiple polar side chains, hydrophilic centers, and a disordered loop (residues 316–322) in the active site of DprE1 led to alternate possible binding modes for these inhibitors (2, Supporting Information Figure S1). The putative binding mode of BI suggests that wider structural diversity can be accommodated for this target.
Figure 2.
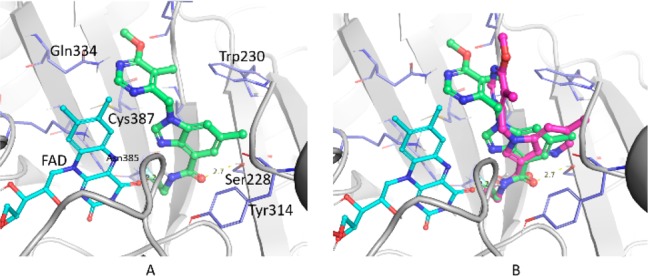
(A) Proposed binding mode of BI (3) in the DprE1 active site. (B) Overlay of docked poses of 1,4-azaindole (1, magenta) and BI (3, green).
From the scaffold morphing output, we decided to test a representative compound for efficacy in a murine model of TB. The microbiological profiling of compound 2 demonstrated a profound bactericidal activity with >3 log10 CFU reduction in 10 days at 1–2× MIC concentration (Figure 3). In an intracellular THP1 macrophage model, ∼1 log10 CFU reduction was observed following treatment with compound 2 in 7 days at MIC concentration (Figure 3). In addition, 2 was active against other mycobacteria like Mycobacterium smegmatis (MIC 0.78–1.56 μM). However, 2 was inactive against a panel of Gram positive and negative bacteria (>200 μM) thereby highlighting their specificity to mycobacterial species. Since compound 2 retained activity against the DprE1 enzyme, the compound was tested for antibacterial activity on available DprE1 related Mtb strains namely—H37Rv overexpressing DprE1; BTZ043 resistant strains: DprE1 with a C387G/S mutation; 1,4-azaindole resistant strain: DprE1 with Y314H mutation. There was a ∼20-fold shift in MIC observed for H37Rv overexpressing DprE1 compared to wild-type Mtb. Also, a similar MIC shift was observed for an Mtb H37Rv strain harboring a DprE1 Y314H mutation, whereas the MIC was retained for H37Rv with DprE1 C387C/S strains (Table 2). Resistant mutants to compound 2 arose at the frequency of 10–7 and 10–8 CFU/mL at 8- and 16-fold MIC concentration, respectively. A ∼10-fold shift in MIC was observed for resistant mutants compared to the wild-type. The dprE1 gene sequenced from clones resistant to compound 2 showed a tyrosine to serine substitution at amino acid position 314.
Figure 3.
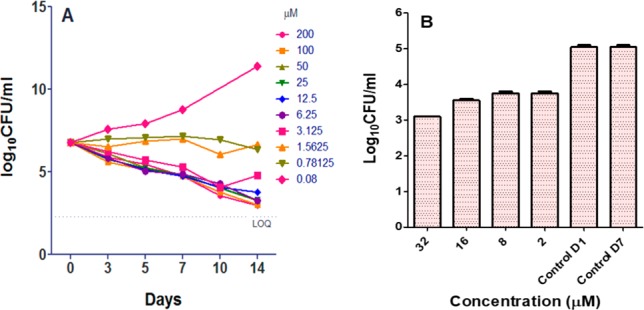
(A) Bactericidality of 2. (B) Intracellular efficacy of 2 in THP1 model.
Table 2. Microbiological Properties of Key Compounds.
| MIC
(μM) |
||||||
|---|---|---|---|---|---|---|
| # | H37Rv | DprE1 OE | DprE1 C387S | DprE1 C387G | DprE1 Y314H | DprE1 Y314S |
| 2 | 1.56–3.12 | 25–50 | 0.78 | 0.78 | 25–50 | 25 |
| BTZ043 | 0.003–0.006 | 12.5 | 25 | 25 | 0.003 | 0.006 |
| isoniazid | 0.44 | 0.22 | 0.44 | 0.44 | 0.44 | 0.22 |
| rifampicin | 0.01 | 0.005 | 0.005 | 0.005 | 0.01 | 0.01 |
The representative compounds from the BI series were profiled for in vitro drug metabolism and pharmacokinetic (DMPK) and in vitro safety properties, as described in Table 4. The measured log D for the BI series ranges between 1.9 and 2.1 (compounds 2 and 9). Measurement of aqueous solubility from DMSO-dried solids for BI compounds (9) was significantly improved compared to the azaindole scaffold (1) (Table 3). The improved solubility could be attributed to the combination of the BI scaffold and 6-(dimethylamino)-5-methylpyrimidine-4-yl group. Moreover, the human plasma protein binding (PPB, % free) for BI compounds 2 and 9 overall shows an improved trend compared to azaindole (Table 3). Based upon in vitro intrinsic clearance (Clint) using human microsomes and rat hepatocytes, the predicted in vivo clearance (CL) for compounds 2 and 9 ranged between 25 and 61% of liver blood flow (% LBF). In contrast, the in vitro intrinsic clearance was higher in the presence of mouse liver microsomes similar to azaindoles (data not shown), suggesting species specific clearance mechanisms. Both series exhibit high permeability with no significant efflux activity as measured in a Caco2 assay and did not show inhibition of cytochrome P450 (CYP) enzymes (Table 3), suggesting a low probability for drug–drug interactions. In vitro safety profiling of representative compound (2) from BI series against a panel of ∼28 high severity human targets and the hERG channel indicated no major safety liabilities (Table 3).
Table 4. Pharmacokinetic Parameters (Mean ± SD, n = 3) of 2 Following Multiple Oral Dose Administration in Mtb Infected Male Balb/C Mice at 10 and 30 mg/kg.
| dose
group (mg/kg) |
||
|---|---|---|
| PK parameter | 10 | 30 |
| Cmax (μM) | 5.1 ± 1.7 | 18.3 ± 6.7 |
| Tmax (h) | 2.0 ± 0.9 | 3.0 ± 0.0 |
| AUCinf (μM.h) | 13.5 ± 1.7 | 78.6 ± 18.9 |
| T1/2 (h) | 4.5 ± 0.2 | 5.9 ± 2.6 |
Table 3. DMPK and Safety Properties of BI Compounds.
| properties | 1 | 2 | 9 | |
|---|---|---|---|---|
| log D | 1.8 | 1.9 | 2 | |
| solubility (μM) | 170 | 152 | >1000 | |
| human CLpred microsomes (% LBF) | 19 | 25 | 61 | |
| rat CLpred hepatocytes (% LBF) | 14 | 25 | 52 | |
| human PPB (% free) | 22 | 32 | 31 | |
| Caco-2 A-B/B-A (1 × 10–6 cm/s) | 11/30 | 37/14 | 26/22 | |
| CYPb inhibition (μM) | >50 | >50 | >50 | |
| hERG (μM) | >33 | >33 | >33 | |
| secondary pharmacology hits | no significant hitsc | NDa | ||
| rat PK | CL (mL/min/kg) | 27 | 48 | ND |
| F (%) | 100 | 114 | ND | |
ND: not determined.
CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4.
Panel of ∼28 high severity targets (binding and functional data): IC50, >100 or >30 μM.
A representative compound from BI series (2) was evaluated for its pharmacokinetic properties in mice and rats. Good oral bioavailability and moderate clearance was observed for compound 2 in rats (Table 3). The exposure in mice was measured in the presence of 1-aminobenzotriazole (ABT), a pan-inhibitor of CYP isoforms used to block mouse specific clearance mechanism (Figure 4A). Significant exposure with plasma levels above MIC for 6–10 h was observed. The bactericidality of compound 2 was assessed in a BALB/c model of “chronic” TB infection, wherein treatment was started 28 days postinfection via the aerosol route.16,17 After 4 weeks of treatment the bacterial burden was reduced by ∼1.5 and ∼1 log10 CFU in lungs and spleen at 30 mg/kg dose, respectively (Figure 4B). The oral exposure of compound 2 assessed from infected animals showed an AUC of 14–79 μM·h (Table 4). No adverse events in the form of body weight loss, organ weight loss or gross pathology were observed following 4 weeks of repeated dosing in the efficacy study.
Figure 4.

(A) Time vs conc. (mean ± SD, n = 3) of 2 following multiple oral dose administration in Mtb infected male Balb/C mice at 10 and 30 mg/kg. (B,C) Efficacy of 2 in chronic TB infection model in BALB/c mice.
We have successfully demonstrated a scaffold morphing approach to discover a novel series of benzimidazoles with potent antimycobacterial activity while retaining DprE1 enzyme inhibition. This novel scaffold exhibited improved solubility and higher free plasma fraction with a representative benzimidazole compound demonstrating efficacy in a chronic model of murine TB.
Acknowledgments
We thank Sudha R. for her contributions in DprE1 overexpression strain. Our sincere thanks to the Global DMPK, Safety, Biosciences (AZ Boston, USA) and Discovery Sciences (AZ, Alderly Park, UK) for technical support in various assays.
Glossary
ABBREVIATIONS
- TB
tuberculosis
- Mtb
Mycobacterium tuberculosis
- DprE1
decaprenylphosphoryl-β-d-ribose2′-epimerase 1
- MDR-TB
multidrug-resistant tuberculosis
- MIC
minimum inhibitory concentration
- SAR
structure–activity relationship
- Msm
Mycobacterium smegmatis
- DMPK
drug metabolism and pharmacokinetic
- Clint
intrinsic clearance
- PPB
plasma protein binding
- CL
clearance
- LBF
liver blood flow
- CYP
cytochrome P450 enzyme
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00343.
Experimental details and 1H and 13C NMR spectra for new synthetic intermediates and products; assay information (PDF)
Author Present Address
○ Biocon Bristol-Myers Squibb Research and Development Centre, Bommasandra Jigani Link Rd, Bangalore-560099, India. Tel: + 91 8698018138.
Author Present Address
∇ Foundation for Neglected Disease Research, 20A, KIADB Industrial Area, Veerapura, Dodda-ballapur, Bengaluru-561203, India.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This research was supported by the Global Alliance for TB Drug Development (GATB).
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization . Global Tuberculosis Report 2018. https://www.who.int/tb/publications/global_report/en/ (accessed September 5, 2019).
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a New Combination Therapy for Tuberculosis with next Generation Benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Möllmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R.; Sadler C.; Naik M.; Hosagrahara V.; Hameed S.; Shinde V.; Bathula C.; Humnabadkar V.; Kumar N.; Reddy J.; Panduga V.; Sharma S.; Ambady A.; Hegde N.; Whiteaker J.; McLaughlin B.; Gardner H.; Madhavapeddi P.; Ramachandran V.; Kaur P.; Narayan A.; Guptha S.; Awasthy D.; Narayan C.; Mahadevaswamy J.; KG V.; Ahuja V.; Srivastava A.; KR P.; Bharath S.; Kale R.; Ramaiah M.; Roy Choudhury N.; Sambandamurthy V.; Solapure S.; Iyer P.; Narayanan S.; Chatterji M. Azaindoles: Noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J. Med. Chem. 2013, 56, 9701–9708. 10.1021/jm401382v. [DOI] [PubMed] [Google Scholar]
- Shirude P. S.; Shandil R.; Manjunatha M. R.; Sadler C.; Panda M.; Panduga V.; Reddy J.; Saralaya R.; Nanduri R.; Ambady A.; Ravishankar S.; Sambandamurthy V.; Humnabadkar V.; Jena L. K.; Rudrapatna S.; Srivastava A.; Prabhakar K. R.; Whiteaker J.; McLaughlin B.; Sharma S.; Cooper C. B.; Mdluli K.; Butler S.; Iyer P.; Narayanan S.; Chatterji M. Lead Optimization of 1,4-Azaindoles as Antimycobacterial Agents. J. Med. Chem. 2014, 57, 5728–5737. 10.1021/jm500571f. [DOI] [PubMed] [Google Scholar]
- Chatterji M.; Shandil R.; Manjunatha M. R.; Solapure S.; Ramachandran V.; Kumar N.; Saralaya R.; Panduga V.; Reddy J.; KR P.; Sharma S.; Sadler C.; Cooper C. B.; Mdluli K.; Iyer P. S.; Narayanan S.; Shirude P. S. 1,4-Azaindole, A Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. 10.1128/AAC.03233-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A Phase 1 Study to Evaluate Safety, Tolerability, PK, and PK Interactions of TBA-7371. https://clinicaltrials.gov/ct2/show/NCT03199339.
- Panda M.; Ramachandran S.; Ramachandran V.; Shirude P. S.; Humnabadkar V.; Nagalapur K.; Sharma S.; Kaur P.; Guptha S.; Narayan A.; Mahadevaswamy J.; Ambady A.; Hegde N.; Rudrapatna S.; Hosagrahara V. P.; Sambandamurthy V.; Raichurkar A. J. Med. Chem. 2014, 57, 4761–4771. 10.1021/jm5002937. [DOI] [PubMed] [Google Scholar]
- Mikušová K.; Huang H.; Yagi T.; Holsters M.; Vereecke D.; D’Haeze W.; Scherman M. S.; Brennan P. J.; McNeil M. R.; Crick D. C. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 2005, 187, 8020–8025. 10.1128/JB.187.23.8020-8025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhale R. V.; Barmade M. A.; Murumkar P. R.; Yadav M. R. Overview of the Development of DprE1 Inhibitors for Combating the Menace of Tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. 10.1021/acs.jmedchem.8b00281. [DOI] [PubMed] [Google Scholar]
- Small-Molecule Drug Discovery Suite 2013–3: Glide, version 6.1; Schrödinger, LLC: New York, 2013.
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Neres J.; Pojer F.; Molteni E.; Chiarelli L. R.; Dhar N.; Boy-Röttger S.; Buroni S.; Fullam E.; Degiacomi G.; Lucarelli A. P.; Read R. J.; Zanoni G.; Edmondson D. E.; De Rossi E.; Pasca M. R.; McKinney J. D.; Dyson P. J.; Riccardi G.; Mattevi A.; Cole S. T.; Binda C. Structural Basis for Benzothiazinone-Mediated Killing of. Sci. Transl. Med. 2012, 4, 150ra121. 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batt S. M.; Jabeen T.; Bhowruth V.; Quill L.; Lund P. A.; Eggeling L.; Alderwick L. J.; Futterer K.; Besra G. S. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11354–11359. 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Sambandan D.; Halder R.; Wang J.; Batt S. M.; Weinrick B.; Ahmad I.; Yang P.; Zhang Y.; Kim J.; Hassani M.; Huszar S.; Trefzer C.; Ma Z.; Kaneko T.; Mdluli K. E.; Franzblau S.; Chatterjee A. K.; Johnsson K.; Mikusova K.; Besra G. S.; Fütterer K.; Robbins S. H.; Barnes S. W.; Walker J. R.; Jacobs W. R. Jr; Schultz P. G. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E2510–E2517. 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram R.; Gaonkar S.; Kaur P.; Suresh B. L.; Mahesh B. N.; Jayashree R.; Nandi V.; Bharat S.; Shandil R. K.; Kantharaj E.; Balasubramanian V. Pharmacokinetics-Pharmacodynamics of Rifampin in an Aerosol Infection Model of Tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 2118–2124. 10.1128/AAC.47.7.2118-2124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groote M. A.; Gilliland J. C.; Wells C. L.; Brooks E. J.; Woolhiser L. K.; Gruppo V.; Peloquin C. A.; Orme I. A.; Lenaerts A. J. Comparative Studies Evaluating Mouse Models Used for Efficacy Testing of Experimental Drugs against. Antimicrob. Agents Chemother. 2011, 55, 1237–1247. 10.1128/AAC.00595-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.